
Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases


Abstract
1. Introduction
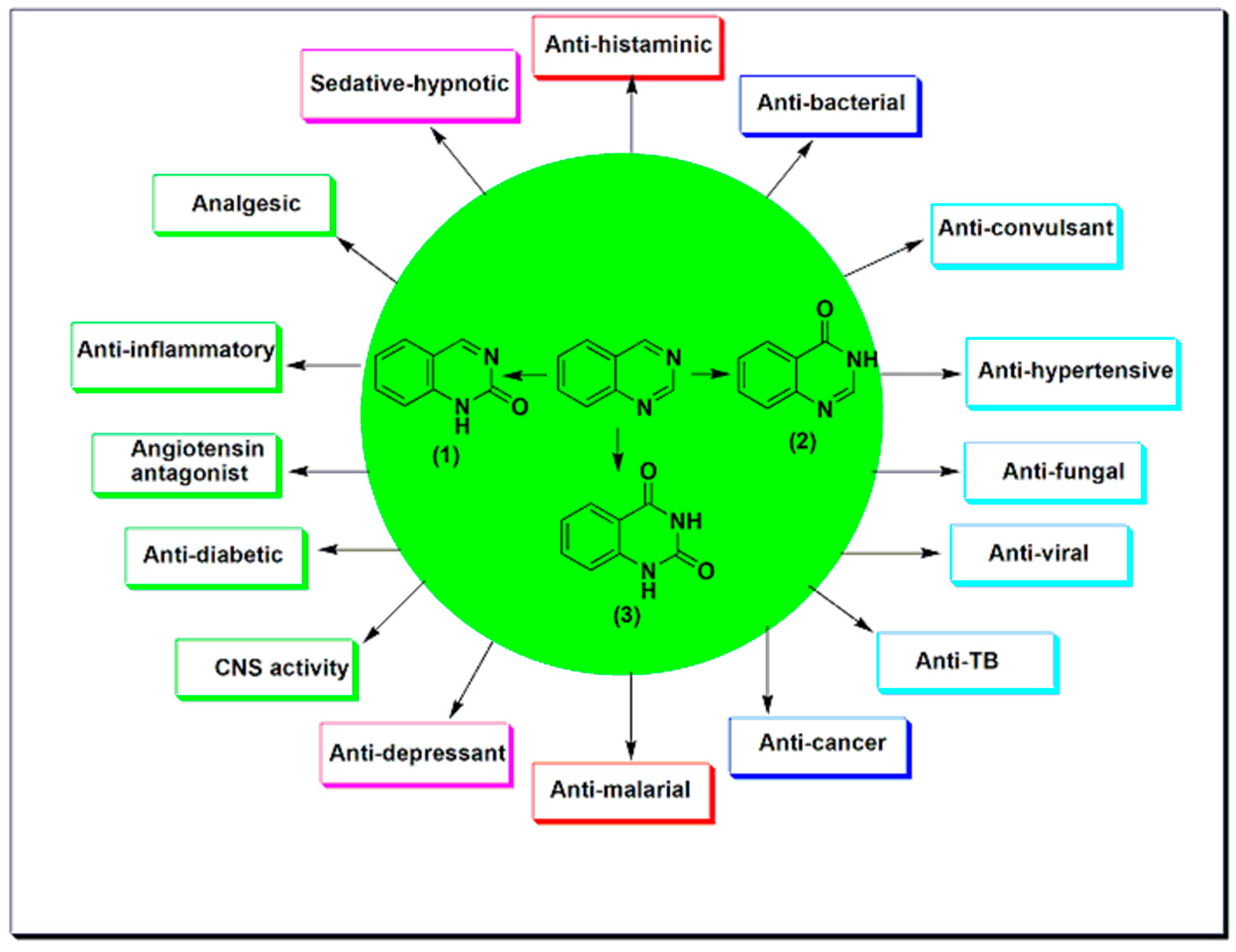
2. Therapeutic Importance of Quinazolines
3. Physicochemical Characters of the Core Structural Feature of Anticancer Quinazolines
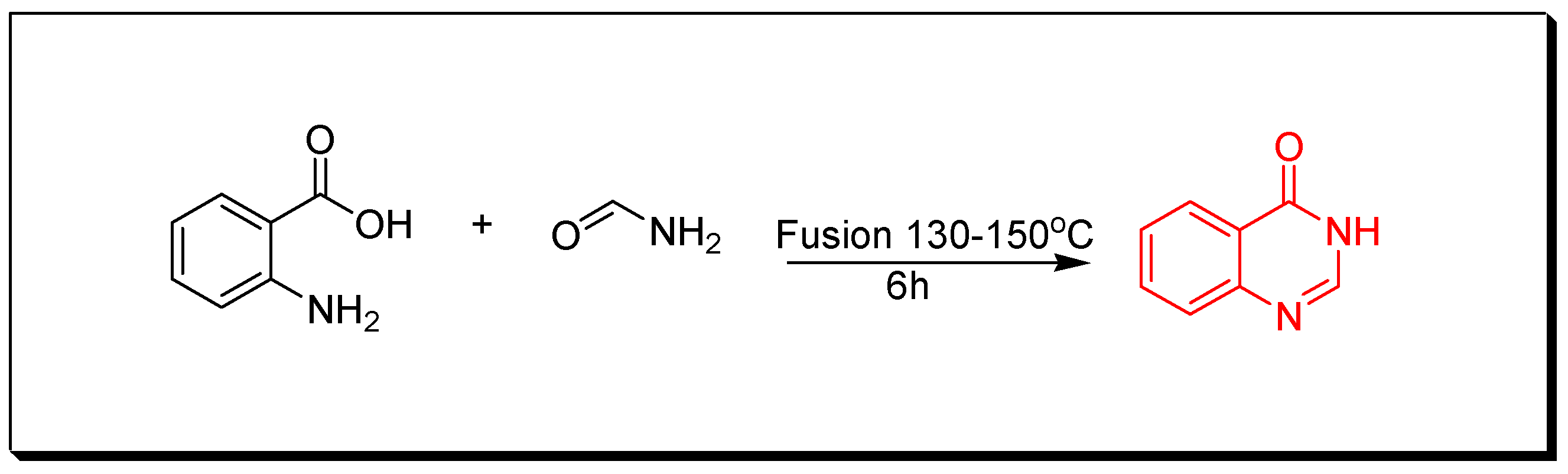
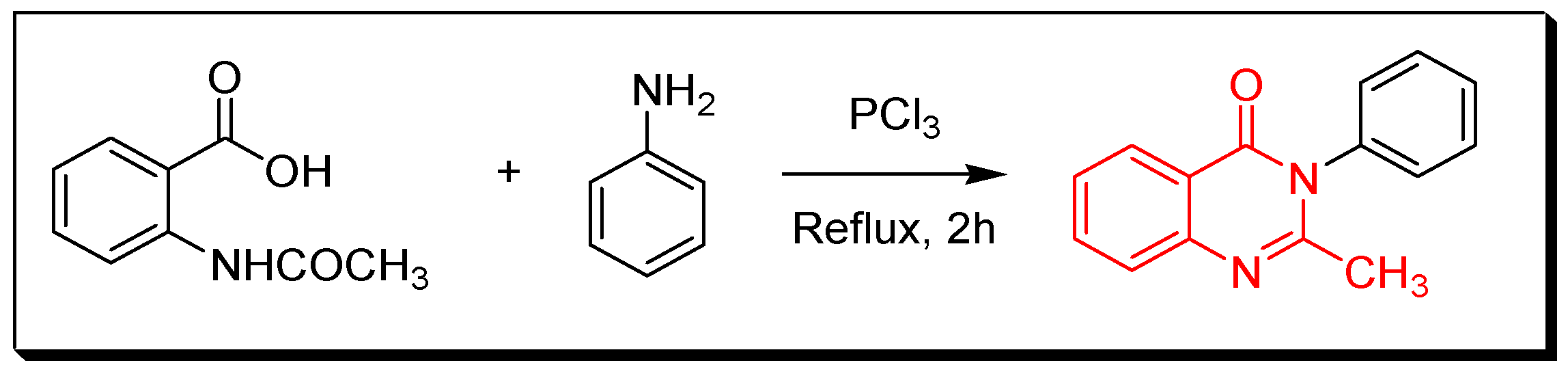
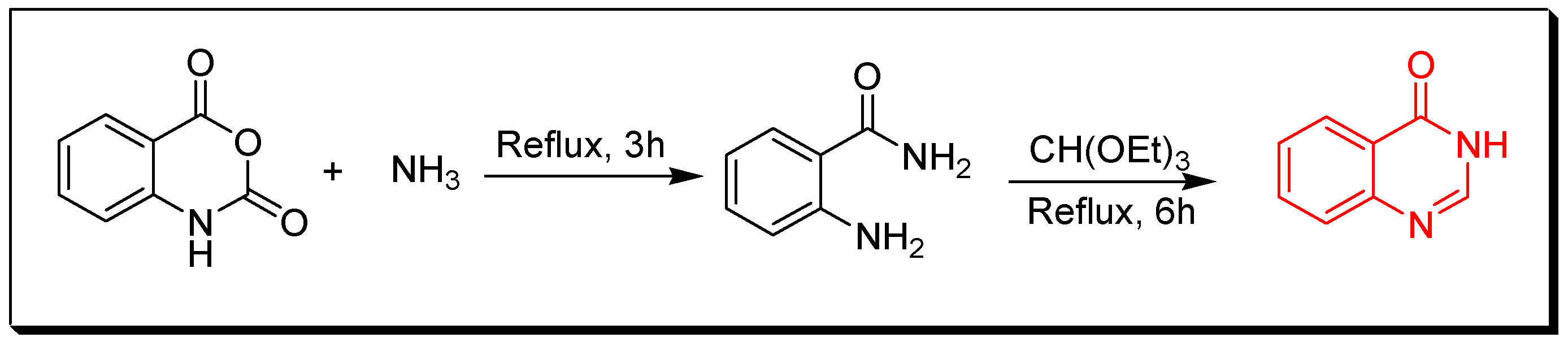
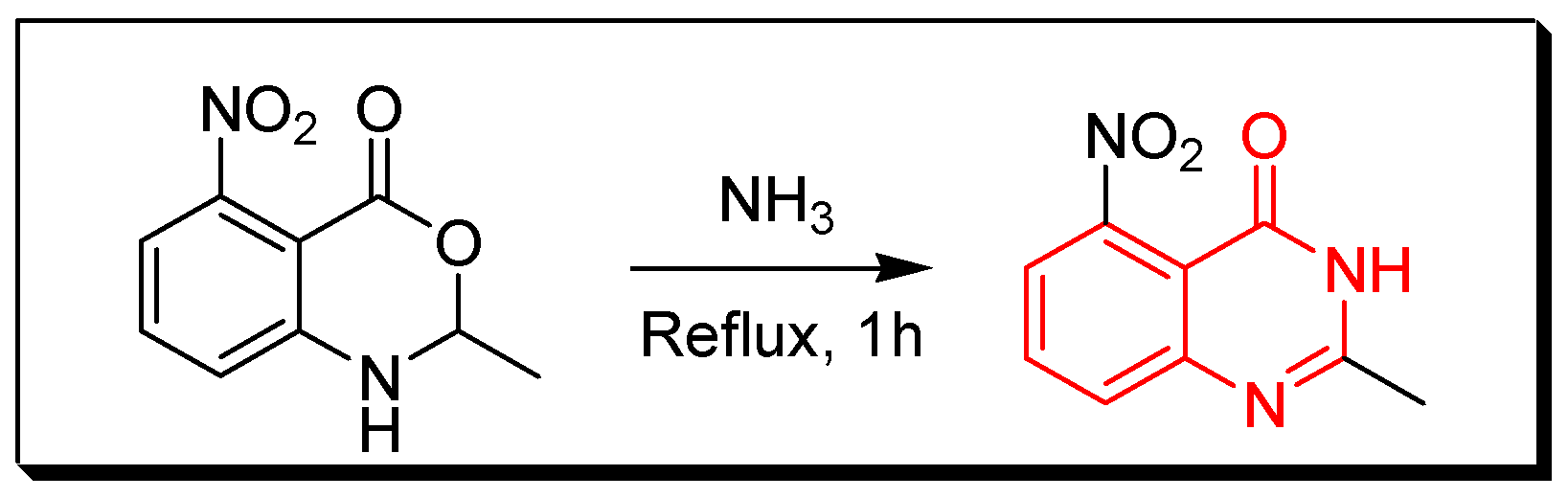
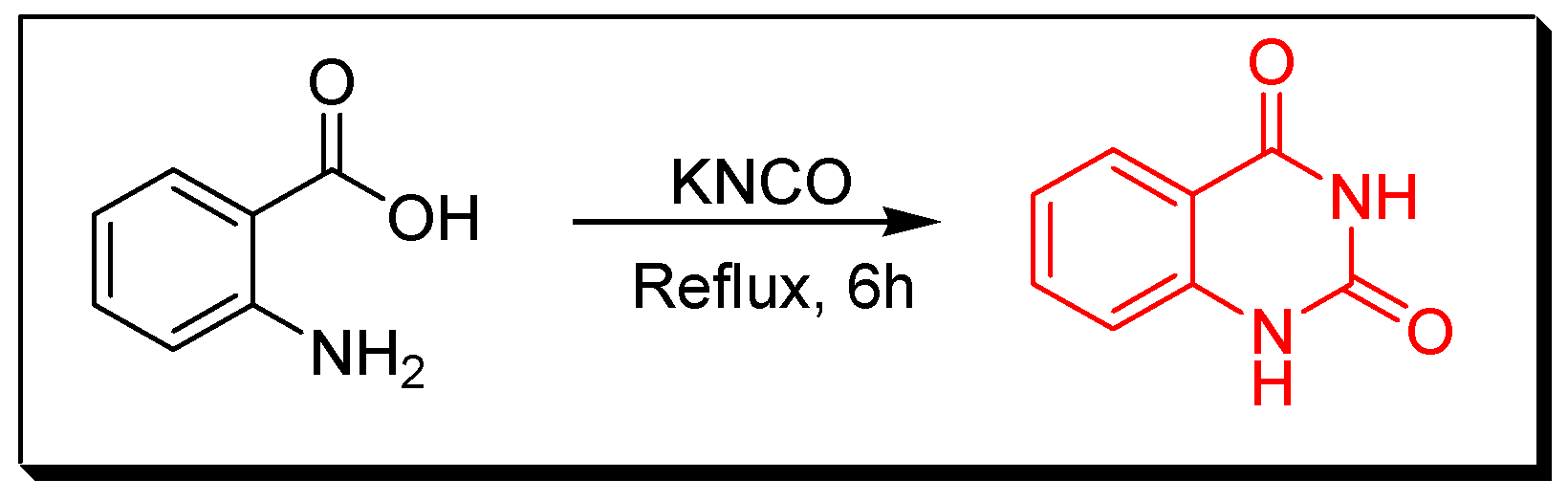
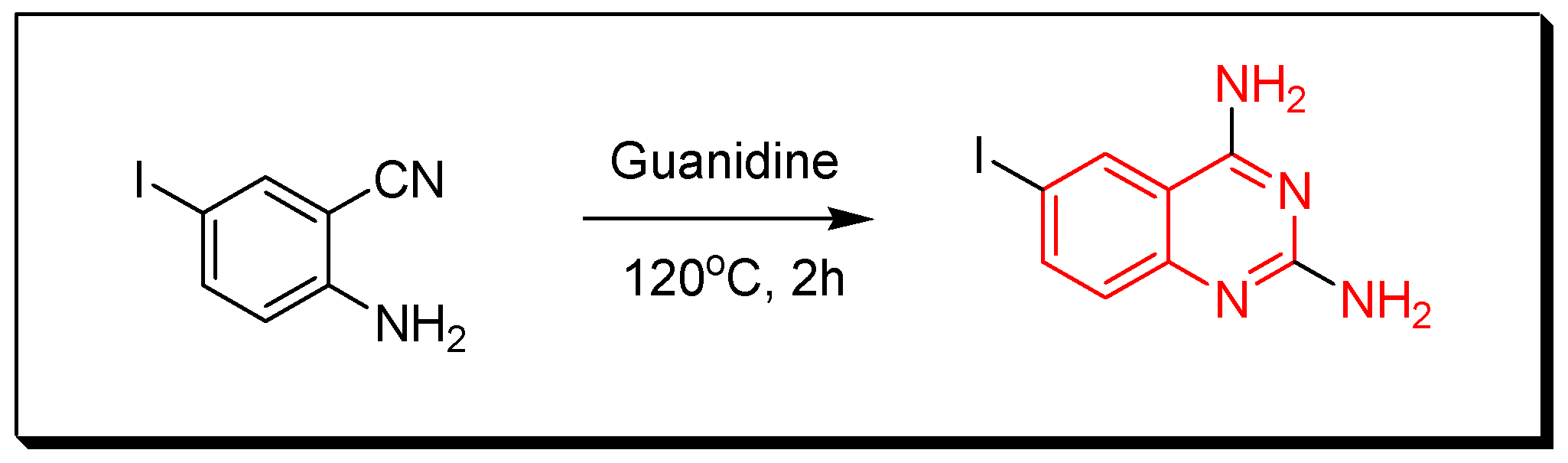
