
Metabolic Versatility of Mycobacterium tuberculosis during Infection and Dormancy


Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
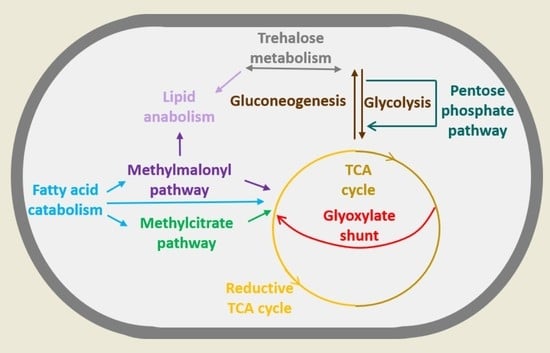
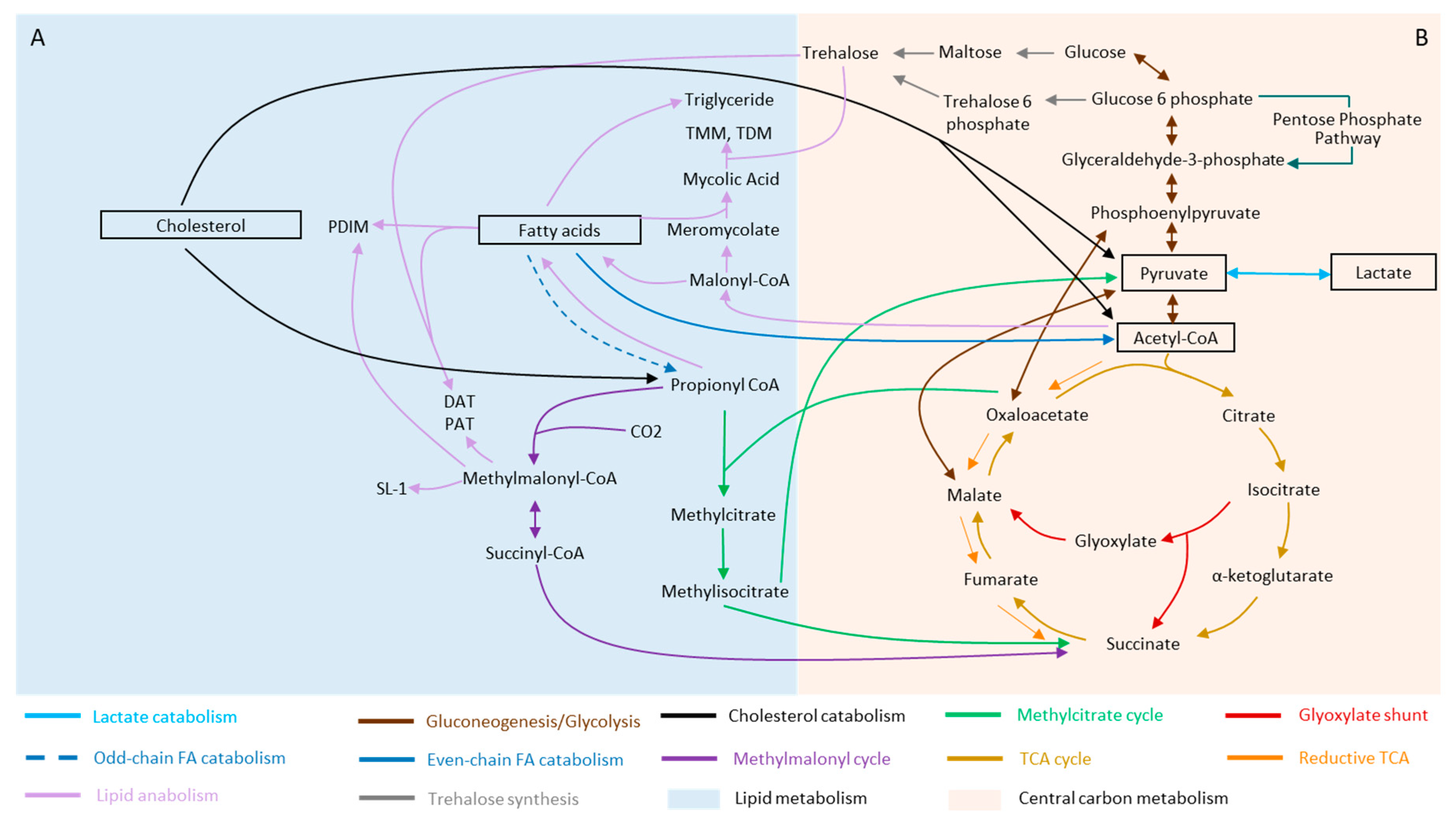
2. Mycobacterium and Its Metabolic Versatility during Infection
2.1. Fatty Acids
2.2. Cholesterol
2.3. Lactate and Pyruvate
3. Remodeling of Metabolism during Stress and Dormancy
3.1. Rewiring of Carbon Metabolism during Stress and Dormancy
3.2. Lipid Metabolism of Mtb and Its Remodeling during Stress and Dormancy
3.2.1. Fatty Acyls (FA)
3.2.2. Glycerolipids (GLs)
3.2.3. Glycerophospholipids (GPs)
3.2.4. Saccharolipids
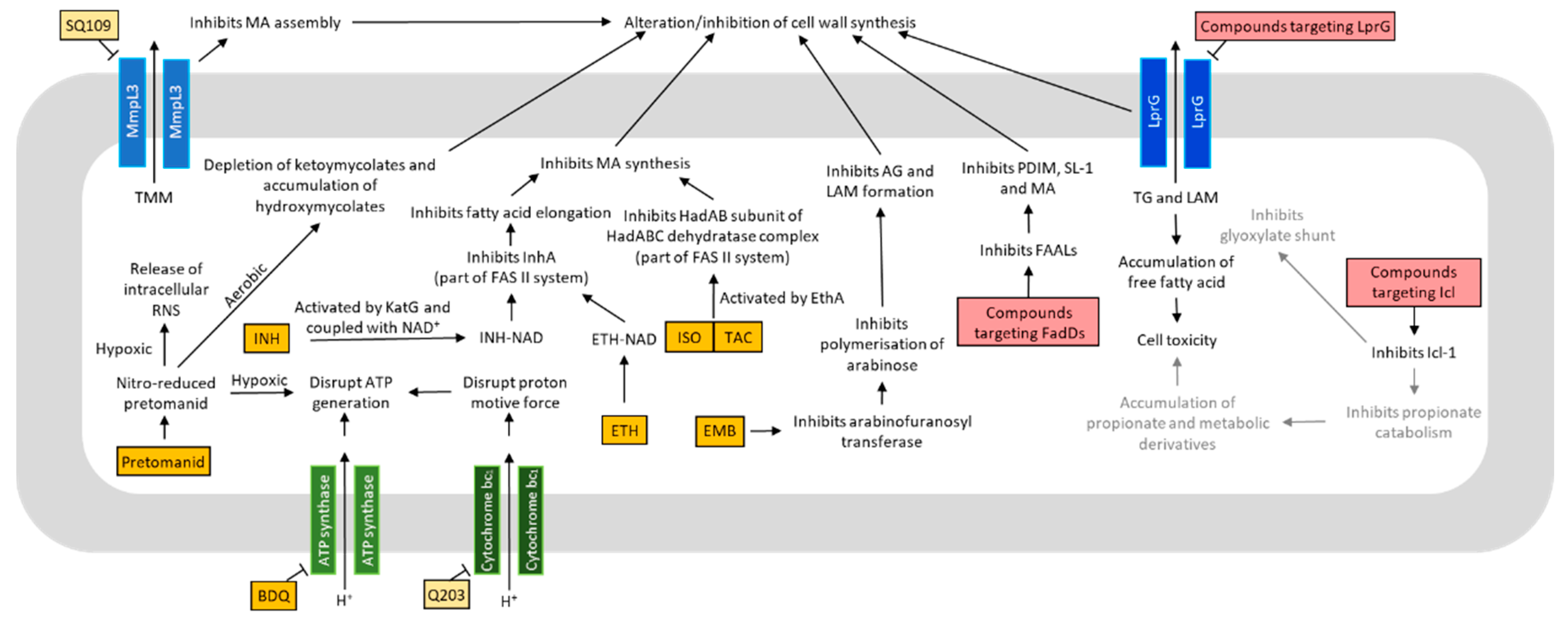
4. Metabolism in Drug Discovery
4.1. Oxidative Phosphorylation and ATP Production
4.2. Lipid Metabolism
5. Systems Biology Methodology and Novel Applications in Mtb Research
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Cambier, C.J.; Falkow, S.; Ramakrishnan, L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell 2014, 159, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Mathiasen, V.D.; Schön, T.; Wejse, C. The global prevalence of latent tuberculosis: A systematic review and meta-analysis. Eur. Respir. J. 2019, 54, 1900655. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Sassetti, C.M. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. USA 2008, 105, 4376–4380. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, L.P.S.; Fischer, S.M.; Marrero, J.; Nathan, C.; Ehrt, S.; Rhee, K.Y. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem. Biol. 2010, 17, 1122–1131. [Google Scholar] [CrossRef]
- Zimmermann, M.; Kogadeeva, M.; Gengenbacher, M.; McEwen, G.; Mollenkopf, H.-J.; Zamboni, N.; Kaufmann, S.H.E.; Sauer, U. Integration of Metabolomics and Transcriptomics Reveals a Complex Diet of Mycobacterium tuberculosis during Early Macrophage Infection. MSystems 2017, 2, 1–18. [Google Scholar] [CrossRef]
- Beste, D.J.V.; Nöh, K.; Niedenführ, S.; Mendum, T.A.; Hawkins, N.D.; Ward, J.L.; Beale, M.H.; Wiechert, W.; McFadden, J. 13C-flux spectral analysis of host-pathogen metabolism reveals a mixed diet for intracellular Mycobacterium tuberculosis. Chem. Biol. 2013, 20, 1012–1021. [Google Scholar] [CrossRef]
- Gengenbacher, M.; Kaufmann, S.H. Mycobacterium tuberculosis: Success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–532. [Google Scholar] [CrossRef]
- Caño-Muñiz, S.; Anthony, R.; Niemann, S.; Alffenaar, J.-W.C. New Approaches and Therapeutic Options for Mycobacterium tuberculosis in a Dormant State. Clin. Microbiol. Rev. 2018, 31, e00060-00017. [Google Scholar] [CrossRef]
- Chao, M.C.; Rubin, E.J. Letting Sleeping dos Lie: Does Dormancy Play a Role in Tuberculosis? Annu. Rev. Microbiol. 2010, 64, 293–311. [Google Scholar] [CrossRef]
- Lipworth, S.; Hammond, R.J.H.; Baron, V.O.; Hu, Y.; Coates, A.; Gillespie, S.H. Defining dormancy in mycobacterial disease. Tuberculosis 2016, 99, 131–142. [Google Scholar] [CrossRef]
- Lewis, K. Persister Cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Ehrt, S.; Schnappinger, D.; Rhee, K.Y. Metabolic principles of persistence and pathogenicity in Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2018, 16, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.M.; Lopatkin, A.J.; Lobritz, M.A.; Collins, J.J. Bacterial Metabolism and Antibiotic Efficacy. Cell Metab. 2019, 30, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Shetye, G.S.; Franzblau, S.G.; Cho, S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl. Res. 2020, 220, 68–97. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Sviriaeva, E.; Pethe, K. Targeting the cytochrome oxidases for drug development in mycobacteria. Prog. Biophys. Mol. Biol. 2020, 152, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Bailo, R.; Bhatt, A.; Aínsa, J.A. Lipid transport in Mycobacterium tuberculosis and its implications in virulence and drug development. Biochem. Pharm. 2015, 96, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.L.; Chan, J.; Lin, P.L. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol. 2011, 4, 271–278. [Google Scholar] [CrossRef]
- Görke, B.; Stülke, J. Carbon catabolite repression in bacteria: Many ways to make the most out of nutrients. Nat. Rev. Microbiol. 2008, 6, 613–624. [Google Scholar] [CrossRef]
- Bloch, H.; Segal, W. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J. Bacteriol. 1956, 72, 132–141. [Google Scholar] [CrossRef]
- Schnappinger, D.; Ehrt, S.; Voskuil, M.I.; Liu, Y.; Mangan, J.A.; Monahan, I.M.; Dolganov, G.; Efron, B.; Butcher, P.D.; Nathan, C.; et al. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J. Exp. Med. 2003, 198, 693–704. [Google Scholar] [CrossRef]
- Talaat, A.M.; Lyons, R.; Howard, S.T.; Johnston, S.A. The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 4602–4607. [Google Scholar] [CrossRef] [PubMed]
- Rohde, K.H.; Veiga, D.F.T.; Caldwell, S.; Balázsi, G.; Russell, D.G. Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog. 2012, 8, e1002769. [Google Scholar] [CrossRef] [PubMed]
