Mining Public Mass Spectrometry Data to Characterize the Diversity and Ubiquity of P. aeruginosa Specialized Metabolites

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
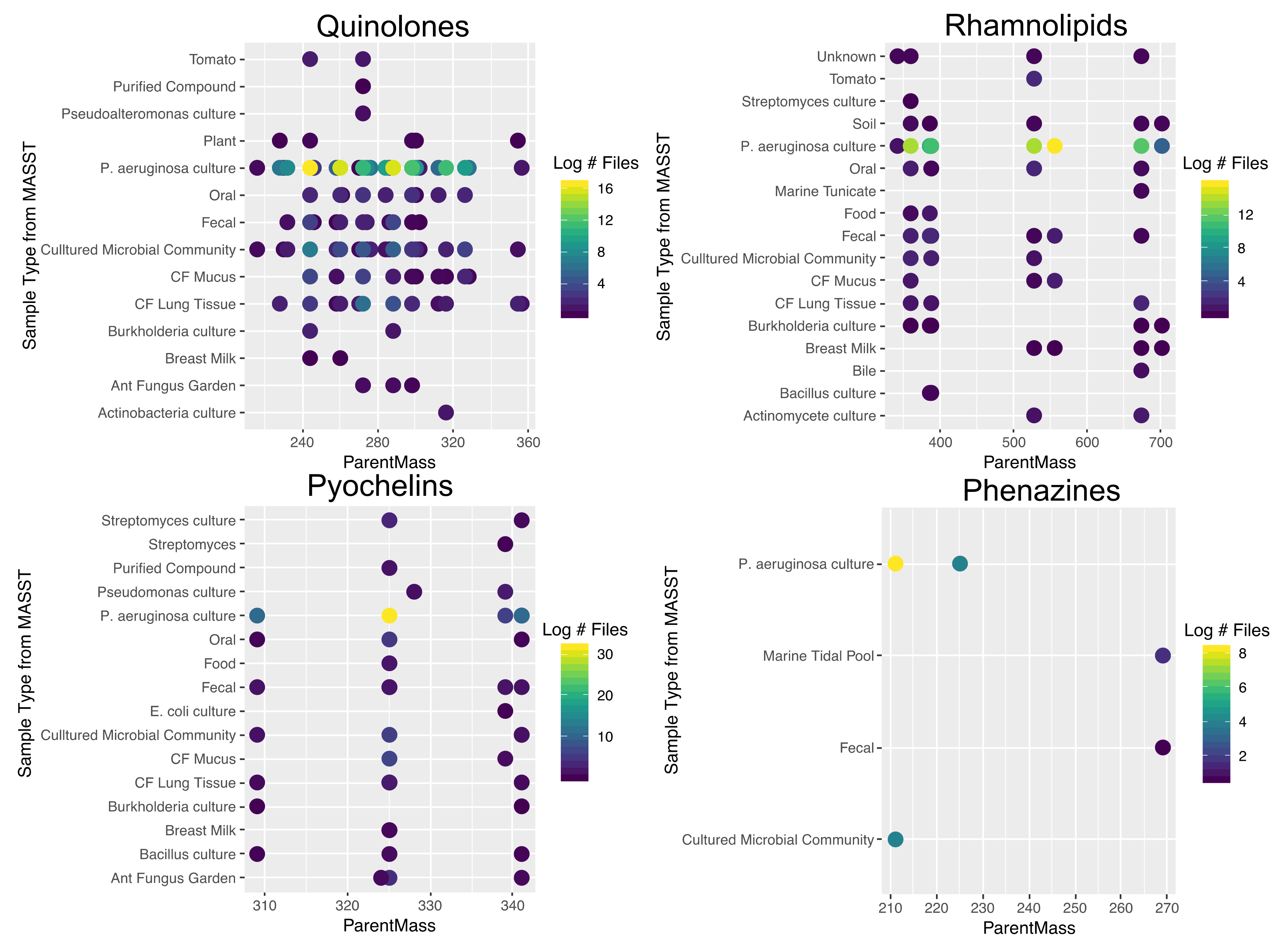
2.1. Quinolone Diversity
2.2. Rhamnolipid Diversity
2.3. Phenazine Diversity
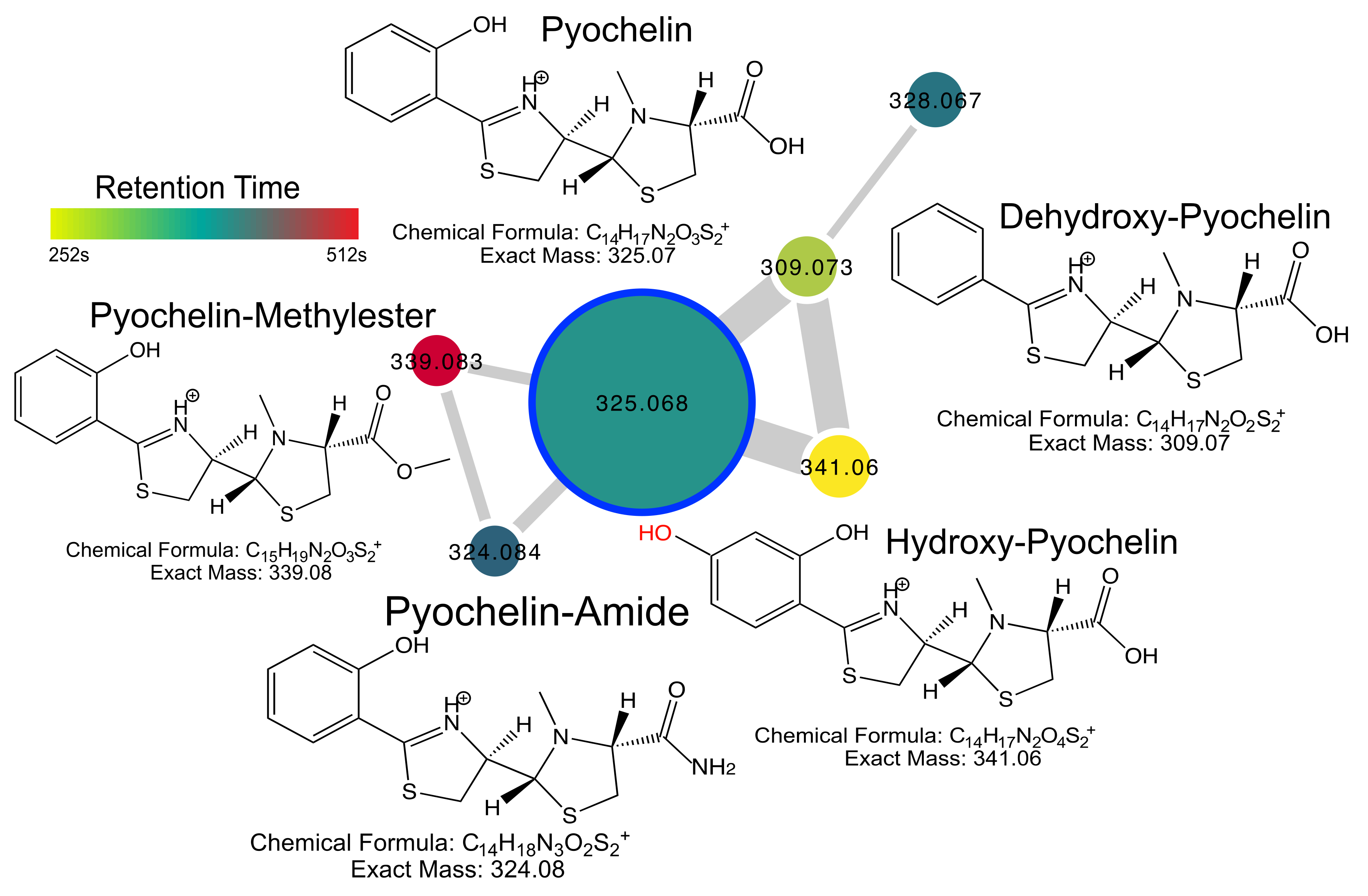
2.4. Pyochelin Diversity
2.5. Ubiquity of P. aeruginosa Specialized Metabolites
3. Discussion
4. Materials and Methods
4.1. P. aeruginosa Strains and Culture Conditions
4.2. Metabolite Extraction
4.3. LC-MS/MS Mass Spectrometry
4.4. Molecular Networking and MASST Searching
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef]
- Watrous, J.; Roach, P.; Alexandrov, T.; Heath, B.S.; Yang, J.Y.; Kersten, R.D.; van der Voort, M.; Pogliano, K.; Gross, H.; Raaijmakers, J.M.; et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. USA 2012, 109, 1743–1752. [Google Scholar] [CrossRef]
- Wang, M.; Jarmusch, A.K.; Vargas, F.; Aksenov, A.A.; Gauglitz, J.M.; Weldon, K.; Petras, D.; da Silva, R.; Quinn, R.; Melnik, A.V.; et al. Mass spectrometry searches using MASST. Nat. Biotechnol. 2020, 38, 23–26. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Quinn, R.A.; Phelan, V.V.; Whiteson, K.L.; Garg, N.; Bailey, B.A.; Lim, Y.W.; Conrad, D.J.; Dorrestein, P.C.; Rohwer, F.L. Microbial, host and xenobiotic diversity in the cystic fibrosis sputum metabolome. ISME J. 2015, 10, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Schaefer, A.L.; Parsek, M.R.; Moninger, T.O.; Welsh, M.J.; Greenberg, E.P. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 2000, 407, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Moree, W.J.; Phelan, V.V.; Wu, C.-H.; Bandeira, N.; Cornett, D.S.; Duggan, B.M.; Dorrestein, P.C. Interkingdom metabolic transformations captured by microbial imaging mass spectrometry. Proc. Natl. Acad. Sci. USA 2012, 109, 13811–13816. [Google Scholar] [CrossRef] [PubMed]