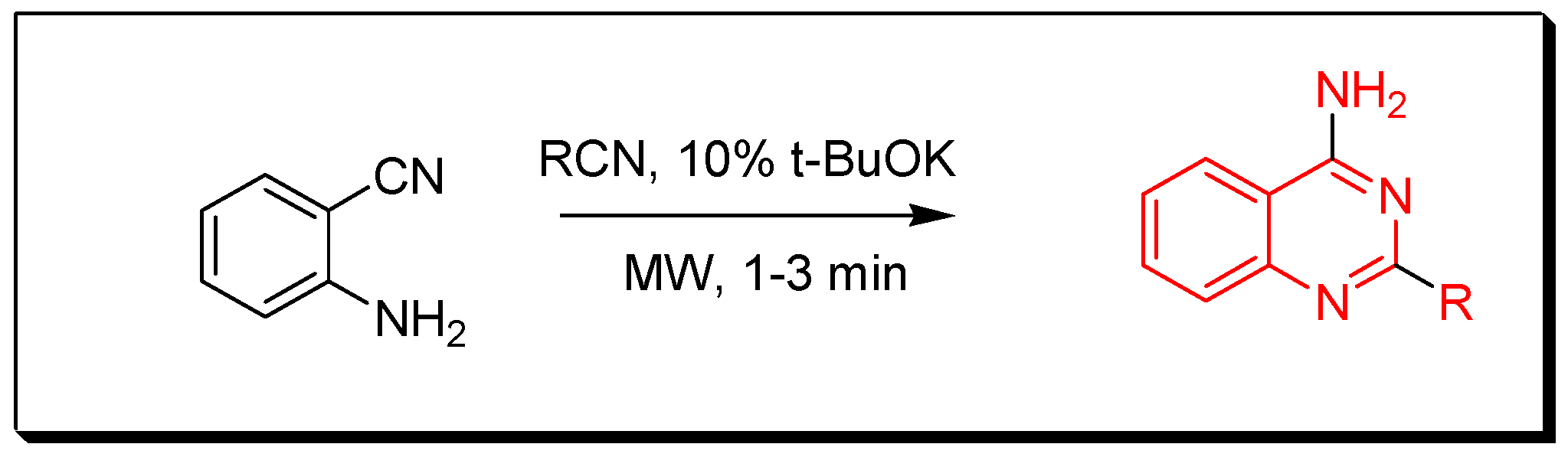
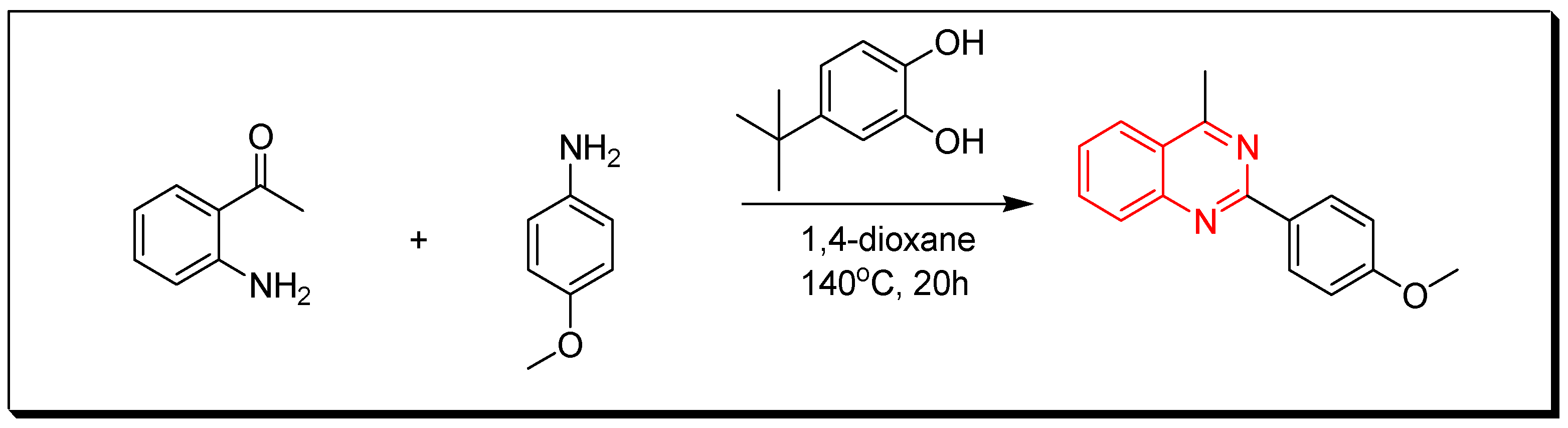
4. Methods of Preparation of Quinazolines
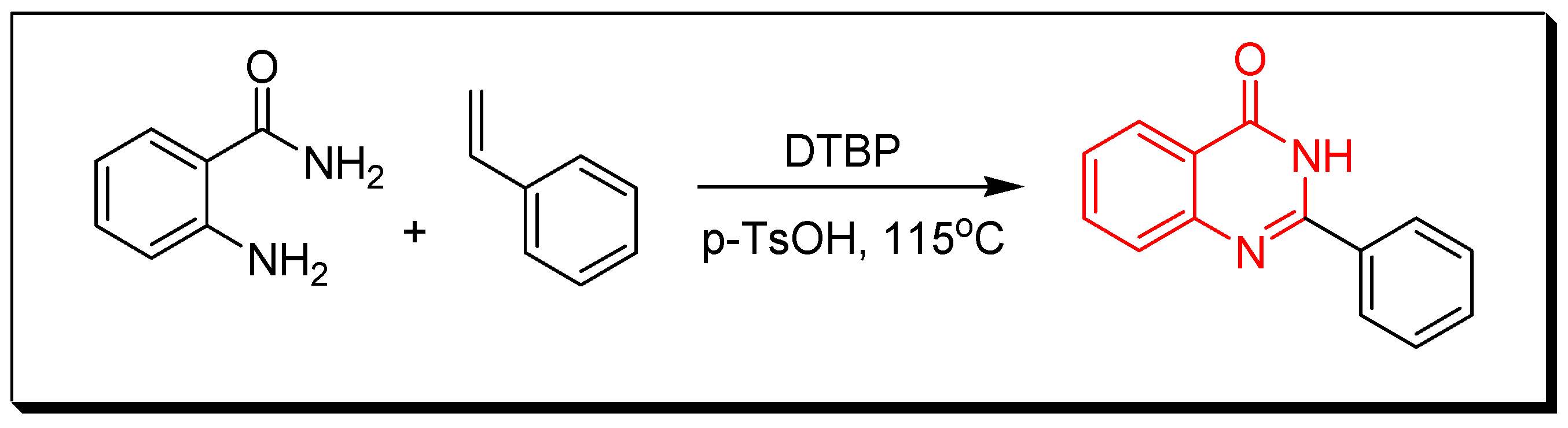
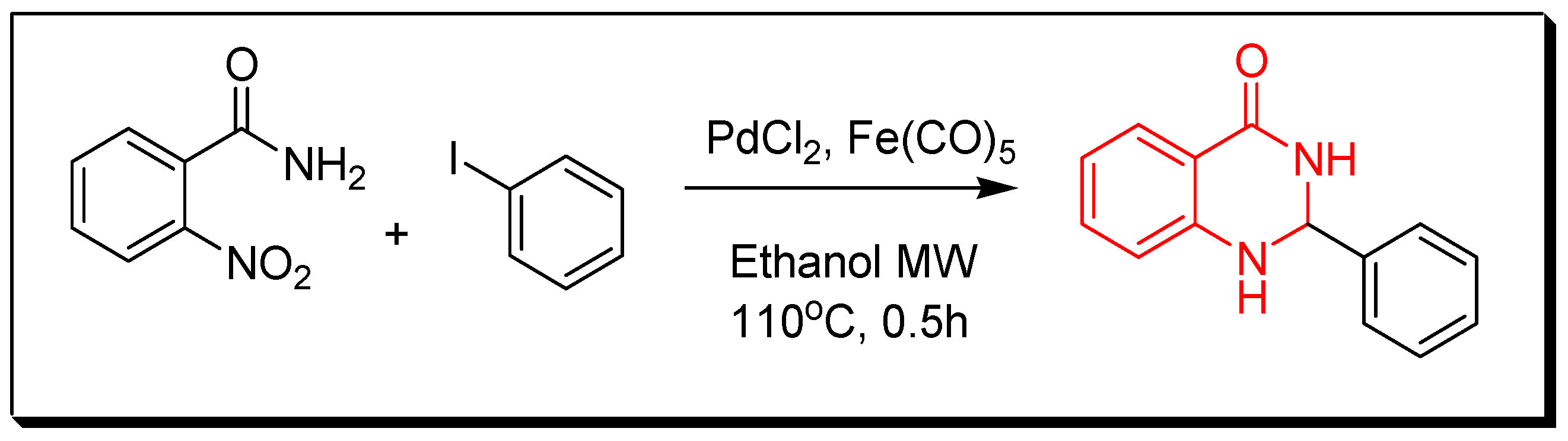
- Transition metals-catalyzed method. The nitrobenzamide derivative is reduced by palladium chloride (PdCl2) and iron pentacarbonyl Fe(CO)5 in presence of iodobenzene to give 2-phenyl-4(3H)-quinazolinone. This reaction is performed by a microwave-assisted reaction at 110 °C for 0.5 h with 85% yield (Figure 9) [36].