- Nazarova, E.V.; Montague, C.R.; La, T.; Wilburn, K.M.; Sukumar, N.; Lee, W.; Caldwell, S.; Russell, D.G.; VanderVen, B.C. Rv3723/LucA coordinates fatty acid and cholesterol uptake in Mycobacterium tuberculosis. eLife 2017, 6, e26969. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Rubin, E.J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 2003, 100, 12989. [Google Scholar] [CrossRef] [PubMed]
- McCann, J.R.; McDonough, J.A.; Sullivan, J.T.; Feltcher, M.E.; Braunstein, M. Genome-wide identification of Mycobacterium tuberculosis exported proteins with roles in intracellular growth. J. Bacteriol. 2011, 193, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.R.; Patel, J.; Robertson, B.D.; Rae, A.; Young, D.B. Mycobacterial mutants with defective control of phagosomal acidification. PLoS Pathog. 2005, 1, 269–278. [Google Scholar] [CrossRef]
- Gioffré, A.; Infante, E.; Aguilar, D.; Santangelo, M.D.l.P.; Klepp, L.; Amadio, A.; Meikle, V.; Etchechoury, I.; Romano, M.I.; Cataldi, A.; et al. Mutation in mce operons attenuates Mycobacterium tuberculosis virulence. Microbes Infect. 2005, 7, 325–334. [Google Scholar] [CrossRef]
- Joshi, S.M.; Pandey, A.K.; Capite, N.; Fortune, S.M.; Rubin, E.J.; Sassetti, C.M. Characterization of mycobacterial virulence genes through genetic interaction mapping. Proc. Natl. Acad. Sci. USA 2006, 103, 11760–11765. [Google Scholar] [CrossRef]
- Queiroz, A.; Medina-Cleghorn, D.; Marjanovic, O.; Nomura, D.K.; Riley, L.W. Comparative metabolic profiling of mce1 operon mutant vs wild-type Mycobacterium tuberculosis strains. Pathog. Dis. 2015, 73. [Google Scholar] [CrossRef]
- Muñoz-Elías, E.J.; Upton, A.M.; Cherian, J.; McKinney, J.D. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol. Microbiol. 2006, 60, 1109–1122. [Google Scholar] [CrossRef]
- Gould, T.A.; van de Langemheen, H.; Muñoz-Elías, E.J.; McKinney, J.D.; Sacchettini, J.C. Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis. Mol Microbiol. 2006, 61, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Savvi, S.; Warner, D.F.; Kana, B.D.; McKinney, J.D.; Mizrahi, V.; Dawes, S.S. Functional Characterization of a Vitamin B12-Dependent Methylmalonyl Pathway in Mycobacterium tuberculosis: Implications for Propionate Metabolism during Growth on Fatty Acids. J. Bacteriol. 2008, 190, 3886. [Google Scholar] [CrossRef] [PubMed]
- Minnikin, D.E.; Kremer, L.; Dover, L.G.; Besra, G.S. The Methyl-Branched Fortifications of Mycobacterium tuberculosis. Chem. Biol. 2002, 9, 545–553. [Google Scholar] [CrossRef]
- Marrero, J.; Rhee, K.Y.; Schnappinger, D.; Pethe, K.; Ehrt, S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc. Natl. Acad. Sci. USA 2010, 107, 9819–9824. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Elías, E.J.; McKinney, J.D. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat. Med. 2005, 11, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Brzostek, A.; Pawelczyk, J.; Rumijowska-Galewicz, A.; Dziadek, B.; Dziadek, J. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J. Bacteriol. 2009, 191, 6584–6591. [Google Scholar] [CrossRef]
- Griffin, J.E.; Pandey, A.K.; Gilmore, S.A.; Mizrahi, V.; McKinney, J.D.; Bertozzi, C.R.; Sassetti, C.M. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem. Biol. 2012, 19, 218–227. [Google Scholar] [CrossRef]
- Hu, Y.; Van Der Geize, R.; Besra, G.S.; Gurcha, S.S.; Liu, A.; Rohde, M.; Singh, M.; Coates, A. 3-Ketosteroid 9α-hydroxylase is an essential factor in the pathogenesis of Mycobacterium tuberculosis. Mol. Microbiol. 2010, 75, 107–121. [Google Scholar] [CrossRef]
- Nesbitt, N.M.; Yang, X.; Fontán, P.; Kolesnikova, I.; Smith, I.; Sampson, N.S.; Dubnau, E. A thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infect. Immun. 2010, 78, 275–282. [Google Scholar] [CrossRef]
- Yam, K.C.; D’Angelo, I.; Kalscheuer, R.; Zhu, H.; Wang, J.X.; Snieckus, V.; Ly, L.H.; Converse, P.J.; Jacobs, W.R.; Strynadka, N.; et al. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 2009, 5, e1000344. [Google Scholar] [CrossRef]
- VanderVen, B.C.; Fahey, R.J.; Lee, W.; Liu, Y.; Abramovitch, R.B.; Memmott, C.; Crowe, A.M.; Eltis, L.D.; Perola, E.; Deininger, D.D.; et al. Novel Inhibitors of Cholesterol Degradation in Mycobacterium tuberculosis Reveal How the Bacterium’s Metabolism Is Constrained by the Intracellular Environment. PLoS Pathog. 2015, 11, e1004679. [Google Scholar] [CrossRef] [PubMed]
- Fineran, P.; Lloyd-Evans, E.; Lack, N.A.; Platt, N.; Davis, L.C.; Morgan, A.J.; Höglinger, D.; Tatituri, R.V.V.; Clark, S.; Williams, I.M.; et al. Pathogenic mycobacteria achieve cellular persistence by inhibiting the Niemann-Pick Type C disease cellular pathway. Wellcome Open Res. 2016, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Billig, S.; Schneefeld, M.; Huber, C.; Grassl, G.A.; Eisenreich, W.; Bange, F.C. Lactate oxidation facilitates growth of Mycobacterium tuberculosis in human macrophages. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Serafini, A.; Tan, L.; Horswell, S.; Howell, S.; Greenwood, D.J.; Hunt, D.M.; Phan, M.D.; Schembri, M.; Monteleone, M.; Montague, C.R.; et al. Mycobacterium tuberculosis requires glyoxylate shunt and reverse methylcitrate cycle for lactate and pyruvate metabolism. Mol. Microbiol. 2019, 112, 1284–1307. [Google Scholar] [CrossRef]