- Lépine, F.; Milot, S.; Déziel, E.; He, J.; Rahme, L.G. Electrospray/mass spectrometric identification and analysis of 4-hydroxy-2-alkylquinolines (HAQs) produced by Pseudomonas aeruginosa. J. Am. Soc. Mass Spectrom. 2004, 15, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Deziel, E.; Lepine, F.; Milot, S.; Villemur, R. rhlA Is Required for the Production of a Novel Biosurfactant Promoting Swarming Motility in Pseudomonas aeruginosa: 3-(3-hydroxyalkanoyloxy)alkanoic Acids (HAAs), the Precursors of Rhamnolipids. Microbiology 2003, 149, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Ankenbauer, R.G.; Toyokuni, T.; Staley, A.; Rinehart, K.L.; Cox, C.D. Synthesis and biological activity of pyochelin, a siderophore of Pseudomonas aeruginosa. J. Bacteriol. 1988, 170, 5344–5351. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.D.; Melnik, A.V.; Koyama, N.; Lu, X.; Schorn, M.; Fang, J.; Aguinaldo, K.; Lincecum, T.L.; Ghequire, M.G.K.; Carrion, V.J.; et al. Indexing the Pseudomonas specialized metabolome enabled the discovery of poaeamide B and the bananamides. Nat. Microbiol. 2016, 2, 16197. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.; Palethorpe, S.; Pesci, E.C. PsrA controls the synthesis of the Pseudomonas aeruginosa quinolone signal via repression of the FadE homolog, PA0506. PLoS ONE 2017, 12, e0189331. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Yang, M.W.; Zhou, A.; Szulc, Z.; Davies, C. Biochemical characterization of PqsD activity in alkylquinolone biosynthesis in Pseudomonas aeruginosa. FASEB J. 2012, 26, 964.7. [Google Scholar] [CrossRef]
- Bredenbruch, F.; Nimtz, M.; Wray, V.; Morr, M.; Müller, R.; Häussler, S. Biosynthetic Pathway of Pseudomonas aeruginosa 4-Hydroxy-2-Alkylquinolines. J. Bacteriol. 2005, 187, 3630 LP–3635 LP. [Google Scholar] [CrossRef]
- Depke, T.; Thöming, J.G.; Kordes, A.; Häussler, S.; Brönstrup, M. Untargeted LC-MS Metabolomics Differentiates between Virulent and Avirulent Clinical Strains of Pseudomonas aeruginosa. Biomolecules 2020, 10, 1041. [Google Scholar] [CrossRef]
- Diggle, S.P.; Lumjiaktase, P.; Dipilato, F.; Winzer, K.; Kunakorn, M.; Barrett, D.A.; Chhabra, S.R.; Cámara, M.; Williams, P. Functional genetic analysis reveals a 2-Alkyl-4-quinolone signaling system in the human pathogen Burkholderia pseudomallei and related bacteria. Chem. Biol. 2006, 13, 701–710. [Google Scholar] [CrossRef]
