The Peptide Chain Release Factor Methyltransferase PrmC Influences the Pseudomonas aeruginosa PA14 Endo- and Exometabolome



Abstract
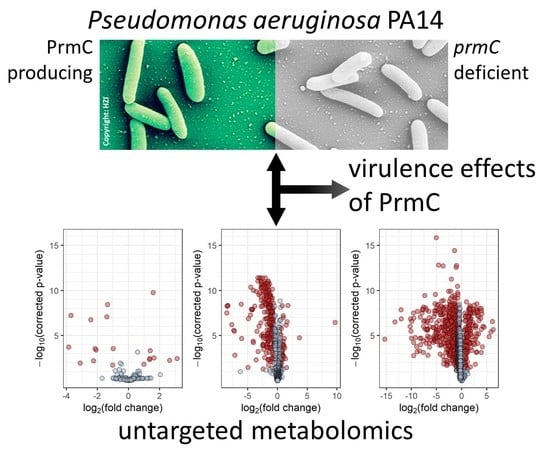
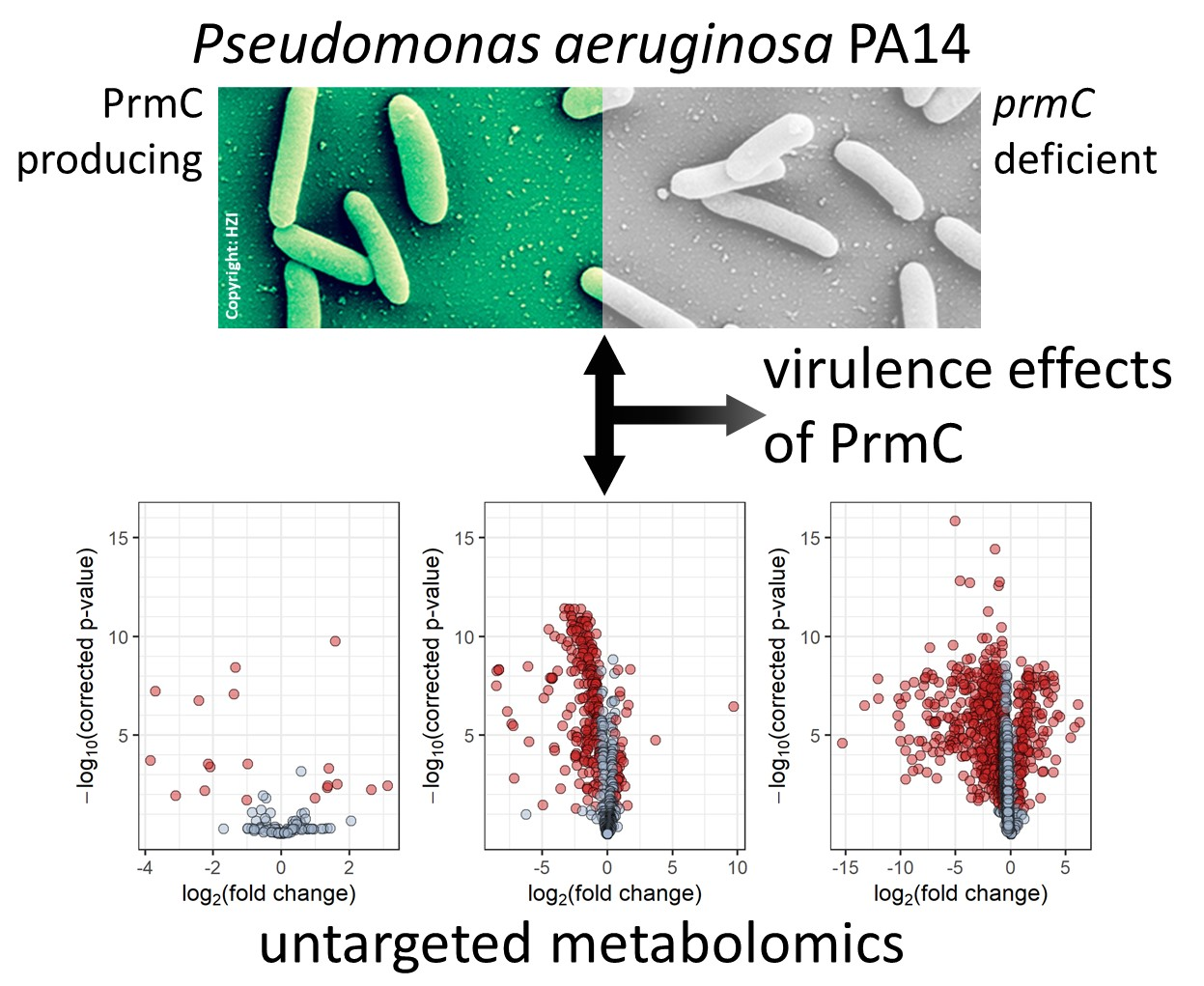
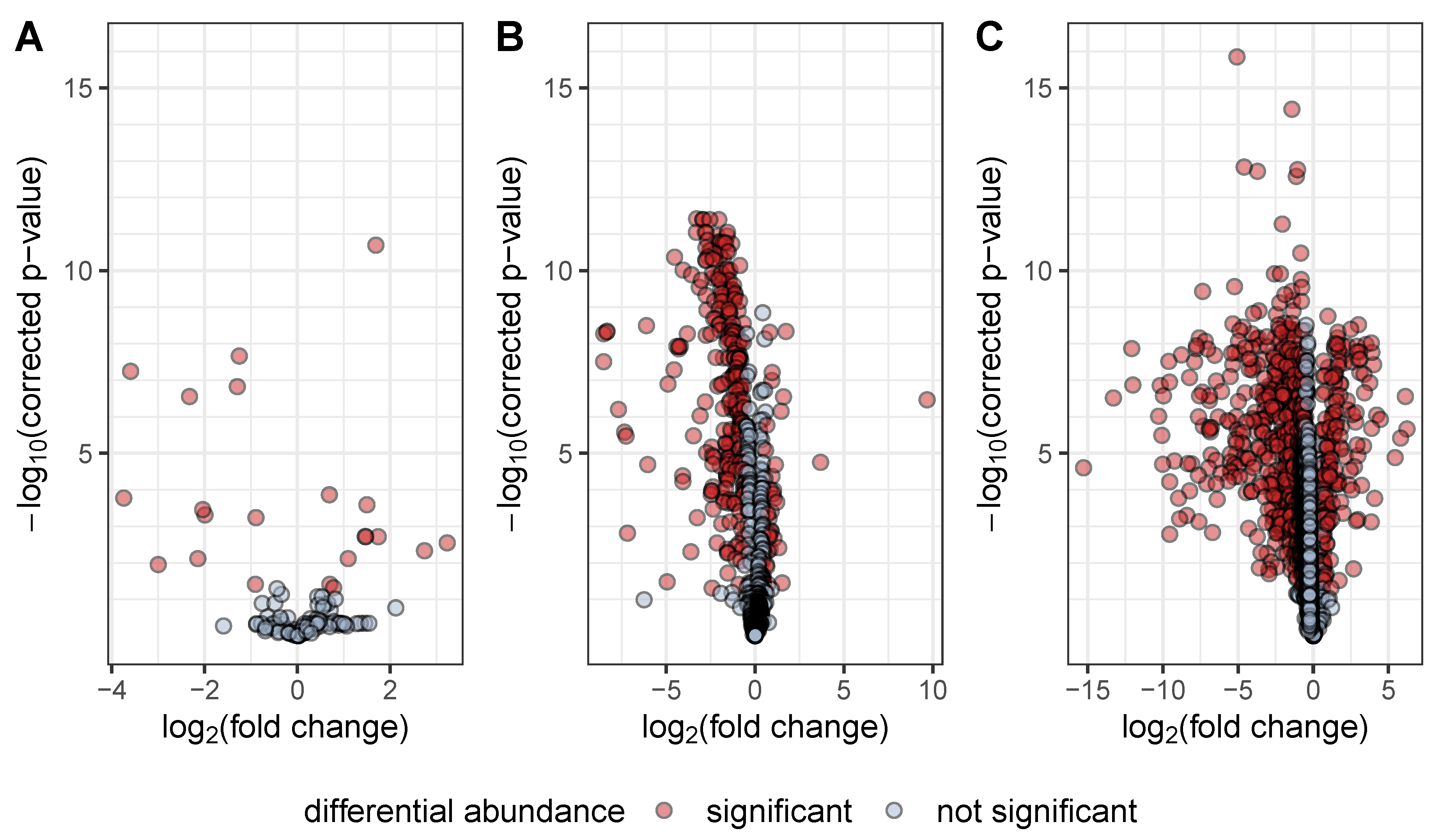
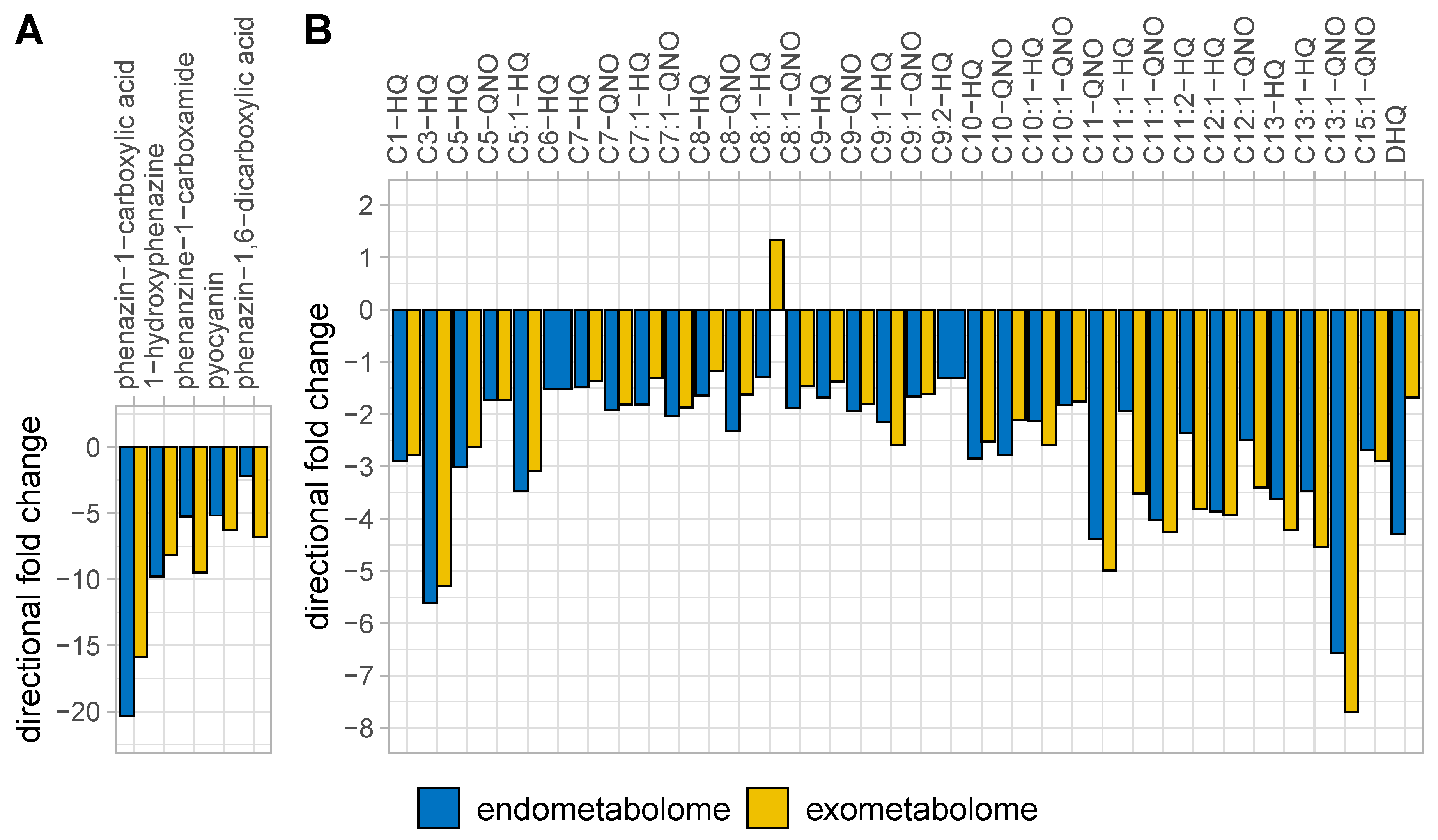
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
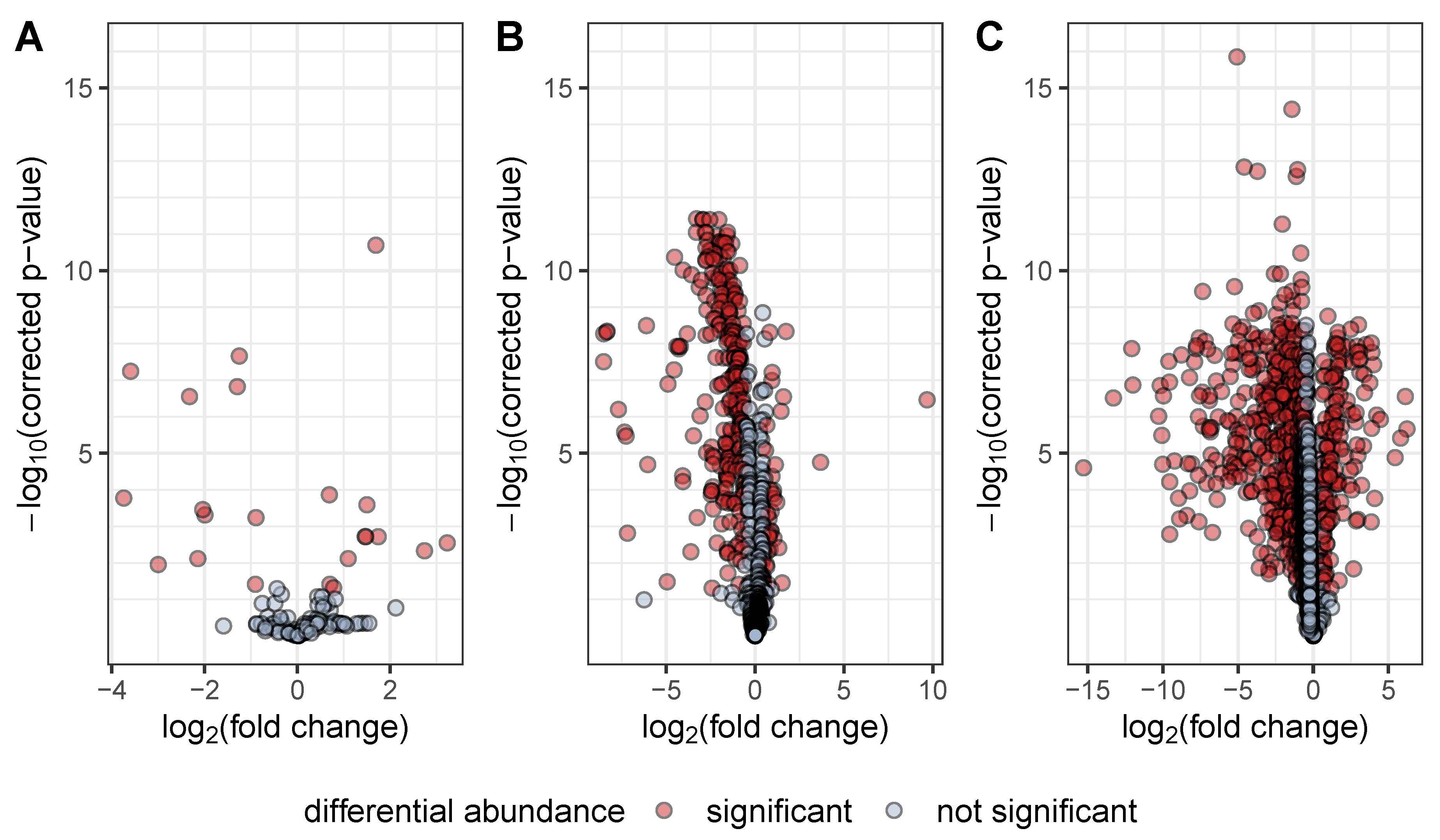
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| WHO | World Health Organisation |
| RF | release factor |
| LC–MS | liquid chromatography–mass spectrometry |
| GC–MS | gas chromatography–mass spectrometry |
| MSTFA | N-methyl-N-(trimethylsilyl)trifluoracetamide |
| DAHP | 3-deoxy-arabino-heptulosonate 7-phosphate |
| AQ | alkylquinolone |
| DHQ | 2,4-dihydroxyquinoline |
| Cn:m | hydrocarbon of chain length n with m double bonds |
| HQ | hydroxyquinoline |
| QNO | quinolone-N-oxide |
References
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunist1. Microbes Infect. 2000, 2, 1051–1060. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Obritsch, M.D.; Fish, D.N.; MacLaren, R.; Jung, R. Nosocomial Infections Due to Multidrug-Resistant Pseudomonas aeruginosa: Epidemiology and Treatment Options. Pharmacotherapy 2005, 25, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- De Bentzmann, S.; Plésiat, P. The Pseudomonas aeruginosa opportunistic pathogen and human infections. Environ. Microbiol. 2011, 13, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, P.N.; Koch, G.; Thompson, J.A.; Xavier, K.B.; Cool, R.H.; Quax, W.J. The Multiple Signaling Systems Regulating Virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2012, 76, 46–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pustelny, C.; Brouwer, S.; Müsken, M.; Bielecka, A.; Dötsch, A.; Nimtz, M.; Häussler, S. The peptide chain release factor methyltransferase PrmC is essential for pathogenicity and environmental adaptation of Pseudomonas aeruginosa PA14. Environ. Microbiol. 2012, 15, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Heurgué-Hamard, V.; Champ, S.; Engström, Å.; Ehrenberg, M.; Buckingham, R.H. The hemK gene in Escherichia coli encodes the N5-glutamine methyltransferase that modifies peptide release factors. EMBO J. 2002, 21, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahigashi, K.; Kubo, N.; Narita, S.I.; Shimaoka, T.; Goto, S.; Oshima, T.; Mori, H.; Maeda, M.; Wada, C.; Inokuchi, H. HemK, a class of protein methyl transferase with similarity to DNA methyl transferases, methylates polypeptide chain release factors, and hemK knockout induces defects in translational termination. Proc. Natl. Acad. Sci. USA 2002, 99, 1473–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, J.; Pohl, S.; Preusse, M.; Kordes, A.; Rugen, N.; Schniederjans, M.; Pich, A.; Häussler, S. Unravelling post-transcriptional PrmC-dependent regulatory mechanisms in Pseudomonas aeruginosa. Environ. Microbiol. 2016, 18, 3583–3592. [Google Scholar] [CrossRef] [Green Version]
- Liberati, N.T.; Urbach, J.M.; Miyata, S.; Lee, D.G.; Drenkard, E.; Wu, G.; Villanueva, J.; Wei, T.; Ausubel, F.M. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. USA 2006, 103, 2833–2838. [Google Scholar] [CrossRef] [Green Version]
- Depke, T.; Franke, R.; Brönstrup, M. Clustering of MS 2 spectra using unsupervised methods to aid the identification of secondary metabolites from Pseudomonas aeruginosa. J. Chromatogr. 2017, 1071, 19–28. [Google Scholar] [CrossRef]