- Keating, L.A.; Wheeler, P.R.; Mansoor, H.; Inwald, J.K.; Dale, J.; Hewinson, R.G.; Gordon, S.V. The pyruvate requirement of some members of the Mycobacterium tuberculosis complex is due to an inactive pyruvate kinase: Implications for in vivo growth. Mol. Microbiol. 2005, 56, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Speer, A.; Sun, J.; Danilchanka, O.; Meikle, V.; Rowland, J.L.; Walter, K.; Buck, B.R.; Pavlenok, M.; Hölscher, C.; Ehrt, S.; et al. Surface hydrolysis of sphingomyelin by the outer membrane protein Rv0888 supports replication of Mycobacterium tuberculosis in macrophages. Mol. Microbiol. 2015, 97, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Van der Wel, N.; Hava, D.; Houben, D.; Fluitsma, D.; van Zon, M.; Pierson, J.; Brenner, M.; Peters, P.J. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 2007, 129, 1287–1298. [Google Scholar] [CrossRef]
- Simeone, R.; Bobard, A.; Lippmann, J.; Bitter, W.; Majlessi, L.; Brosch, R.; Enninga, J. Phagosomal Rupture by Mycobacterium tuberculosis Results in Toxicity and Host Cell Death. PLoS Pathog. 2012, 8, e1002507. [Google Scholar] [CrossRef]
- Simeone, J.C.; Ward, A.J.; Rotella, P.; Collins, J.; Windisch, R. An evaluation of variation in published estimates of schizophrenia prevalence from 1990–2013: A systematic literature review. BMC Psychiatry 2015, 15, 193. [Google Scholar] [CrossRef]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002, 43, 717–731. [Google Scholar] [CrossRef]
- Wayne, L.G.; Hayes, L.G. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 1996, 64, 2062–2069. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.P.S.; Alonso, S.; Rand, L.; Dick, T.; Pethe, K. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2008, 105, 11945–11950. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Guidry, L.; Narasimhulu, K.V.; Mai, D.; Trombley, J.; Redding, K.E.; Giles, G.I.; Lancaster, J.R., Jr.; Steyn, A.J. Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe-4S] cluster and is essential for nutrient starvation survival. Proc. Natl. Acad. Sci. USA 2007, 104, 11562–11567. [Google Scholar] [CrossRef] [PubMed]
- Rustad, T.R.; Harrell, M.I.; Liao, R.; Sherman, D.R. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS ONE 2008, 3, e1502. [Google Scholar] [CrossRef]
- Leistikow, R.L.; Morton, R.A.; Bartek, I.L.; Frimpong, I.; Wagner, K.; Voskuil, M.I. The Mycobacterium tuberculosis DosR Regulon Assists in Metabolic Homeostasis and Enables Rapid Recovery from Nonrespiring Dormancy. J. Bacteriol. 2010, 192, 1662. [Google Scholar] [CrossRef]
- Schubert, O.T.; Ludwig, C.; Kogadeeva, M.; Zimmermann, M.; Rosenberger, G.; Gengenbacher, M.; Gillet, L.C.; Collins, B.C.; Röst, H.L.; Kaufmann, S.H.; et al. Absolute Proteome Composition and Dynamics during Dormancy and Resuscitation of Mycobacterium tuberculosis. Cell Host Microbe 2015, 18, 96–108. [Google Scholar] [CrossRef]
- Peterson, E.J.R.; Abidi, A.A.; Arrieta-Ortiz, M.L.; Aguilar, B.; Yurkovich, J.T.; Kaur, A.; Pan, M.; Srinivas, V.; Shmulevich, I.; Baliga, N.S. Intricate Genetic Programs Controlling Dormancy in Mycobacterium tuberculosis. Cell Rep. 2020, 31, 107577. [Google Scholar] [CrossRef]
- Tian, J.; Bryk, R.; Itoh, M.; Suematsu, M.; Nathan, C. Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: Identification of alpha-ketoglutarate decarboxylase. Proc. Natl. Acad. Sci. USA 2005, 102, 10670–10675. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Tian, J.; Bryk, R.; Shi, S.; Erdjument-Bromage, H.; Tempst, P.; Nathan, C. Mycobacterium tuberculosis appears to lack alpha-ketoglutarate dehydrogenase and encodes pyruvate dehydrogenase in widely separated genes. Mol. Microbiol. 2005, 57, 859–868. [Google Scholar] [CrossRef]
- Gest, H. Evolution of the citric acid cycle and respiratory energy conversion in prokaryotes. FEMS Microbiol. Lett. 1981, 12, 209–215. [Google Scholar] [CrossRef]
- Eoh, H.; Rhee, K.Y. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, 6554–6559. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Elías, E.J.; McKinney, J.D. Carbon metabolism of intracellular bacteria. Cell. Microbiol. 2006, 8, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, M.; Nathan, C.; Rhee, K.Y. Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Zimmermann, M.; Goodwin, M.B.; Sauer, U.; Barry, C.E.; Boshoff, H.I. Fumarate reductase activity maintains an energized membrane in anaerobic Mycobacterium tuberculosis. PLoS Pathog. 2011, 7, e1002287. [Google Scholar] [CrossRef] [PubMed]
- Larrouy-Maumus, G.; Marino, L.B.; Madduri, A.V.R.; Ragan, T.J.; Hunt, D.M.; Bassano, L.; Gutierrez, M.G.; Moody, D.B.; Pavan, F.R.; De Carvalho, L.P.S. Cell-envelope remodeling as a determinant of phenotypic antibacterial tolerance in Mycobacterium tuberculosis. ACS Infect. Dis. 2016, 2, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Eoh, H.; Wang, Z.; Layre, E.; Rath, P.; Morris, R.; Branch Moody, D.; Rhee, K.Y. Metabolic anticipation in Mycobacterium tuberculosis. Nat. Microbiol. 2017, 2, 1–7. [Google Scholar] [CrossRef]
- Zhong, W.; Guo, J.; Cui, L.; Chionh, Y.H.; Li, K.; El Sahili, A.; Cai, Q.; Yuan, M.; Michels, P.A.M.; Fothergill-Gilmore, L.A.; et al. Pyruvate Kinase Regulates the Pentose-Phosphate Pathway in Response to Hypoxia in Mycobacterium tuberculosis. J. Mol. Biol. 2019, 431, 3690–3705. [Google Scholar] [CrossRef]