5. Mode of Action of Quinazolines as Anticancer Agents
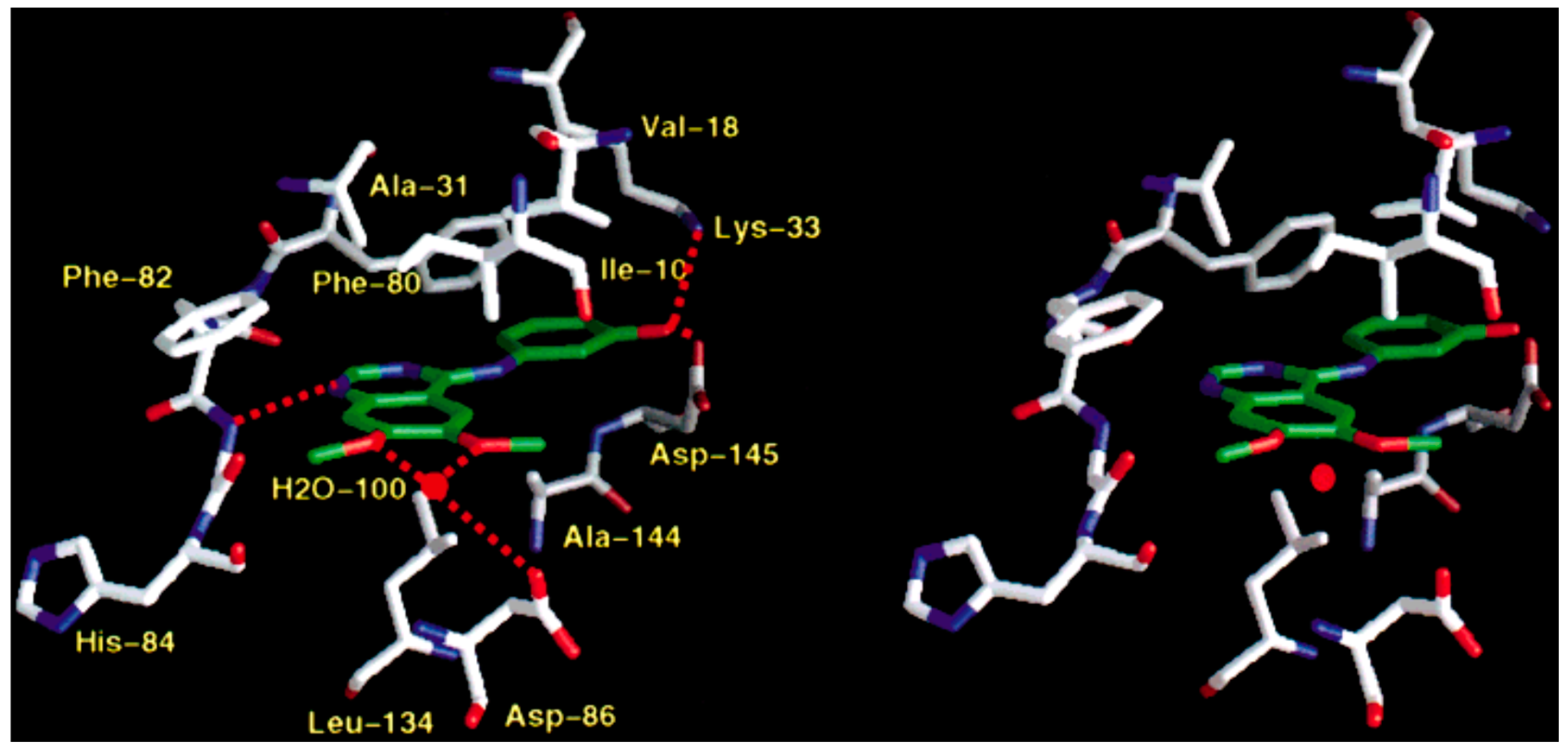
5.1. Crystallographic Studies of Quinazolines
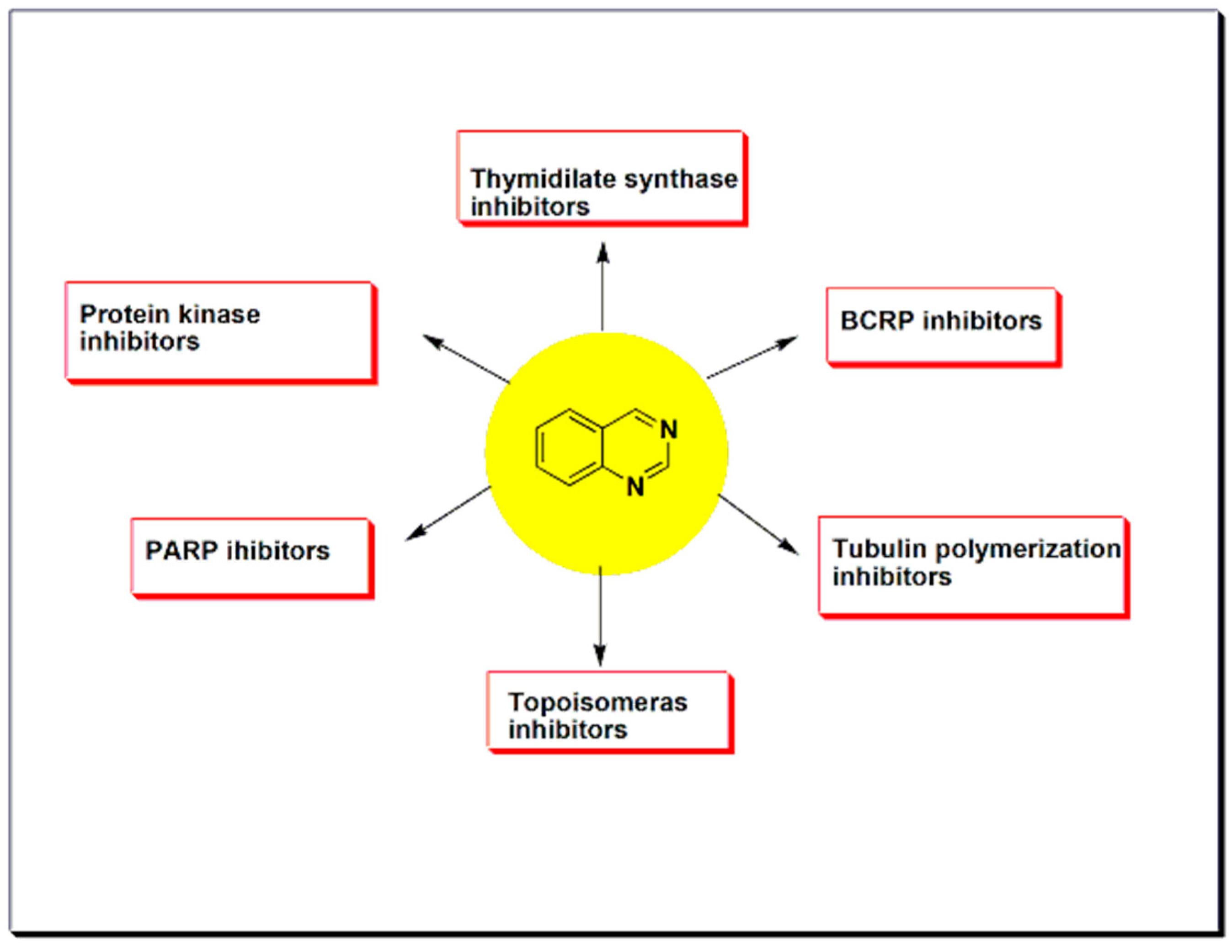
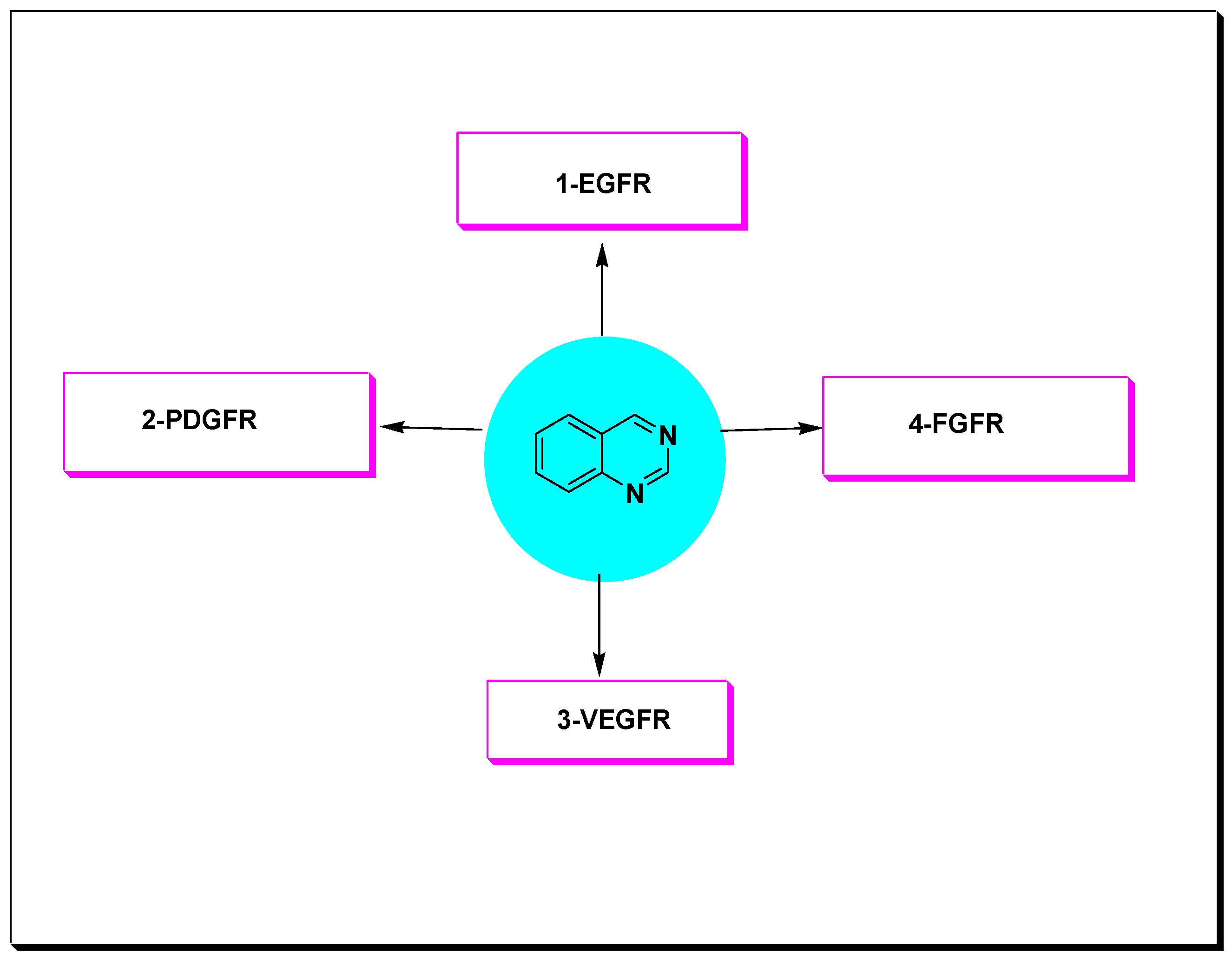
5.2. Protein Kinases Inhibitors
- Tyrosine kinases responsible for phosphorylation of phenolic hydroxyl (OH) group.
- Serine-threonine kinases responsible for phosphorylation of serine and threonine amino acids.
- Histidine-kinases responsible for phosphorylation of nitrogen in histidine residues.
- Epidermal growth factor receptor (EGFR);
- Platelet derived growth factor receptor (PDGFR);
- Vascular endothelial growth factor receptor (VEGFR);
- Fibroblast growth factor receptor (FGFR).