- Kim, W.J.; Kim, Y.O.; Kim, J.H.; Nam, B.-H.; Kim, D.-G.; An, C.M.; Lee, J.S.; Kim, P.S.; Lee, H.M.; Oh, J.-S.; et al. Liquid Chromatography-Mass Spectrometry-Based Rapid Secondary-Metabolite Profiling of Marine Pseudoalteromonas sp. M2. Mar. Drugs 2016, 14, 24. [Google Scholar] [CrossRef]
- Markou, P.; Apidianakis, Y. Pathogenesis of intestinal Pseudomonas aeruginosa infection in patients with cancer. Front. Cell. Infect. Microbiol. 2014, 3, 115. [Google Scholar] [CrossRef] [PubMed]
- Raghuvanshi, R.; Vasco, K.; Vázquez-Baeza, Y.; Jiang, L.; Morton, J.T.; Li, D.; Gonzalez, A.; DeRight Goldasich, L.; Humphrey, G.; Ackermann, G.; et al. High-Resolution Longitudinal Dynamics of the Cystic Fibrosis Sputum Microbiome and Metabolome through Antibiotic Therapy. mSystems 2020, 5, e00292-20. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.A.; Adem, S.; Mills, R.H.; Comstock, W.; DeRight Goldasich, L.; Humphrey, G.; Aksenov, A.A.; Melnik, A.V.; da Silva, R.; Ackermann, G.; et al. Neutrophilic proteolysis in the cystic fibrosis lung correlates with a pathogenic microbiome. Microbiome 2019, 7, 23. [Google Scholar] [CrossRef]
- Quinn, R.A.; Navas-Molina, J.A.; Hyde, E.R.; Song, S.J.; Vázquez-Baeza, Y.; Humphrey, G.; Gaffney, J.; Minich, J.J.; Melnik, A.V.; Herschend, J.; et al. From sample to multi-omics conclusions in under 48 h. mSystems 2016, 1, e00038-16. [Google Scholar] [CrossRef] [PubMed]
- Seipke, R.F.; Song, L.; Bicz, J.; Laskaris, P.; Yaxley, A.M.; Challis, G.L.; Loria, R. The plant pathogen Streptomyces scabies 87-22 has a functional pyochelin biosynthetic pathway that is regulated by TetR- and AfsR-family proteins. Microbiology 2011, 157, 2681–2693. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.N.; Li, Q. Microbial production of rhamnolipids using sugars as carbon sources. Microb. Cell Fact. 2018, 17, 89. [Google Scholar] [CrossRef] [PubMed]
- Brillet, K.; Reimmann, C.; Mislin, G.L.A.; Noël, S.; Rognan, D.; Schalk, I.J.; Cobessi, D. Pyochelin Enantiomers and Their Outer-Membrane Siderophore Transporters in Fluorescent Pseudomonads: Structural Bases for Unique Enantiospecific Recognition. J. Am. Chem. Soc. 2011, 133, 16503–16509. [Google Scholar] [CrossRef]