- Baker, J.J.; Johnson, B.K.; Abramovitch, R.B. Slow growth of Mycobacterium tuberculosis at acidic pH is regulated by phoPR and host-associated carbon sources. Mol. Microbiol. 2014, 94, 56–69. [Google Scholar] [CrossRef]
- Rizvi, A.; Shankar, A.; Chatterjee, A.; More, T.H.; Bose, T.; Dutta, A.; Balakrishnan, K.; Madugulla, L.; Rapole, S.; Mande, S.S.C.S.S.; et al. Rewiring of Metabolic Network in Mycobacterium tuberculosis During Adaptation to Different Stresses. Front. Microbiol. 2019, 10, 2417. [Google Scholar] [CrossRef]
- Kalscheuer, R.; Koliwer-Brandl, H. Genetics of Mycobacterial Trehalose Metabolism. Microbiol. Spectr. 2014, 2, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Elbein, A.D.; Pan, Y.T.; Pastuszak, I.; Carroll, D. New insights on trehalose: A multifunctional molecule. Glycobiology 2003, 13, 17R–27R. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Lee, S.K.; Song, N.; Nathan, T.O.; Swarts, B.M.; Eum, S.Y.; Ehrt, S.; Cho, S.N.; Eoh, H. Transient drug-tolerance and permanent drug-resistance rely on the trehalose-catalytic shift in Mycobacterium tuberculosis. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Galagan, J.E.; Minch, K.; Peterson, M.; Lyubetskaya, A.; Azizi, E.; Sweet, L.; Gomes, A.; Rustad, T.; Dolganov, G.; Glotova, I.; et al. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 2013, 499, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.-H.; Li, A.H.; Sassetti, C.M. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. 2011, 9, e1001065. [Google Scholar] [CrossRef]
- Cunningham, A.F.; Spreadbury, C.L. Mycobacterial stationary phase induced by low oxygen tension: Cell wall thickening and localization of the 16-kilodalton alpha-crystallin homolog. J. Bacteriol. 1998, 180, 801–808. [Google Scholar] [CrossRef]
- Fahy, E.; Cotter, D.; Sud, M.; Subramaniam, S. Lipid classification, structures and tools. Biochim. Biophys. Acta 2011, 1811, 637–647. [Google Scholar] [CrossRef]
- Sartain, M.J.; Dick, D.L.; Rithner, C.D.; Crick, D.C.; Belisle, J.T. Lipidomic analyses of Mycobacterium tuberculosis based on accurate mass measurements and the novel “Mtb LipidDB”. J. Lipid Res. 2011, 52, 861–872. [Google Scholar] [CrossRef]
- Rodríguez, J.G.; Hernández, A.C.; Helguera-Repetto, C.; Aguilar Ayala, D.; Guadarrama-Medina, R.; Anzóla, J.M.; Bustos, J.R.; Zambrano, M.M.; González-y-Merchand, J.; García, M.J.; et al. Global Adaptation to a Lipid Environment Triggers the Dormancy-Related Phenotype of Mycobacterium tuberculosis. mBio 2014, 5, e01125-01114. [Google Scholar] [CrossRef]
- Shleeva, M.; Goncharenko, A.; Kudykina, Y.; Young, D.; Young, M.; Kaprelyants, A. Cyclic Amp-Dependent Resuscitation of Dormant Mycobacteria by Exogenous Free Fatty Acids. PLoS ONE 2013, 8, e82914. [Google Scholar] [CrossRef]
- McGillivray, A.; Golden, N.A.; Kaushal, D. The Mycobacterium tuberculosis Clp gene regulator is required for in vitro reactivation from hypoxia-induced dormancy. J. Biol. Chem. 2015, 290, 2351–2367. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E., Jr.; Bailo, R.; Rothchild, A.C.; Arrieta-Ortiz, M.L.; Kaur, A.; Pan, M.; Mai, D.; Abidi, A.A.; Cooper, C.; Aderem, A.; et al. Path-seq identifies an essential mycolate remodeling program for mycobacterial host adaptation. Mol. Syst. Biol. 2019, 15, e8584. [Google Scholar] [CrossRef] [PubMed]
- Deb, C.; Daniel, J.; Sirakova, T.D.; Abomoelak, B.; Dubey, V.S.; Kolattukudy, P.E. A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. J. Biol. Chem. 2006, 281, 3866–3875. [Google Scholar] [CrossRef] [PubMed]
- Cotes, K.; Bakala N’goma, J.C.; Dhouib, R.; Douchet, I.; Maurin, D.; Carriere, F.; Canaan, S. Lipolytic enzymes in Mycobacterium tuberculosis. Appl. Microbiol. Biotechnol. 2008, 78, 741–749. [Google Scholar] [CrossRef]
- Singh, G.; Singh, G.; Jadeja, D.; Kaur, J. Lipid hydrolizing enzymes in virulence: Mycobacterium tuberculosis as a model system. Crit. Rev. Microbiol. 2010, 36, 259–269. [Google Scholar] [CrossRef]
- Santucci, P.; Diomandé, S.; Poncin, I.; Alibaud, L.; Viljoen, A.; Kremer, L.; de Chastellier, C.; Canaan, S. Delineating the Physiological Roles of the PE and Catalytic Domains of LipY in Lipid Consumption in Mycobacterium-Infected Foamy Macrophages. Infect. Immun. 2018, 86, e00394-00318. [Google Scholar] [CrossRef]
- Deb, C.; Lee, C.-M.; Dubey, V.S.; Daniel, J.; Abomoelak, B.; Sirakova, T.D.; Pawar, S.; Rogers, L.; Kolattukudy, P.E. A Novel In Vitro Multiple-Stress Dormancy Model for Mycobacterium tuberculosis Generates a Lipid-Loaded, Drug-Tolerant, Dormant Pathogen. PLoS ONE 2009, 4, e6077. [Google Scholar] [CrossRef]
- Low, K.L.; Rao, P.S.S.; Shui, G.; Bendt, A.K.; Pethe, K.; Dick, T.; Wenk, M.R. Triacylglycerol Utilization Is Required for Regrowth of In Vitro Hypoxic Nonreplicating Mycobacterium bovis Bacillus Calmette-Guerin. J. Bacteriol. 2009, 191, 5037. [Google Scholar] [CrossRef]
- Sirakova, T.D.; Dubey, V.S.; Deb, C.; Daniel, J.; Korotkova, T.A.; Abomoelak, B.; Kolattukudy, P.E. Identification of a diacylglycerol acyltransferase gene involved in accumulation of triacylglycerol in Mycobacterium tuberculosis under stress. Microbiol. Read. Engl. 2006, 152, 2717–2725. [Google Scholar] [CrossRef]