5.2.1. Epidermal Growth Factor Receptor (EGFR) Inhibitors
- Tyrosine kinase inhibitors molecules which act as a competitive inhibitor on EGFR;
- Monoclonal antibodies which interfere with the binding of EGF and TGF-α.
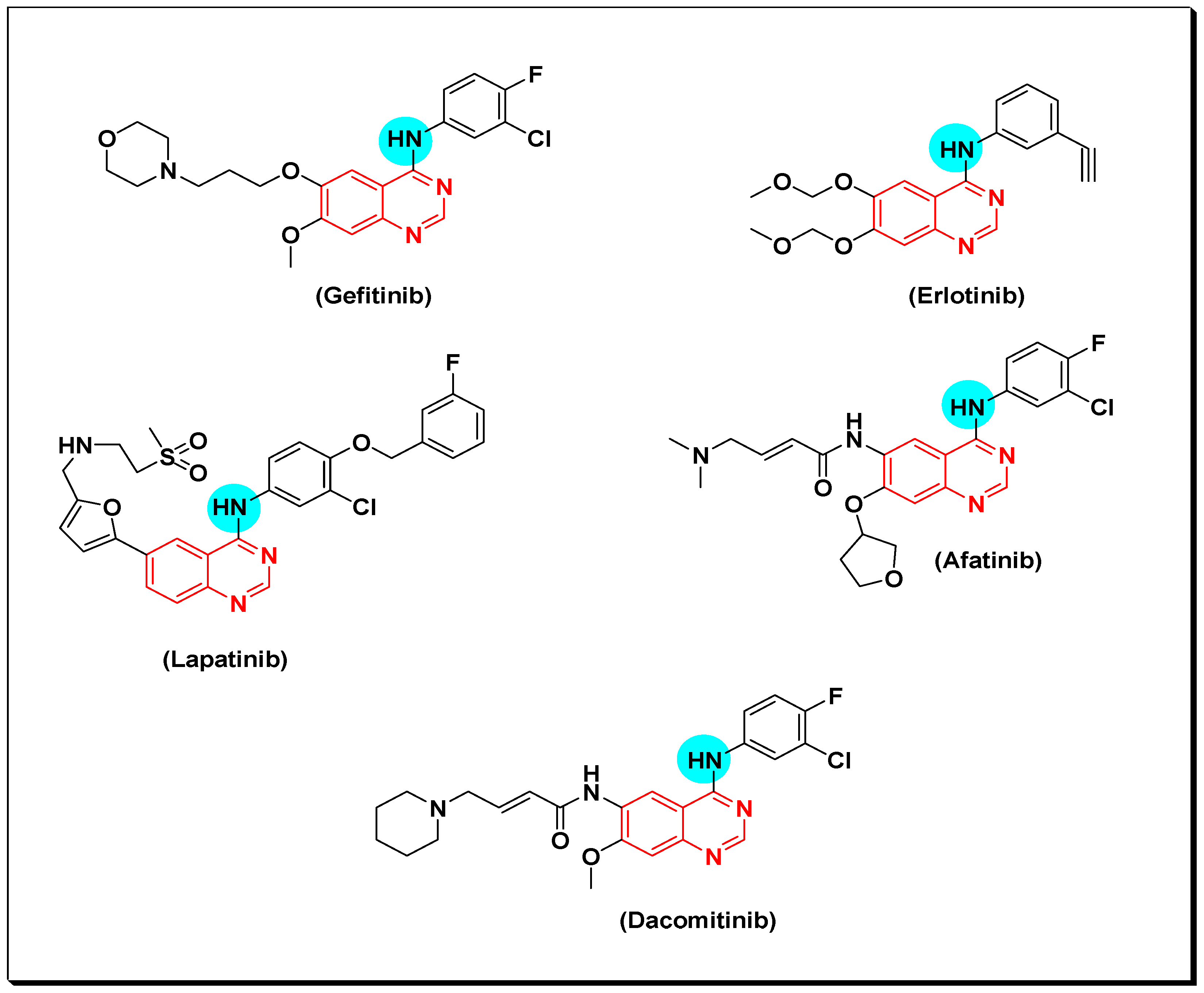
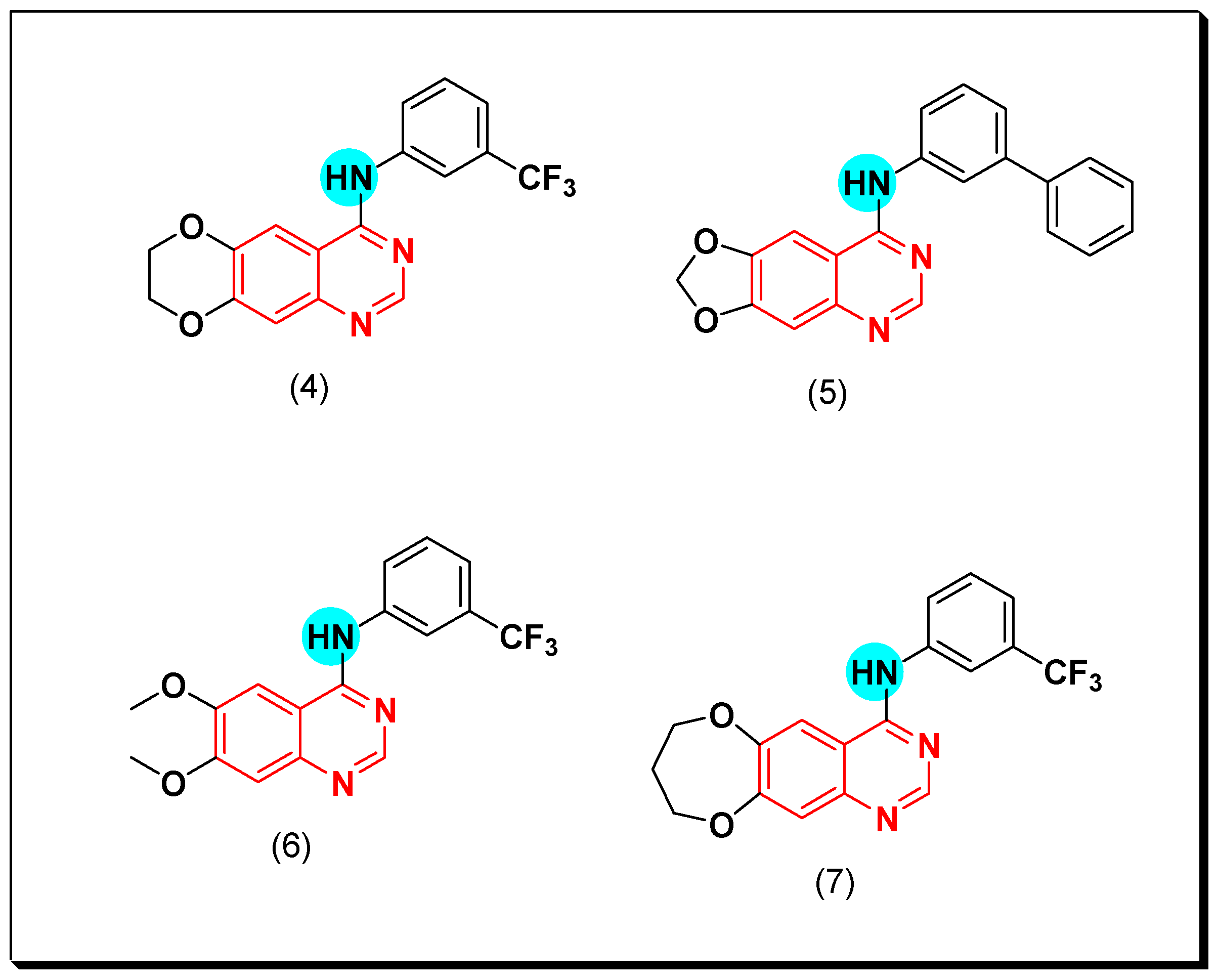
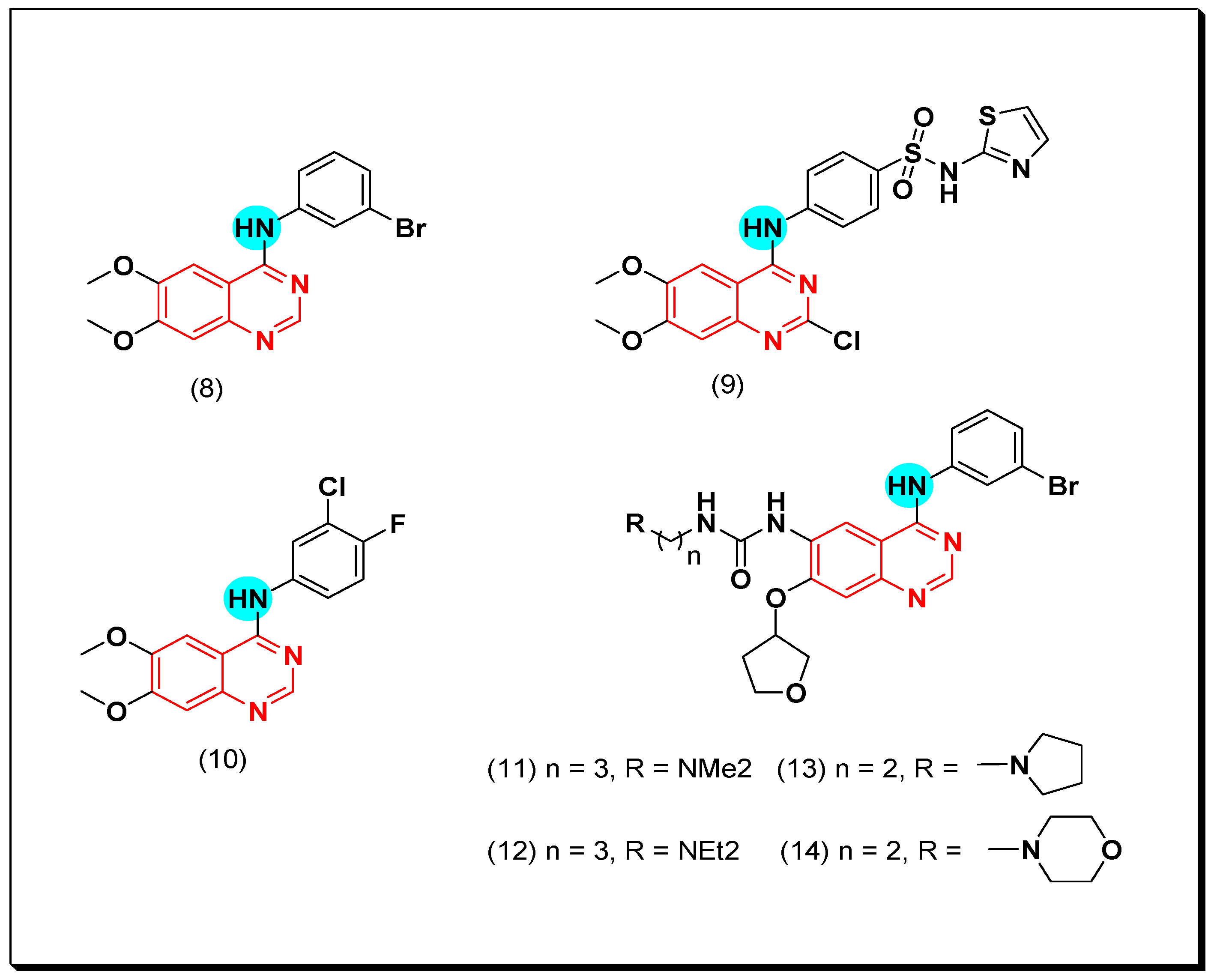
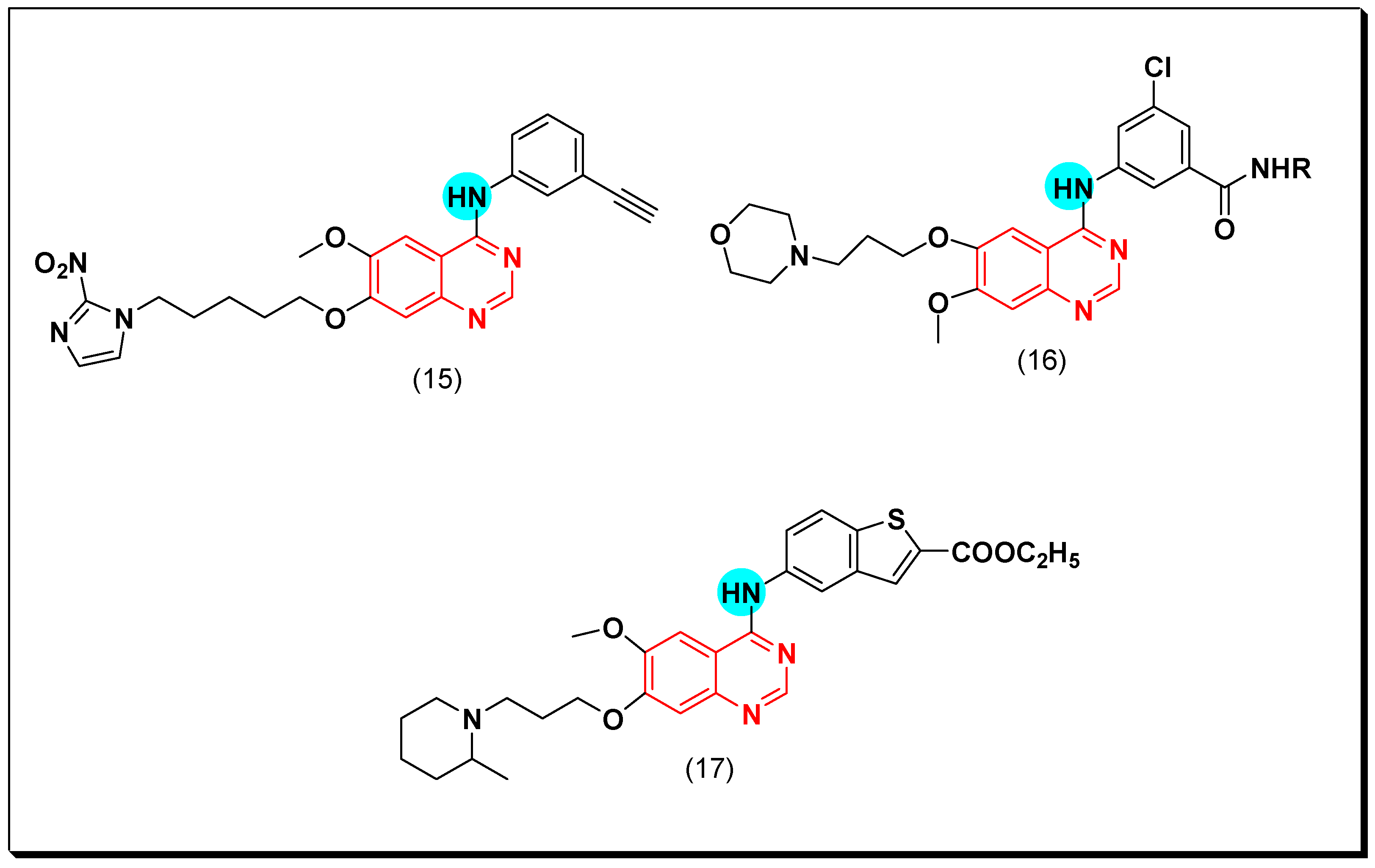
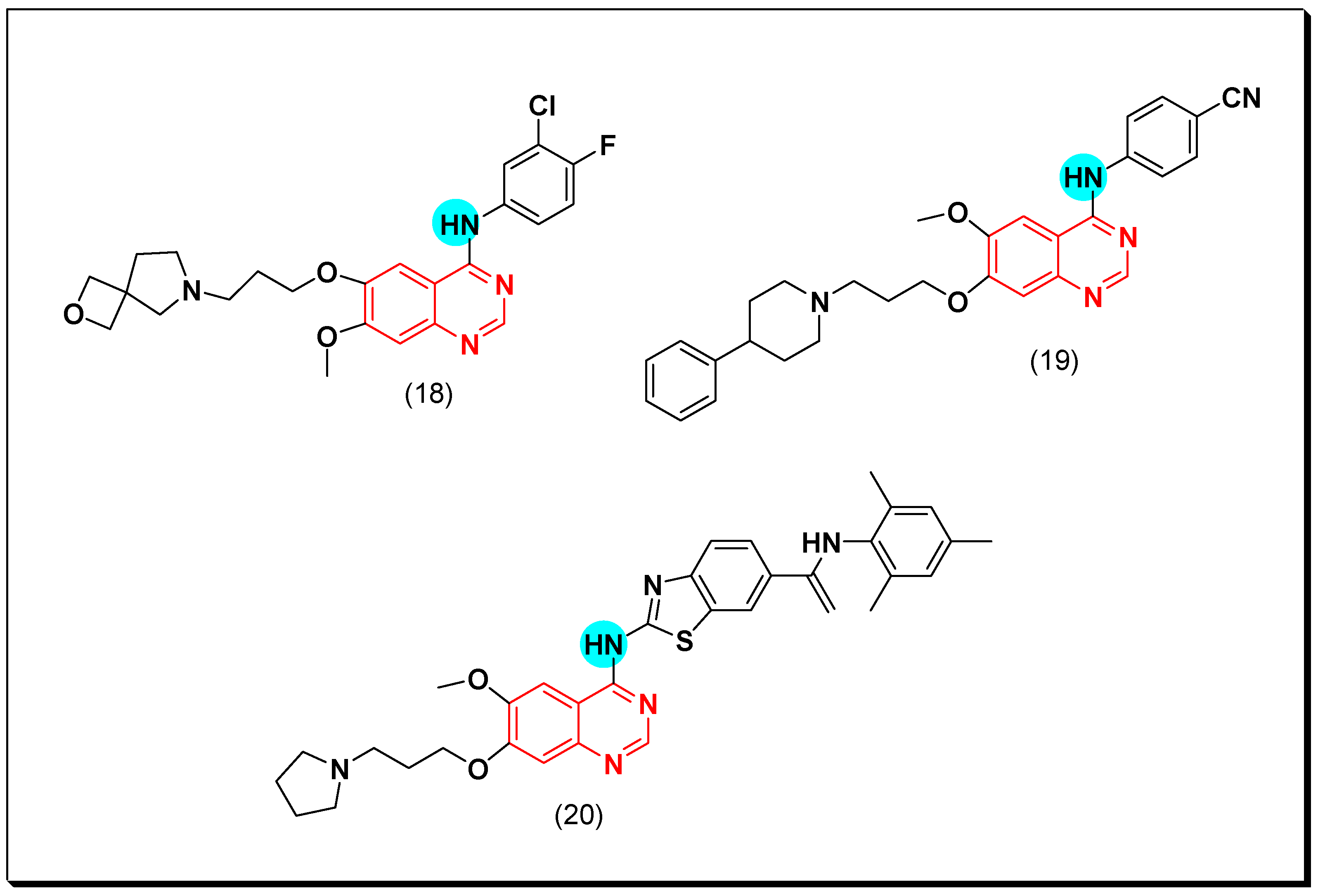
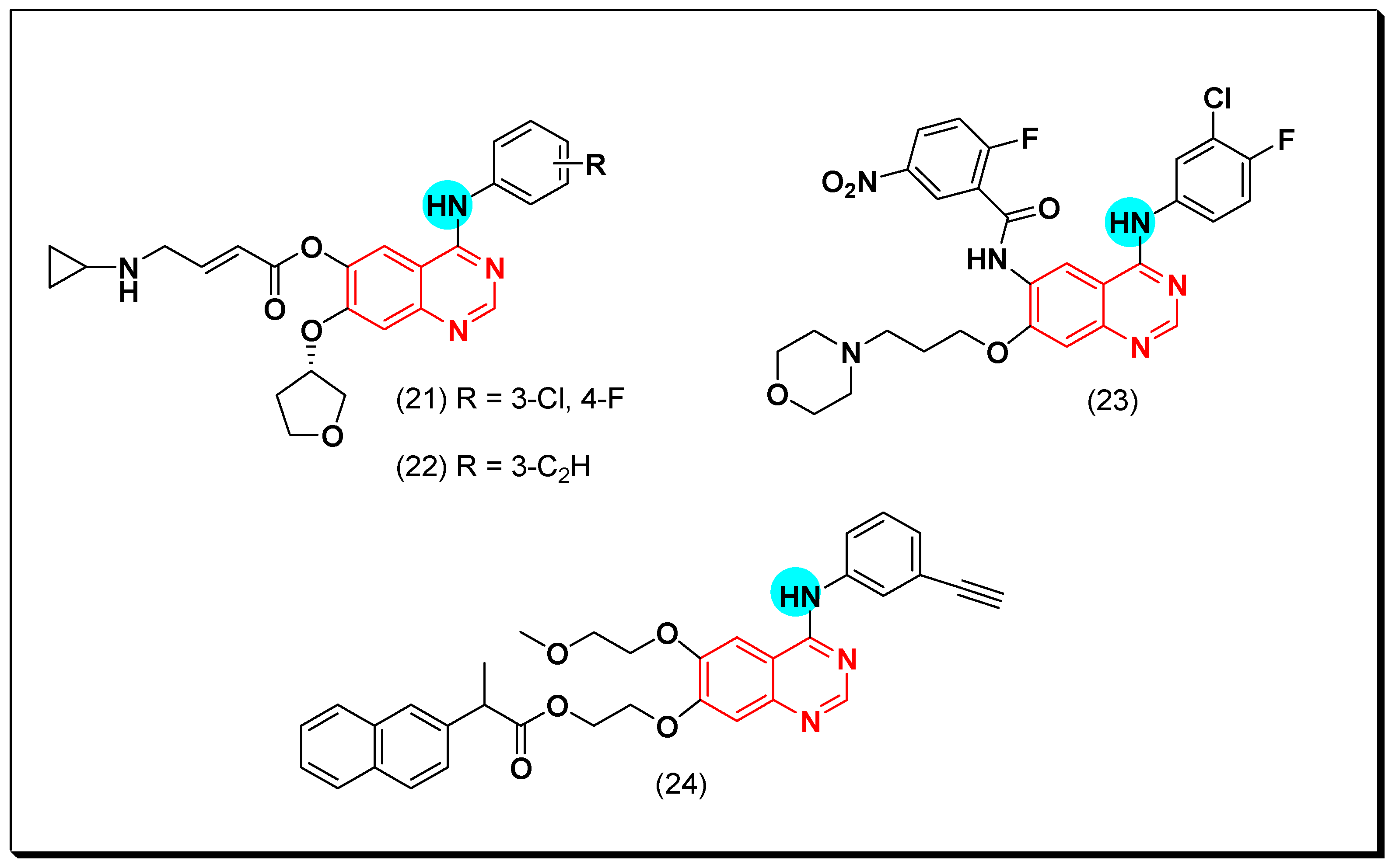
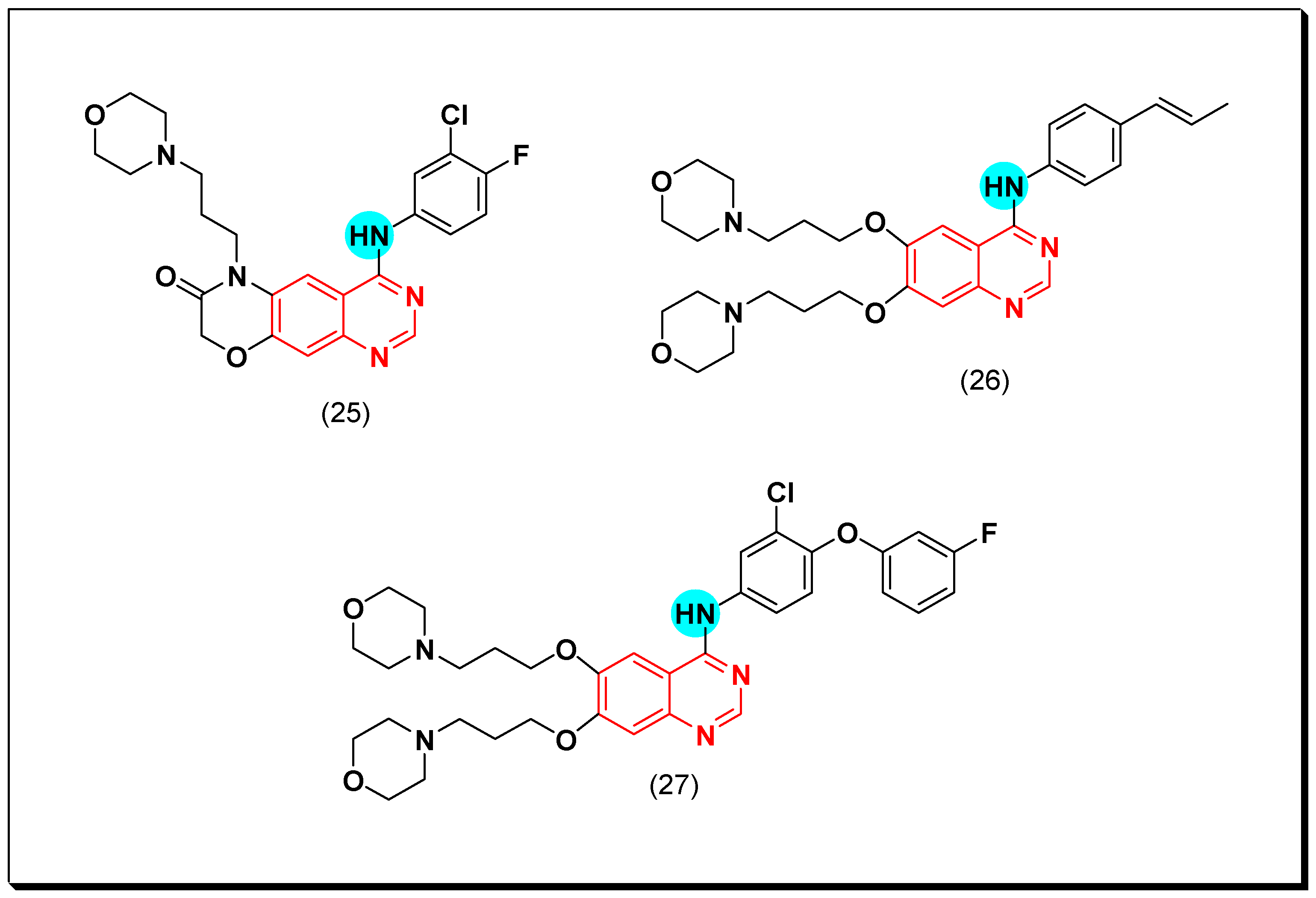
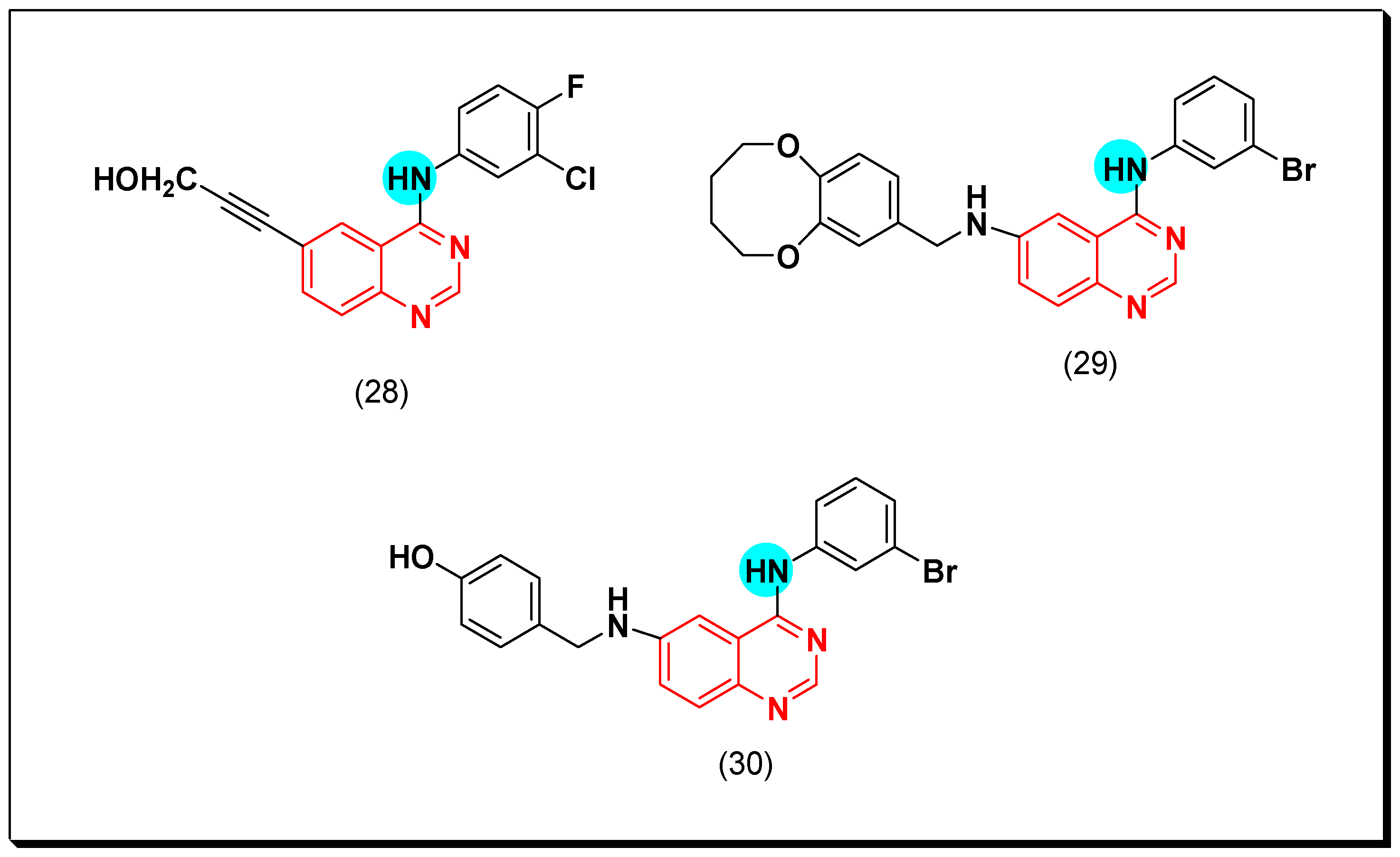
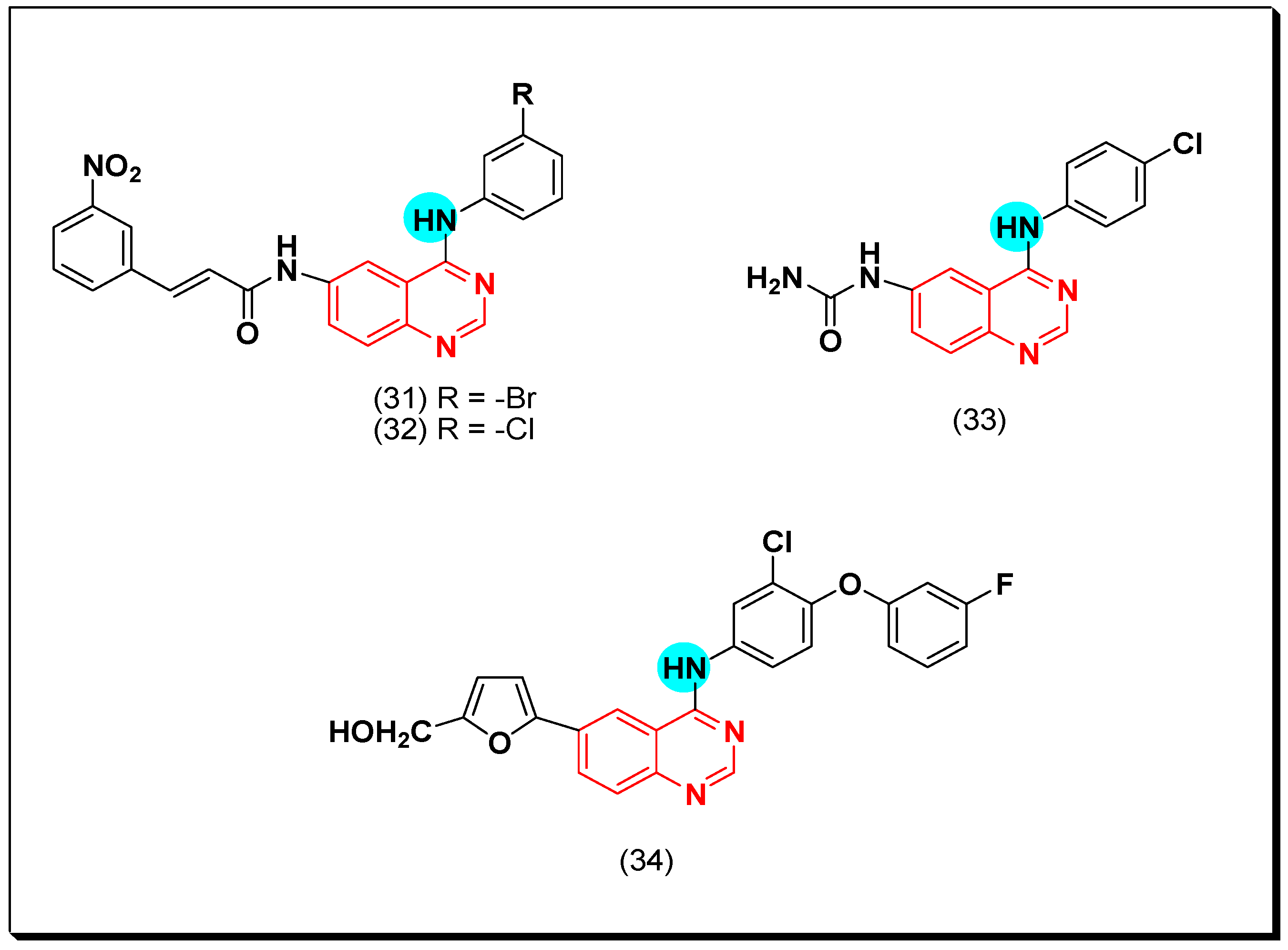
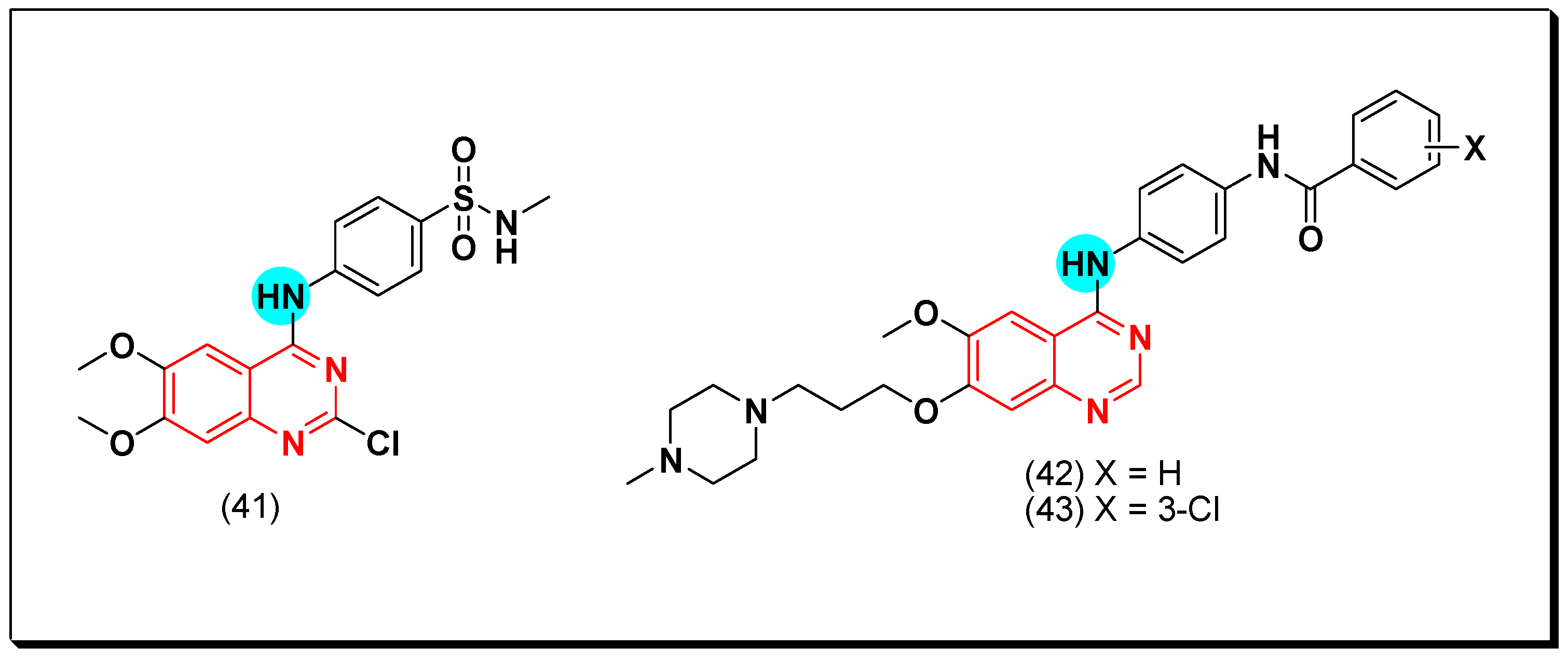
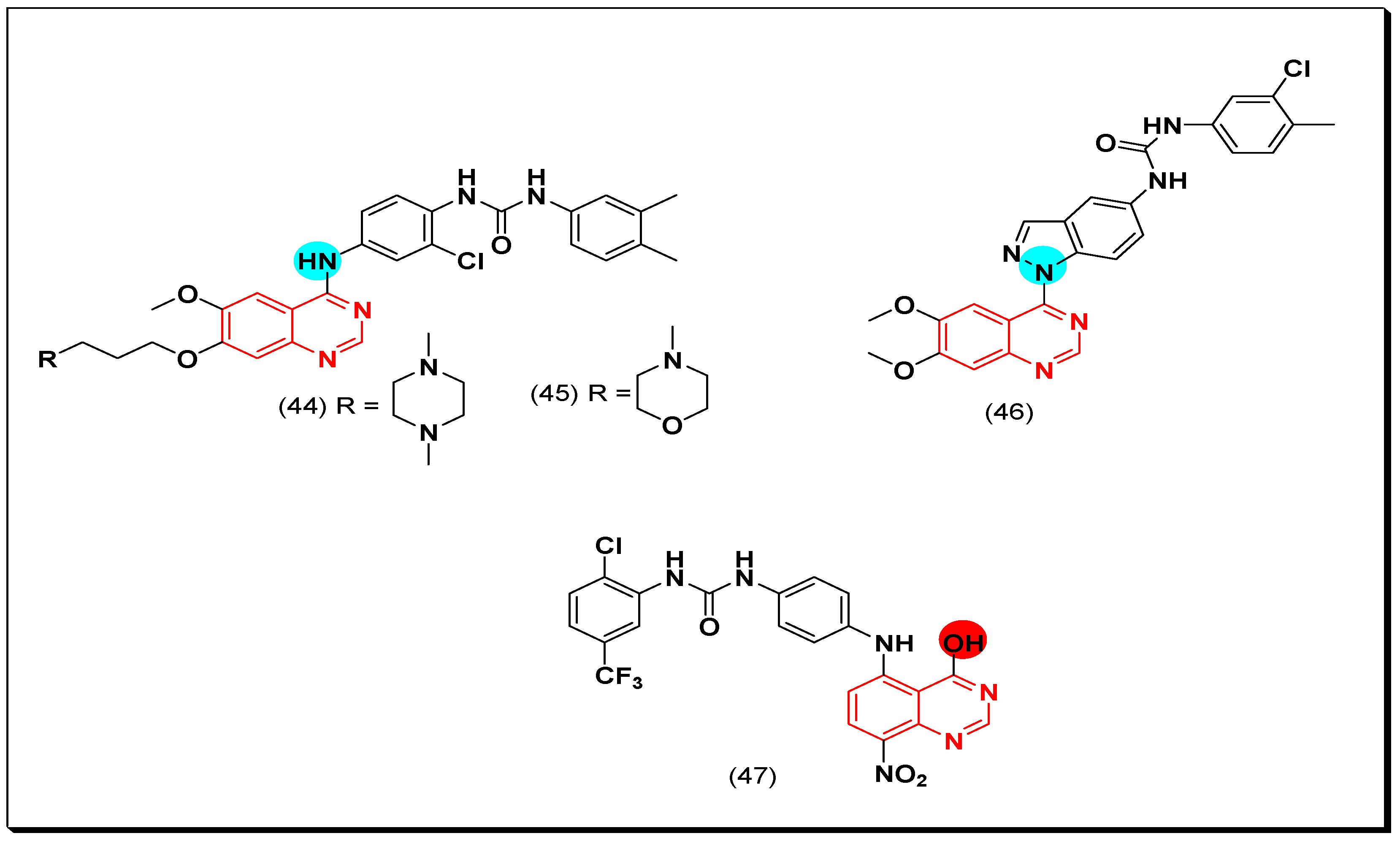
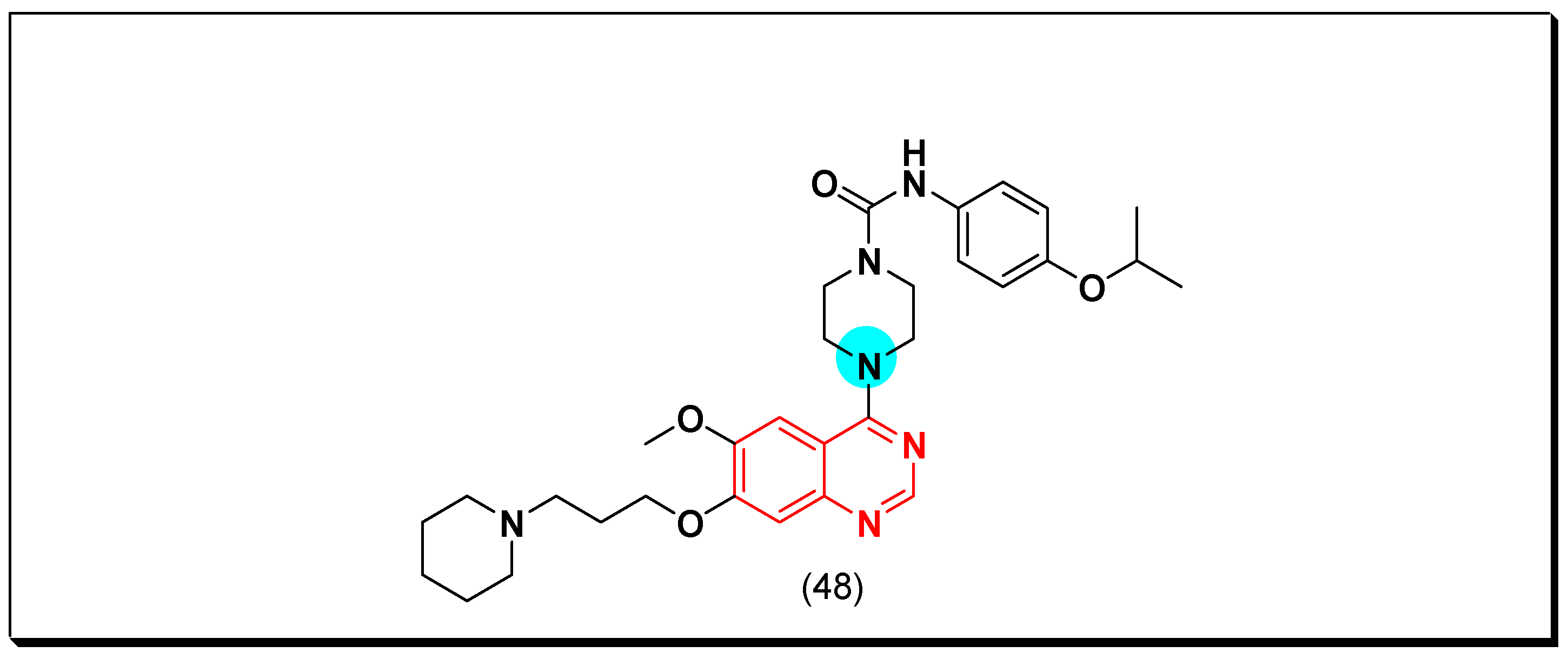