- Berndt, V.; Beckstette, M.; Volk, M.; Dersch, P.; Brönstrup, M. Metabolome and transcriptome-wide effects of the carbon storage regulator A in enteropathogenic Escherichia coli. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiller, K.; Hangebrauk, J.; Jäger, C.; Spura, J.; Schreiber, K.; Schomburg, D. MetaboliteDetector: Comprehensive Analysis Tool for Targeted and Nontargeted GC/MS Based Metabolome Analysis. Anal. Chem. 2009, 81, 3429–3439. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depke, T.; Thöming, J.G.; Kordes, A.; Häussler, S.; Brönstrup, M. Untargeted LC-MS Metabolomics Differentiates Between Virulent and Avirulent Clinical Strains of Pseudomonas aeruginosa. Biomolecules 2020, 10, 1041. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 2011, 84, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Bredenbruch, F.; Nimtz, M.; Wray, V.; Morr, M.; Muller, R.; Haussler, S. Biosynthetic Pathway of Pseudomonas aeruginosa 4-Hydroxy-2-Alkylquinolines. J. Bacteriol. 2005, 187, 3630–3635. [Google Scholar] [CrossRef] [Green Version]
- Mentel, M.; Ahuja, E.G.; Mavrodi, D.V.; Breinbauer, R.; Thomashow, L.S.; Blankenfeldt, W. Of Two Make One: The Biosynthesis of Phenazines. ChemBioChem 2009, 10, 2295–2304. [Google Scholar] [CrossRef]
- Witzgall, F.; Depke, T.; Hoffmann, M.; Empting, M.; Brönstrup, M.; Müller, R.; Blankenfeldt, W. The Alkylquinolone Repertoire of Pseudomonas aeruginosa is Linked to Structural Flexibility of the FabH-like 2-Heptyl-3-hydroxy-4(1H)-quinolone (PQS) Biosynthesis Enzyme PqsBC. ChemBioChem 2018, 19, 1531–1544. [Google Scholar] [CrossRef]
- Cornelis, P.; Dingemans, J. Pseudomonas aeruginosa adapts its iron uptake strategies in function of the type of infections. Front. Cell. Infect. Microbiol. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Hare, N.J.; Soe, C.Z.; Rose, B.; Harbour, C.; Codd, R.; Manos, J.; Cordwell, S.J. Proteomics of Pseudomonas aeruginosa Australian Epidemic Strain 1 (AES-1) Cultured under Conditions Mimicking the Cystic Fibrosis Lung Reveals Increased Iron Acquisition via the Siderophore Pyochelin. J. Proteome Res. 2011, 11, 776–795. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.D. Effect of pyochelin on the virulence of Pseudomonas aeruginosa. Infect. Immun. 1982, 36, 17–23. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Depke, T.; Häussler, S.; Brönstrup, M. The Peptide Chain Release Factor Methyltransferase PrmC Influences the Pseudomonas aeruginosa PA14 Endo- and Exometabolome. Metabolites 2020, 10, 417. https://doi.org/10.3390/metabo10100417
Depke T, Häussler S, Brönstrup M. The Peptide Chain Release Factor Methyltransferase PrmC Influences the Pseudomonas aeruginosa PA14 Endo- and Exometabolome. Metabolites. 2020; 10(10):417. https://doi.org/10.3390/metabo10100417
Chicago/Turabian StyleDepke, Tobias, Susanne Häussler, and Mark Brönstrup. 2020. "The Peptide Chain Release Factor Methyltransferase PrmC Influences the Pseudomonas aeruginosa PA14 Endo- and Exometabolome" Metabolites 10, no. 10: 417. https://doi.org/10.3390/metabo10100417
APA StyleDepke, T., Häussler, S., & Brönstrup, M. (2020). The Peptide Chain Release Factor Methyltransferase PrmC Influences the Pseudomonas aeruginosa PA14 Endo- and Exometabolome. Metabolites, 10(10), 417. https://doi.org/10.3390/metabo10100417