- Garton, N.J.; Waddell, S.J.; Sherratt, A.L.; Lee, S.M.; Smith, R.J.; Senner, C.; Hinds, J.; Rajakumar, K.; Adegbola, R.A.; Besra, G.S.; et al. Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PLoS Medicine 2008, 5, 634–645. [Google Scholar] [CrossRef]
- Daniel, J.; Sirakova, T.; Kolattukudy, P. An acyl-CoA synthetase in Mycobacterium tuberculosis involved in triacylglycerol accumulation during dormancy. PLoS ONE 2014, 9, e114877. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.; Maamar, H.; Deb, C.; Sirakova, T.D.; Kolattukudy, P.E. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011, 7, e1002093. [Google Scholar] [CrossRef] [PubMed]
- Caire-Brändli, I.; Papadopoulos, A.; Malaga, W.; Marais, D.; Canaan, S.; Thilo, L.; de Chastellier, C. Reversible Lipid Accumulation and Associated Division Arrest of Mycobacterium avium in Lipoprotein-Induced Foamy Macrophages May Resemble Key Events during Latency and Reactivation of Tuberculosis. Infect. Immun. 2014, 82, 476. [Google Scholar] [CrossRef] [PubMed]
- Barisch, C.; Soldati, T. Mycobacterium marinum Degrades Both Triacylglycerols and Phospholipids from Its Dictyostelium Host to Synthesise Its Own Triacylglycerols and Generate Lipid Inclusions. PLoS Pathog. 2017, 13, e1006095. [Google Scholar] [CrossRef]
- Alvarez, H.M.; Steinbuchel, A. Triacylglycerols in prokaryotic microorganisms. Appl. Microbiol. Biotechnol. 2002, 60, 367–376. [Google Scholar] [CrossRef]
- Banoub, J.H.; Aneed, A.E.; Cohen, A.M.; Joly, N. Structural investigation of bacterial lipopolysaccharides by mass spectrometry and tandem mass spectrometry. Mass Spectrom. Rev. 2010, 29, 606–650. [Google Scholar] [CrossRef]
- Sirakova, T.D.; Thirumala, A.K.; Dubey, V.S.; Sprecher, H.; Kolattukudy, P.E. The Mycobacterium tuberculosis pks2 Gene Encodes the Synthase for the Hepta- and Octamethyl-branched Fatty Acids Required for Sulfolipid Synthesis. J. Biol. Chem. 2001, 276, 16833–16839. [Google Scholar] [CrossRef]
- Foo, C.S.-Y.; Pethe, K.; Lupien, A. Oxidative Phosphorylation—An Update on a New, Essential Target Space for Drug Discovery in Mycobacterium tuberculosis. Appl. Sci. 2020, 10, 2339. [Google Scholar] [CrossRef]
- Mdluli, K.; Kaneko, T.; Upton, A. The tuberculosis drug discovery and development pipeline and emerging drug targets. Cold Spring Harb. Perspect. Med. 2015, 5, a021154. [Google Scholar] [CrossRef]
- North, E.J.; Jackson, M.; Lee, R.E. New approaches to target the mycolic acid biosynthesis pathway for the development of tuberculosis therapeutics. Curr. Pharm. Des. 2014, 20, 4357–4378. [Google Scholar] [CrossRef]
- Bald, D.; Villellas, C.; Lu, P.; Koul, A. Targeting Energy Metabolism in Mycobacterium tuberculosis, a New Paradigm in Antimycobacterial Drug Discovery. mBio 2017, 8, e00272-17. [Google Scholar] [CrossRef] [PubMed]
- Förster, A.H.; Gescher, J. Metabolic Engineering of Escherichia coli for Production of Mixed-Acid Fermentation End Products. Front. Bioeng. Biotechnol. 2014, 2, 16. [Google Scholar] [PubMed]
- Clark, D.P. The fermentation pathways of Escherichia coli. FEMS Microbiol. Lett. 1989, 63, 223–234. [Google Scholar] [CrossRef]
- Dhandayuthapani, S.; Zhang, Y.; Mudd, M.H.; Deretic, V.; Cook, G.M.; Hards, K.; Dunn, E.; Heikal, A.; Nakatani, Y.; Greening, C.; et al. Oxidative Phosphorylation as a Target Space for Tuberculosis: Success, Caution, and Future Directions. Microbiol. Spectr. 2017, 5, 295–316. [Google Scholar] [CrossRef]
- Pym, A.S.; Diacon, A.H.; Tang, S.-J.; Conradie, F.; Danilovits, M.; Chuchottaworn, C.; Vasilyeva, I.; Andries, K.; Bakare, N.; De Marez, T.; et al. Bedaquiline in the treatment of multidrug- and extensively drug-resistant tuberculosis. Eur. Respir. J. 2016, 47, 564–574. [Google Scholar] [CrossRef]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.H.; Neefs, J.-M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A Diarylquinoline Drug Active on the ATP Synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef]
- Cox, E.; Laessig, K. FDA approval of bedaquiline—the benefit–risk balance for drug-resistant tuberculosis. N. Engl. J. Med. 2014, 371, 689–691. [Google Scholar] [CrossRef]
- Koul, A.; Vranckx, L.; Dendouga, N.; Balemans, W.; Van den Wyngaert, I.; Vergauwen, K.; Göhlmann, H.W.; Willebrords, R.; Poncelet, A.; Guillemont, J.; et al. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 2008, 283, 25273–25280. [Google Scholar] [CrossRef]
- Harbut, M.B.; Yang, B.; Liu, R.; Yano, T.; Vilchèze, C.; Cheng, B.; Lockner, J.; Guo, H.; Yu, C.; Franzblau, S.G.; et al. Small Molecules Targeting Mycobacterium tuberculosis Type II NADH Dehydrogenase Exhibit Antimycobacterial Activity. Angew. Chem. 2018, 57, 3478–3482. [Google Scholar] [CrossRef]
- Murugesan, D.; Ray, P.C.; Bayliss, T.; Prosser, G.A.; Harrison, J.R.; Green, K.; Soares de Melo, C.; Feng, T.-S.; Street, L.J.; Chibale, K.; et al. 2-Mercapto-Quinazolinones as Inhibitors of Type II NADH Dehydrogenase and Mycobacterium tuberculosis: Structure-Activity Relationships, Mechanism of Action and Absorption, Distribution, Metabolism, and Excretion Characterization. ACS Infect. Dis. 2018, 4, 954–969. [Google Scholar] [CrossRef]