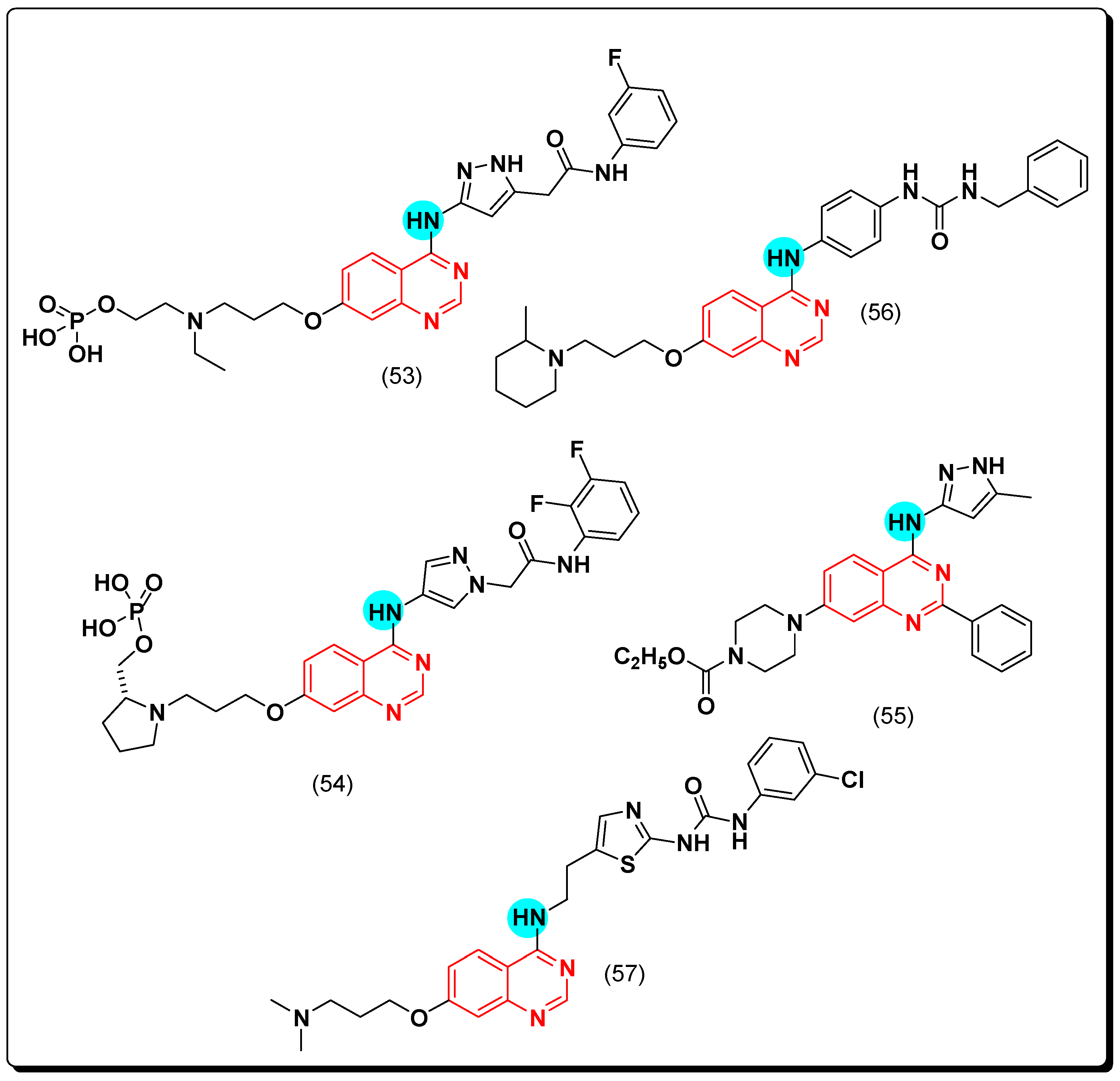
6,7-Substituted-4-anilinoquiazolines as Anticancer Agents
6-Substituted-4-anilinoquinazoline Derivatives
Structural activity relationship studies (SAR)
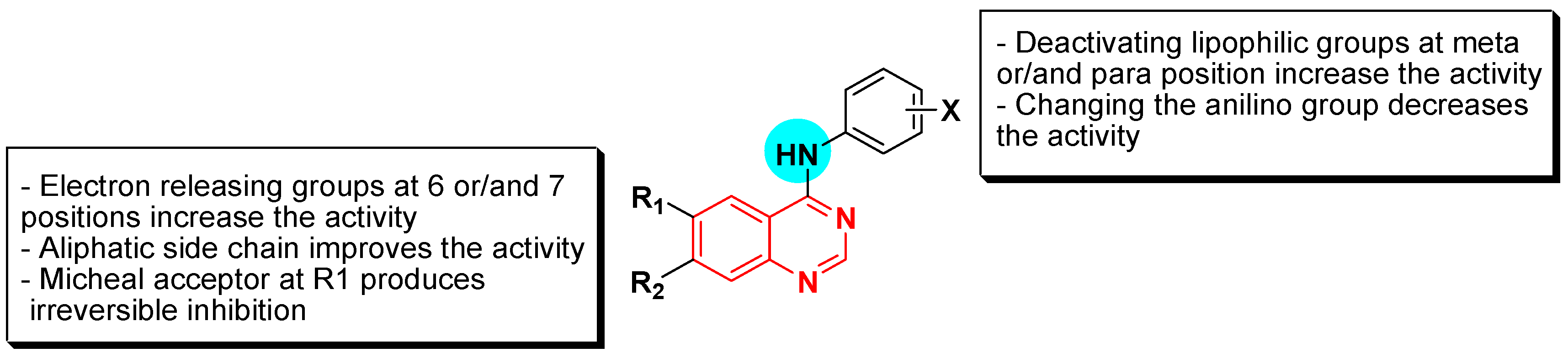
- The 4-anilinoquinazoline with substitution at the C-6 and/or the C-7 positions is the general pharmacophoric group required for the EGFR inhibitory activity. These structural requirements are shown by the common tyrosine-kinase inhibitors such as gefitinib, erlotinib, and other anticancer pharmaceutically marketed products.
- The electron-withdrawing groups such as fluoro, bromo, chloro, and ethylene at the aniline ring is advantageous for the antiproliferative activity.
- The 3-bromo substituted quinazoline molecules displayed potent activity.
- The 3-chloro-4-fluoro-aniline substituted quinazoline molecules showed strong activity.
- Changing the aniline moiety at the 4-position with other groups decreased the activity.
- The electron donating groups at the 6 and/or the 7-positions improved the binding activity of N1 and N3 of quinazoline system with the binding pocket.
- The propoxy linker at the C-6 and/or the C-7 of quinazoline moiety showed stronger activity than the methoxy group.
- Dioxygenated groups at the 6 and the 7 positions of quinazoline moiety improved the cytotoxic activity.
- The Michael addition group at 6-position of quinazoline leads to irreversible binding with the receptor-site.