- Terano, H.; Nomoto, K.; Takase, S. Siderophore production and induction of iron-regulated proteins by a microorganism from rhizosphere of barley. Biosci. Biotechnol. Biochem. 2002, 66, 2471–2473. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cox, C.D.; Rinehart, K.L.J.; Moore, M.L.; Cook, J.C.J. Pyochelin: Novel structure of an iron-chelating growth promoter for Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1981, 78, 4256–4260. [Google Scholar] [CrossRef] [PubMed]
- Quadri, L.E.; Keating, T.A.; Patel, H.M.; Walsh, C.T. Assembly of the Pseudomonas aeruginosa nonribosomal peptide siderophore pyochelin: In vitro reconstitution of aryl-4, 2-bisthiazoline synthetase activity from PchD, PchE, and PchF. Biochemistry 1999, 38, 14941–14954. [Google Scholar] [CrossRef]
- Raghuvanshi, R.; Grayson, A.G.; Schena, I.; Amanze, O.; Suwintono, K.; Quinn, R.A. Microbial Transformations of Organically Fermented Foods. Metabolites 2019, 9, 165. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lybbert, A.C.; Williams, J.L.; Raghuvanshi, R.; Jones, A.D.; Quinn, R.A. Mining Public Mass Spectrometry Data to Characterize the Diversity and Ubiquity of P. aeruginosa Specialized Metabolites. Metabolites 2020, 10, 445. https://doi.org/10.3390/metabo10110445
Lybbert AC, Williams JL, Raghuvanshi R, Jones AD, Quinn RA. Mining Public Mass Spectrometry Data to Characterize the Diversity and Ubiquity of P. aeruginosa Specialized Metabolites. Metabolites. 2020; 10(11):445. https://doi.org/10.3390/metabo10110445
Chicago/Turabian StyleLybbert, Andrew C., Justin L. Williams, Ruma Raghuvanshi, A. Daniel Jones, and Robert A. Quinn. 2020. "Mining Public Mass Spectrometry Data to Characterize the Diversity and Ubiquity of P. aeruginosa Specialized Metabolites" Metabolites 10, no. 11: 445. https://doi.org/10.3390/metabo10110445
APA StyleLybbert, A. C., Williams, J. L., Raghuvanshi, R., Jones, A. D., & Quinn, R. A. (2020). Mining Public Mass Spectrometry Data to Characterize the Diversity and Ubiquity of P. aeruginosa Specialized Metabolites. Metabolites, 10(11), 445. https://doi.org/10.3390/metabo10110445