- Kurosu, M.; Crick, D.C. MenA is a promising drug target for developing novel lead molecules to combat Mycobacterium tuberculosis. Med. Chem. 2009, 5, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Sukheja, P.; Kumar, P.; Mittal, N.; Li, S.-G.; Singleton, E.; Russo, R.; Perryman, A.L.; Shrestha, R.; Awasthi, D.; Husain, S.; et al. A Novel Small-Molecule Inhibitor of the Mycobacterium tuberculosis Demethylmenaquinone Methyltransferase MenG Is Bactericidal to Both Growing and Nutritionally Deprived Persister Cells. mBio 2017, 8, e02022-02016. [Google Scholar] [CrossRef] [PubMed]
- Cleghorn, L.A.T.; Ray, P.C.; Odingo, J.; Kumar, A.; Wescott, H.; Korkegian, A.; Masquelin, T.; Lopez Moure, A.; Wilson, C.; Davis, S.; et al. Identification of Morpholino Thiophenes as Novel Mycobacterium tuberculosis Inhibitors, Targeting QcrB. J. Med. Chem. 2018, 61, 6592–6608. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekera, N.S.; Berube, B.J.; Shetye, G.; Chettiar, S.; O’Malley, T.; Manning, A.; Flint, L.; Awasthi, D.; Ioerger, T.R.; Sacchettini, J.; et al. Improved Phenoxyalkylbenzimidazoles with Activity against Mycobacterium tuberculosis Appear to Target QcrB. ACS Infect. Dis. 2017, 3, 898–916. [Google Scholar] [CrossRef] [PubMed]
- de Jager, V.R.; Dawson, R.; van Niekerk, C.; Hutchings, J.; Kim, J.; Vanker, N.; van der Merwe, L.; Choi, J.; Nam, K.; Diacon, A.H. Telacebec (Q203), a New Antituberculosis Agent. N. Engl. J. Med. 2020, 382, 1280–1281. [Google Scholar] [CrossRef]
- Koul, A.; Vranckx, L.; Dhar, N.; Göhlmann, H.W.H.; Özdemir, E.; Neefs, J.-M.; Schulz, M.; Lu, P.; Mørtz, E.; McKinney, J.D.; et al. Delayed bactericidal response of Mycobacterium tuberculosis to bedaquiline involves remodelling of bacterial metabolism. Nat. Commun. 2014, 5, 3369. [Google Scholar] [CrossRef]
- Greenwood, D.J.; Dos Santos, M.S.; Huang, S.; Russell, M.R.G.; Collinson, L.M.; MacRae, J.I.; West, A.; Jiang, H.; Gutierrez, M.G. Subcellular antibiotic visualization reveals a dynamic drug reservoir in infected macrophages. Science 2019, 364, 1279. [Google Scholar] [CrossRef]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef]
- Kalia, N.P.; Shi Lee, B.; Ab Rahman, N.B.; Moraski, G.C.; Miller, M.J.; Pethe, K. Carbon metabolism modulates the efficacy of drugs targeting the cytochrome bc 1:aa 3 in Mycobacterium tuberculosis. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Huang, L.; Kushner, N.L.; Theriault, M.E.; Pisu, D.; Tan, S.; McNamara, C.W.; Petrassi, H.M.; Russell, D.G.; Brown, A.C. The Deconstructed Granuloma: A Complex High-Throughput Drug Screening Platform for the Discovery of Host-Directed Therapeutics Against Tuberculosis. Front. Cell. Infect. Microbiol. 2018, 8, 275. [Google Scholar] [CrossRef]
- Isoniazid trials. Lancet 1952, 2, 471–472. [CrossRef]
- Clark, C.M.; Elmendorf, D.F., Jr.; Cawthon, W.U.; Muschenheim, C.; McDermott, W. Isoniazid (isonicotinic acid hydrazide) in the treatment of miliary and meningeal tuberculosis. Am. Rev. Tuberc. 1952, 66, 391–415. [Google Scholar] [CrossRef] [PubMed]
- Winder, F.G.; Collins, P.; Rooney, S.A. Effects of isoniazid on mycolic acid synthesis in Mycobacterium tuberculosis and on its cell envelope. Biochem. J. 1970, 117, 27P. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; de Lisle, G.; Jacobs, W.R., Jr. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 1994, 263, 227–230. [Google Scholar] [CrossRef]
- Phetsuksiri, B.; Jackson, M.; Scherman, H.; McNeil, M.; Besra, G.S.; Baulard, A.R.; Slayden, R.A.; DeBarber, A.E.; Barry, C.E., 3rd; Baird, M.S.; et al. Unique mechanism of action of the thiourea drug isoxyl on Mycobacterium tuberculosis. J. Biol. Chem. 2003, 278, 53123–53130. [Google Scholar] [CrossRef]
- Grzegorzewicz, A.E.; Kordulakova, J.; Jones, V.; Born, S.E.; Belardinelli, J.M.; Vaquie, A.; Gundi, V.A.; Madacki, J.; Slama, N.; Laval, F.; et al. A common mechanism of inhibition of the Mycobacterium tuberculosis mycolic acid biosynthetic pathway by isoxyl and thiacetazone. J. Biol. Chem. 2012, 287, 38434–38441. [Google Scholar] [CrossRef]
- Baran, M.; Grimes, K.D.; Sibbald, P.A.; Fu, P.; Boshoff, H.I.M.; Wilson, D.J.; Aldrich, C.C. Development of small-molecule inhibitors of fatty acyl-AMP and fatty acyl-CoA ligases in Mycobacterium tuberculosis. Eur. J. Med. Chem. 2020, 201, 112408. [Google Scholar] [CrossRef]
- Mohanty, D.; Sankaranarayanan, R.; Gokhale, R.S. Fatty acyl-AMP ligases and polyketide synthases are unique enzymes of lipid biosynthetic machinery in Mycobacterium tuberculosis. Tuberculosis 2011, 91, 448–455. [Google Scholar] [CrossRef]
- Sharma, V.; Sharma, S.; zu Bentrup, K.H.; McKinney, J.D.; Russell, D.G.; Jacobs, W.R.; Sacchettini, J.C. Structure of isocitrate lyase, a persistence factor of Mycobacterium tuberculosis. Nat. Struct. Biol. 2000, 7, 663–668. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Novotná, E.; Mandíková, J.; Wsól, V.; Trejtnar, F.; Ulmann, V.; Stolaříková, J.; Fernandes, S.; Bhat, S.; et al. Salicylanilide derivatives block Mycobacterium tuberculosis through inhibition of isocitrate lyase and methionine aminopeptidase. Tuberculosis 2012, 92, 434–439. [Google Scholar] [CrossRef]