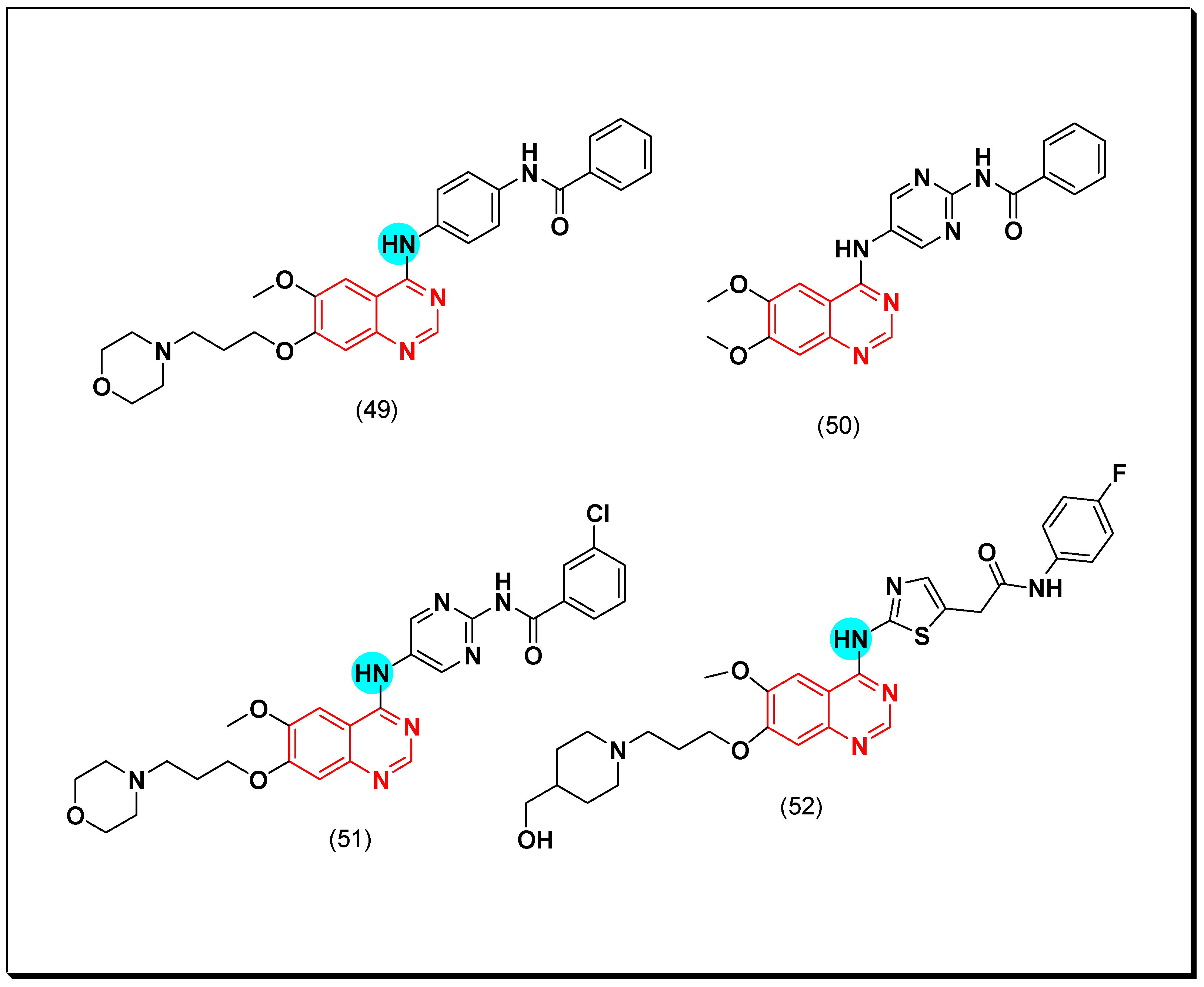
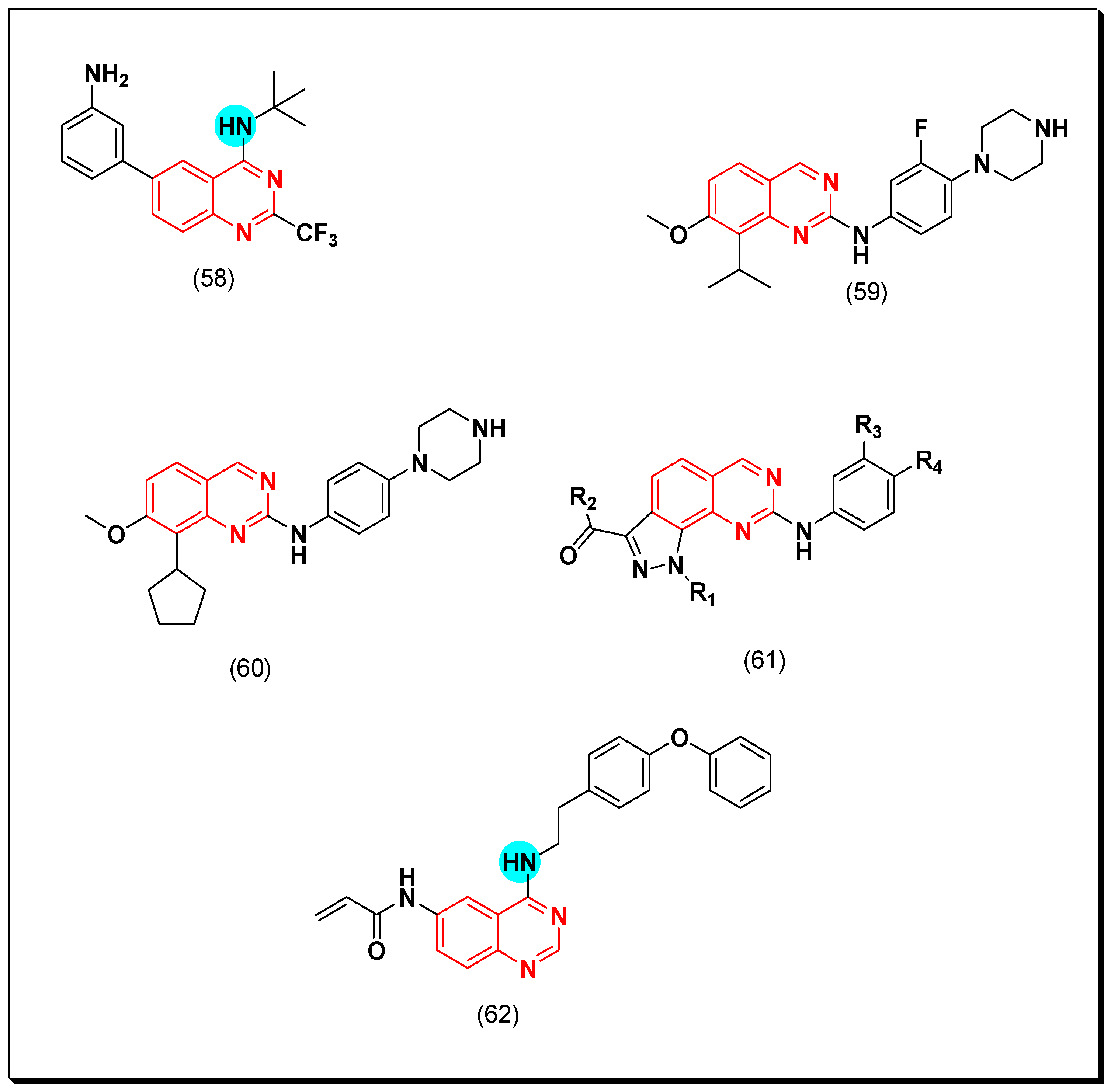
5.2.2. Vascular Endothelial Growth Factor Receptors (VEGFR) Inhibitors
SAR of VEGFR Inhibitors
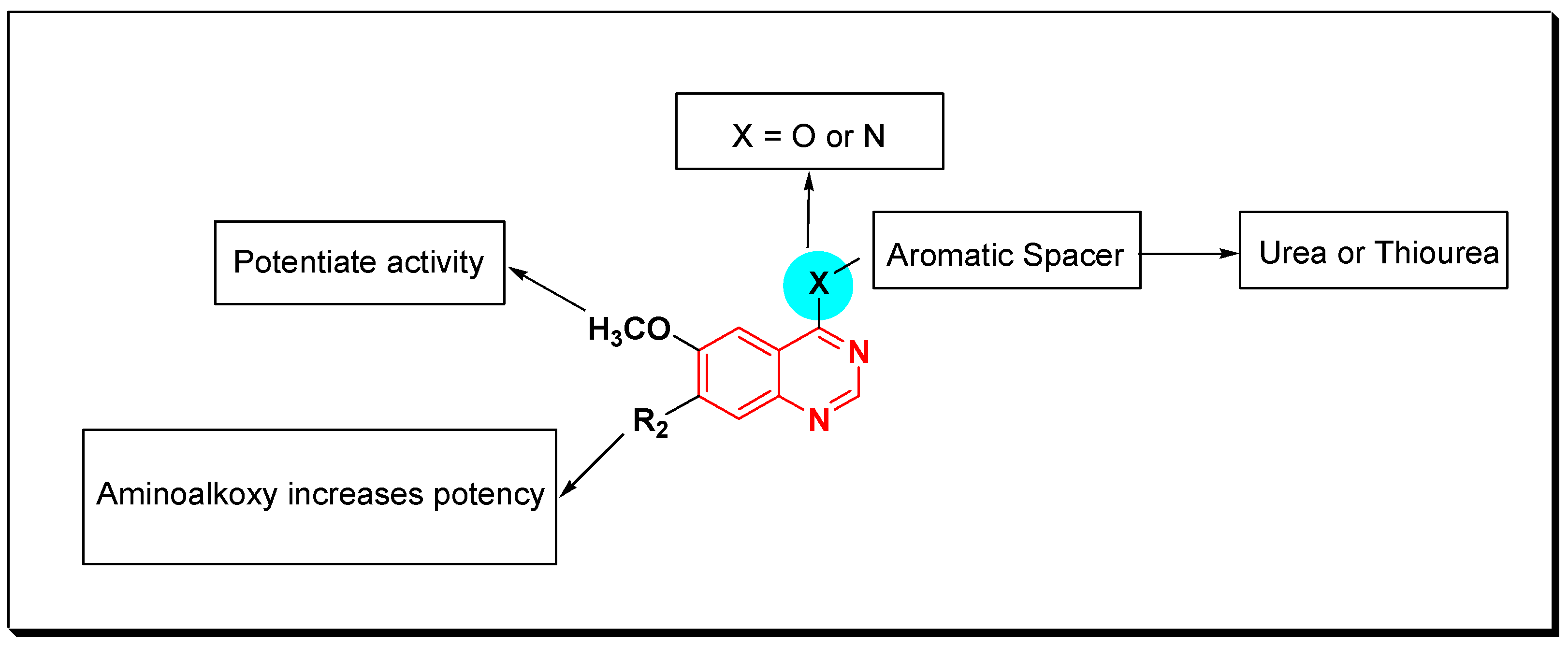
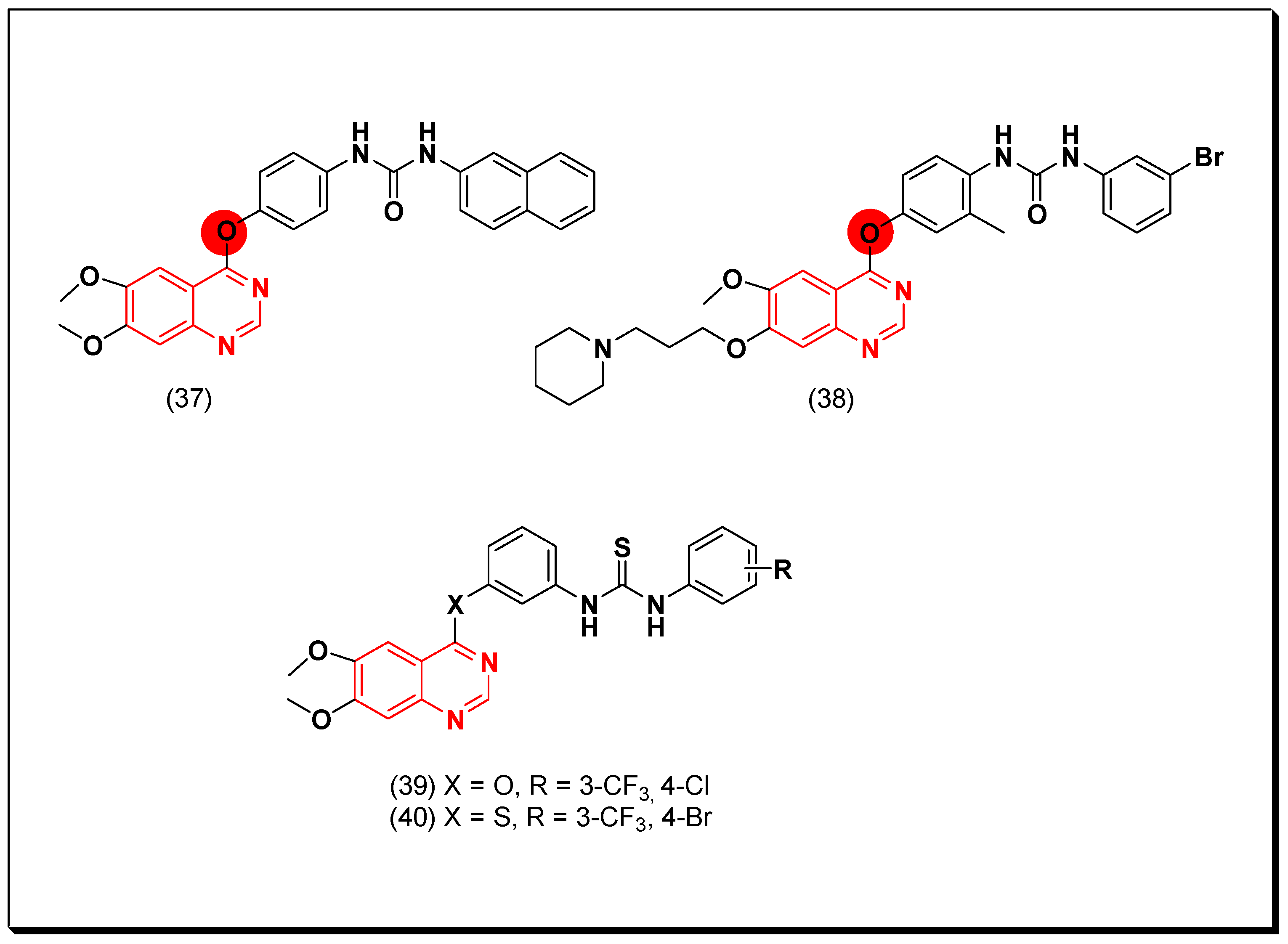
- Quinazolin-4-aniloino or quinazoline-4-oxyaryl scaffold is required for VEGFR inhibitory activity.
- Substitution at the 6-position of the quinazoline moiety with an electron-releasing group enhances the activity.
- Substitution at the 7-position of the quinazoline with an aminoalkoxy group increases the activity.
- Aromatic spacer between urea or thiourea and N or O at the 4-position of quinazoline is necessary for the activity.