- Sriram, D.; Aubry, A.; Yogeeswari, P.; Fisher, L.M. Gatifloxacin derivatives: Synthesis, antimycobacterial activities, and inhibition of Mycobacterium tuberculosis DNA gyrase. Bioorg. Med. Chem. Lett. 2006, 16, 2982–2985. [Google Scholar] [CrossRef] [PubMed]
- Sriram, D.; Yogeeswari, P.; Vyas, D.R.; Senthilkumar, P.; Bhat, P.; Srividya, M. 5-Nitro-2-furoic acid hydrazones: Design, synthesis and in vitro antimycobacterial evaluation against log and starved phase cultures. Bioorg. Med. Chem. Lett. 2010, 20, 4313–4316. [Google Scholar] [CrossRef] [PubMed]
- Sriram, D.; Yogeeswari, P.; Senthilkumar, P.; Naidu, G.; Bhat, P. 5-Nitro-2,6-dioxohexahydro-4-pyrimidinecarboxamides: Synthesis, in vitro antimycobacterial activity, cytotoxicity, and isocitrate lyase inhibition studies. J. Enzym. Inhib. Med. Chem. 2010, 25, 765–772. [Google Scholar] [CrossRef]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E.; et al. SQ109 Targets MmpL3, a Membrane Transporter of Trehalose Monomycolate Involved in Mycolic Acid Donation to the Cell Wall Core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Meshcheryakov, V.A.; Poce, G.; Chng, S.-S. MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 7993–7998. [Google Scholar] [CrossRef]
- Borisov, S.; Bogorodskaya, E.; Volchenkov, G.; Kulchavenya, E.; Maryandyshev, A.; Skornyakov, S.; Talibov, O.; Tikhonov, A.; Vasilyeva, I. Efficiency and safety of chemotherapy regimen with SQ109 in those suffering from multiple drug resistant tuberculosis. Tuberc. Lung. Dis. 2018, 96, 6–18. [Google Scholar] [CrossRef]
- Martinot, A.J.; Farrow, M.; Bai, L.; Layre, E.; Cheng, T.-Y.; Tsai, J.H.; Iqbal, J.; Annand, J.W.; Sullivan, Z.A.; Hussain, M.M.; et al. Mycobacterial Metabolic Syndrome: LprG and Rv1410 Regulate Triacylglyceride Levels, Growth Rate and Virulence in Mycobacterium tuberculosis. PLoS Pathog. 2016, 12, e1005351. [Google Scholar] [CrossRef]
- Gaur, R.L.; Ren, K.; Blumenthal, A.; Bhamidi, S.; Gibbs, S.; Jackson, M.; Zare, R.N.; Ehrt, S.; Ernst, J.D.; Banaei, N. LprG-Mediated Surface Expression of Lipoarabinomannan Is Essential for Virulence of Mycobacterium tuberculosis. PLoS Pathog. 2014, 10, e1004376. [Google Scholar] [CrossRef]
- Pisu, D.; Huang, L.; Grenier, J.K.; Russell, D.G. Dual RNA-Seq of Mtb-Infected Macrophages In Vivo Reveals Ontologically Distinct Host-Pathogen Interactions. Cell Rep. 2020, 30, 335.e334–350.e334. [Google Scholar] [CrossRef]
- Vrieling, F.; Kostidis, S.; Spaink, H.P.; Haks, M.C.; Mayboroda, O.A.; Ottenhoff, T.H.M.; Joosten, S.A. Analyzing the impact of Mycobacterium tuberculosis infection on primary human macrophages by combined exploratory and targeted metabolomics. Sci. Rep. 2020, 10, 7085. [Google Scholar] [CrossRef]
- Beste, D.J.V.; Bonde, B.; Hawkins, N.; Ward, J.L.; Beale, M.H.; Noack, S.; Nöh, K.; Kruger, N.J.; Ratcliffe, R.G.; McFadden, J. 13C Metabolic Flux Analysis Identifies an Unusual Route for Pyruvate Dissimilation in Mycobacteria which Requires Isocitrate Lyase and Carbon Dioxide Fixation. PLoS Pathog. 2011, 7, e1002091. [Google Scholar] [CrossRef]
- Carninci, P.; Kvam, C.; Kitamura, A.; Ohsumi, T.; Okazaki, Y.; Itoh, M.; Kamiya, M.; Shibata, K.; Sasaki, N.; Izawa, M.; et al. High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics 1996, 37, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Duffy, F.J.; Weiner, J., 3rd; Hansen, S.; Tabb, D.L.; Suliman, S.; Thompson, E.; Maertzdorf, J.; Shankar, S.; Tromp, G.; Parida, S.; et al. Immunometabolic Signatures Predict Risk of Progression to Active Tuberculosis and Disease Outcome. Front Immunol. 2019, 10, 527. [Google Scholar] [CrossRef]
- Crick, P.J.; Guan, X.L. Lipid metabolism in mycobacteria--Insights using mass spectrometry-based lipidomics. Biochim. Biophys. Acta 2016, 1861, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Kuehne, A.; Boshoff, H.I.; Barry, C.E., 3rd; Zamboni, N.; Sauer, U. Dynamic exometabolome analysis reveals active metabolic pathways in non-replicating mycobacteria. Env. Microbiol. 2015, 17, 4802–4815. [Google Scholar] [CrossRef] [PubMed]
- Layre, E.; Sweet, L.; Hong, S.; Madigan, C.A.; Desjardins, D.; Young, D.C.; Cheng, T.-Y.; Annand, J.W.; Kim, K.; Shamputa, I.C.; et al. A comparative lipidomics platform for chemotaxonomic analysis of Mycobacterium tuberculosis. Chem. Biol. 2011, 18, 1537–1549. [Google Scholar] [CrossRef]
- Emwas, A.H. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol. Biol. 2015, 1277, 161–193. [Google Scholar] [CrossRef]
- Fernández-García, M.; Rey-Stolle, F.; Boccard, J.; Reddy, V.P.; García, A.; Cumming, B.M.; Steyn, A.J.C.; Rudaz, S.; Barbas, C. Comprehensive Examination of the Mouse Lung Metabolome Following Mycobacterium tuberculosis Infection Using a Multiplatform Mass Spectrometry Approach. J. Proteome Res. 2020, 19, 2053–2070. [Google Scholar] [CrossRef]
- Zampieri, M.; Szappanos, B.; Buchieri, M.V.; Trauner, A.; Piazza, I.; Picotti, P.; Gagneux, S.; Borrell, S.; Gicquel, B.; Lelievre, J.; et al. High-throughput metabolomic analysis predicts mode of action of uncharacterized antimicrobial compounds. Sci. Transl. Med. 2018, 10, eaal3973. [Google Scholar] [CrossRef]
- Diaz, C.; Perez Del Palacio, J.; Valero-Guillen, P.L.; Mena Garcia, P.; Perez, I.; Vicente, F.; Martin, C.; Genilloud, O.; Sanchez Pozo, A.; Gonzalo-Asensio, J. Comparative Metabolomics between Mycobacterium tuberculosis and the MTBVAC Vaccine Candidate. ACS Infect. Dis. 2019, 5, 1317–1326. [Google Scholar] [CrossRef]
- Drapal, M.; Fraser, P.D. Metabolite Profiling: A Tool for the Biochemical Characterisation of Mycobacterium sp. Microorganisms 2019, 7, 148. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Haseley, N.; Brown, D.; Penaranda, C.; Jijon, H.B.; Trombetta, J.J.; Satija, R.; Shalek, A.K.; Xavier, R.J.; Regev, A.; et al. Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses. Cell 2015, 162, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.-E.; Li, L.; Westermann, A.J.; Appenzeller, S.; Stapels, D.A.C.; Schulte, L.N.; Helaine, S.; Vogel, J. Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat. Microbiol. 2016, 2, 16206. [Google Scholar] [CrossRef]
- Maglica, Ž.; Özdemir, E.; McKinney, J.D. Single-Cell Tracking Reveals Antibiotic-Induced Changes in Mycobacterial Energy Metabolism. mBio 2015, 6, e02236-02214. [Google Scholar] [CrossRef]
- Wakamoto, Y.; Dhar, N.; Chait, R.; Schneider, K.; Signorino-Gelo, F.; Leibler, S.; McKinney, J.D. Dynamic Persistence of Antibiotic-Stressed Mycobacteria. Science 2013, 339, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Kirschman, J.C.; Ogongo, P.; Ernst, J.; Altin, J. Single cell transcriptomics of the T cell response to Mycobacterium tuberculosis reveals phenotypic diversity within and between infected individuals. J. Immunol. 2020, 204, 225.221. [Google Scholar]
- Cai, Y.; Dai, Y.; Wang, Y.; Yang, Q.; Guo, J.; Wei, C.; Chen, W.; Huang, H.; Zhu, J.; Zhang, C.; et al. Single-cell transcriptomics of blood reveals a natural killer cell subset depletion in tuberculosis. EBioMedicine 2020, 53, 102686. [Google Scholar] [CrossRef]
- Gagneux, S. Ecology and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2018, 16, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Liu, Q.; Wu, J.; Jiang, Y.; Takiff, H.E.; Gao, Q. Mycobacterium tuberculosis strains of the modern Beijing sublineage excessively accumulate triacylglycerols in vitro. Tuberculosis 2020, 120, 101892. [Google Scholar] [CrossRef]
- Rose, G.; Cortes, T.; Comas, I.; Coscolla, M.; Gagneux, S.; Young, D.B. Mapping of genotype-phenotype diversity among clinical isolates of Mycobacterium tuberculosis by sequence-based transcriptional profiling. Genome Biol. Evol. 2013, 5, 1849–1862. [Google Scholar] [CrossRef]
- Oyas, O.; Borrell, S.; Trauner, A.; Zimmermann, M.; Feldmann, J.; Liphardt, T.; Gagneux, S.; Stelling, J.; Sauer, U.; Zampieri, M. Model-based integration of genomics and metabolomics reveals SNP functionality in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2020, 117, 8494–8502. [Google Scholar] [CrossRef] [PubMed]
- Portevin, D.; Sukumar, S.; Coscolla, M.; Shui, G.; Li, B.; Guan, X.L.; Bendt, A.K.; Young, D.; Gagneux, S.; Wenk, M.R. Lipidomics and genomics of Mycobacterium tuberculosis reveal lineage-specific trends in mycolic acid biosynthesis. Microbiologyopen 2014, 3, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rebollo-Ramirez, S.; Larrouy-Maumus, G. Metabolomics reveals that the cAMP receptor protein regulates nitrogen and peptidoglycan synthesis in Mycobacterium tuberculosis. RSC Adv. 2020, 10, 26212–26219. [Google Scholar] [CrossRef]
- Kahramanoglou, C.; Cortes, T.; Matange, N.; Hunt, D.M.; Visweswariah, S.S.; Young, D.B.; Buxton, R.S. Genomic mapping of cAMP receptor protein (CRPMt) in Mycobacterium tuberculosis: Relation to transcriptional start sites and the role of CRPMt as a transcription factor. Nucleic Acids Res. 2014, 42, 8320–8329. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef]
- Kapopoulou, A.; Lew, J.M.; Cole, S.T. The MycoBrowser portal: A comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis 2011, 91, 8–13. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef]
- Furness, L.M. Bridging the gap: The need for genomic and clinical -omics data integration and standardization in overcoming the bottleneck of variant interpretation. Expert Rev. Precis. Med. Drug Dev. 2017, 2, 79–89. [Google Scholar] [CrossRef]
- Borrell, S.; Trauner, A.; Brites, D.; Rigouts, L.; Loiseau, C.; Coscolla, M.; Niemann, S.; De Jong, B.; Yeboah-Manu, D.; Kato-Maeda, M.; et al. Reference set of Mycobacterium tuberculosis clinical strains: A tool for research and product development. PLoS ONE 2019, 14, e0214088. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, D.P.S.; Guan, X.L. Metabolic Versatility of Mycobacterium tuberculosis during Infection and Dormancy. Metabolites 2021, 11, 88. https://doi.org/10.3390/metabo11020088
Chang DPS, Guan XL. Metabolic Versatility of Mycobacterium tuberculosis during Infection and Dormancy. Metabolites. 2021; 11(2):88. https://doi.org/10.3390/metabo11020088
Chicago/Turabian StyleChang, Dorothy Pei Shan, and Xue Li Guan. 2021. "Metabolic Versatility of Mycobacterium tuberculosis during Infection and Dormancy" Metabolites 11, no. 2: 88. https://doi.org/10.3390/metabo11020088
APA StyleChang, D. P. S., & Guan, X. L. (2021). Metabolic Versatility of Mycobacterium tuberculosis during Infection and Dormancy. Metabolites, 11(2), 88. https://doi.org/10.3390/metabo11020088