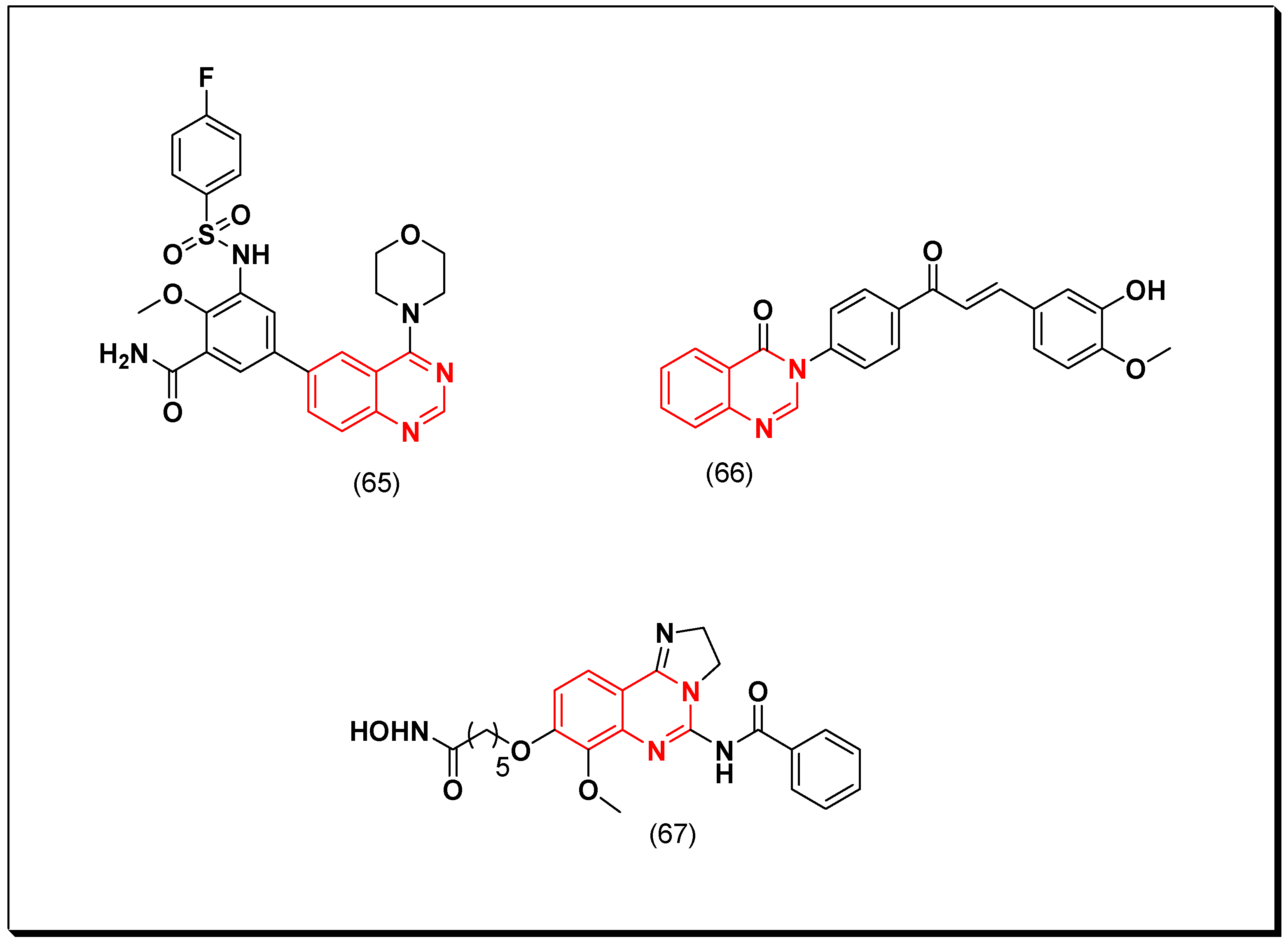
5.2.3. PDGFR Inhibitors
5.2.4. Serine-Threonine Kinase Inhibitors
- Serine-threonine receptor type kinase (TGFBR).
- Serine-threonine non-receptor type kinas (aurora kinases, CDK, and PI3K).
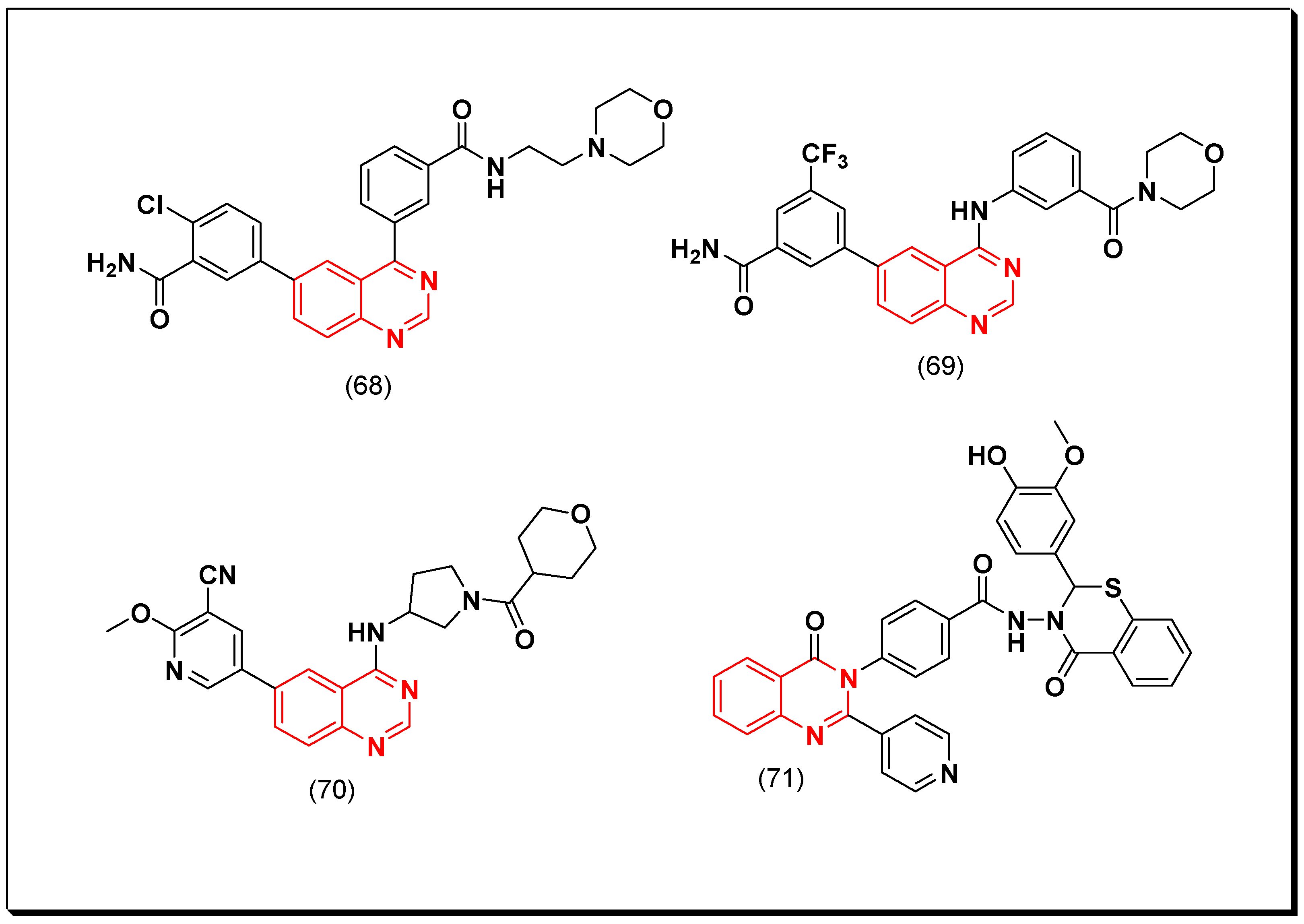
Aurora Kinase Inhibitors
SAR of Aurora Kinase Inhibitors
- Quinazoline with an aminoalkyl or an aminoaryl moiety at the 4-position of the quinazoline is required for the anticancer activity.
- Substitution at the 5 and the 6-positions of the quinazoline with an electron releasing group increases the activity.
- A lipophilic aromatic group attached to the 4-aminoaryl group increases the activity.
Cyclin-Dependent Kinase (CDK) Inhibitors
Phosphoinositid-3-Kinase (PI3K) Inhibitors
6. Pharmaceutical Marketed Anticancer Quinazolines
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zayed, M.F.; Ahmed, H.E.A.; Ihmaid, S.K.; Omar, A.M.; Abdelrahim, A.S. Synthesis and screening of some new fluorinated quinazolinone–sulphonamide hybrids as anticancer agents. J. Taibah Univ. Sci. 2015, 10, 333–339. [Google Scholar] [CrossRef]
- Bansal, R.; Malhotra, A. Therapeutic progression of quinazolines as targeted chemotherapeutic agents. Eur. J. Med. Chem. 2021, 211, 113016. [Google Scholar] [CrossRef]
- Zayed, M.F.; Rateb, H.; Ahmed, S.; Khaled, O.; Ibrahim, S. Quinazolinone-amino acid hybrids as Dual Inhibitors of EGFR Kinase and Tubulin Polymerization. Molecules 2018, 23, 1699. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Al-Rashida, M.; Uroos, M.; Ali, S.A.; Arshia; Ishtiaq, M.; Khan, K.M. Quinazoline and quinazolinone as important medicinal scaffolds: A comparative patent review (2011–2016). Expert Opin. Ther. Pat. 2018, 28, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F.; Ahmed, S.; Ihmaid, S.; Ahmed, H.E.; Rateb, H.; Ibrahim, S. Design, Synthesis, Cytotoxic Evaluation and Molecular Docking of New Fluoroquinazolinones as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Int. J. Mol. Sci. 2018, 19, 1731. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F.; Ahmed, H.E.A.; Ihmaid, S.; El-Adl, K.; Asiri, A.; Omar, A.M. Modelling and Anticonvulsant Studies of New Quinazolines Showing Three Highly Active Compounds with Low Toxicity and High Affinity to the GABA-A Receptor. Molecules 2017, 22, 188. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F.; Hassan, M.H. Design, Synthesis and Biological Evaluation Studies of Novel Quinazoline Derivatives as Cytotoxic Agents. Drug Res. 2013, 63, 210–215. [Google Scholar] [CrossRef]
- Barreca, M.; Spanò, V.; Rocca, R.; Bivacqua, R.; Abel, A.; Maruca, A.; Montalbano, A.; Raimondi, V.M.; Tarantelli, C.; Gaudio, E.; et al. Development of [1,2]oxazoloisoindoles tubulin polymerization inhibitors: Further chemical modifications and potential therapeutic effects against lymphomas. Eur. J. Med. Chem. 2022, 243, 114744. [Google Scholar] [CrossRef]
- Barreca, M.; Ingarra, M.A.; Raimondi, V.M.; Spanò, V.; Piccionello, P.A.; Franco, D.M.; Menilli, L.; Gandin, V.; Miolo, G.; Barraja, P.; et al. New tricyclic systems as photosensitizers towards triple negative breast cancer cells. Arch. Pharm. Res. 2022, 45, 806–821. [Google Scholar] [CrossRef]
- Duró, C.; Jernei, T.; Szekeres, K.J.; Láng, G.G.; Oláh-Szabó, R.; Bősze, S.; Szabó, I.; Hudecz, F.; Csámpai, A. Synthesis and SAR Analysis of Novel 4-Hydroxytamoxifen Analogues Based on Their Cytotoxic Activity and Electron-Donor Character. Molecules 2022, 27, 6758. [Google Scholar] [CrossRef]
- Fan, C.; Zhong, T.; Yang, H.; Yang, Y.; Wang, D.; Yang, X.; Xu, Y.; Fan, Y. Design, synthesis, biological evaluation of 6-(2-amino-1H-benzo[d]imidazole-6-yl)quinazolin-4(3H)-one derivatives as novel anticancer agents with Aurora kinase inhibition. Eur. J. Med. Chem. 2020, 190, 112108. [Google Scholar] [CrossRef]
- Shagufta; Ahmad, I. An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. Med. Chem. Commun. 2017, 8, 871–885. [Google Scholar] [CrossRef]
- Haider, K.; Das, S.; Joseph, A.; Yar, M.S. An appraisal of anticancer activity with structure-activity relationship of quinazoline and quinazolinone analogues through EGFR and VEGFR inhibition: A review. Drug Dev. Res. 2022, 83, 859–890. [Google Scholar] [CrossRef] [PubMed]
- Auti, P.S.; George, G.; Paul, A.T. Recent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids. RSC Adv. 2020, 10, 41353–41392. [Google Scholar] [CrossRef]
- Sunil Kumar, A.; Kudva, J.; Lahtinen, M.; Peuronen, A.; Sadashiva, R.; Naral, D. Synthesis, characterization, crystal structures and biological screening of 4-amino quinazoline sulfonamide derivatives. J. Mol. Struct. 2019, 1190, 29–36. [Google Scholar] [CrossRef]
- El-Zahabia, M.A.; Bamanie, H.F.; Ghareeb, S.; Alshaeri, K.H.; Alasmari, M.M.; Muostafa, M.; Al-Marzoki, Z.; Zayed, M.F. Design, Synthesis, Molecular Modeling andAnti-Hyperglycemic Evaluation of Quinazoline-Sulfonylurea Hybrids as Peroxisome Proliferator-Activated Receptor Gamma (PPAR) and Sulfonylurea Receptor (SUR) Agonists. Int. J. Mol. Sci. 2022, 23, 9605. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F.; Ibrahim, S.; Habib, E.E.; Hassan, M.H.; Ahmed, S.; Rateb, H.S. Design, synthesis, antimicrobial and anti-biofilm evaluation, and molecular docking of new substituted fluoroquinazolinones. J. Med. Chem. 2019, 15, 657–673. [Google Scholar]
- Zhang, J.; Liu, J.; Ma, Y.; Ren, D.; Cheng, P.; Zhao, J.; Zhang, F.; Yao, Y. One-pot synthesis and antifungal activity against plant pathogens of quinazolinone derivatives containing an amide moiety. Bioorg. Med. Chem. Lett. 2016, 26, 2273–2277. [Google Scholar] [CrossRef] [PubMed]
- Santos-Ballardo, L.; García-Páez, F.; Picos-Corrales, L.A.; Ochoa-Terán, A.; Bastidas, P.; Calderón-Zamora, L.; Rendón-Maldonado, G.; Osuna-Martínez, U.; Sarmiento-Sánchez, J.I. Synthesis, biological evaluation and molecular docking of 3-substituted quinazoline-2,4(1H, 3H)-diones. J. Chem. Sci. 2020, 132, 100. [Google Scholar] [CrossRef]
- Jain, R.K.; Kashaw, V. Design, synthesis and evaluation of novel 2,3-disubstituted-4-(3H) quinazolinone derivatives. Asian J. Pharm. Pharmacol. 2018, 4, 644–656. [Google Scholar] [CrossRef]
- Hricoviniova, J.; Hricoviniova, Z.; Kozica, K. Antioxidant, Cytotoxic, Genotoxic, and DNA Protective Potential of 2,3-Substituted Quinazolinones: Structure—Activity Relationship Study. Int. J. Mol. Sci. 2021, 22, 610. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F. New fluorinated quinazolinone derivatives as anticonvulsant agents. J. Taibah Univ. Sci. 2014, 9, 104–109. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Abdel-Kader, M.S.; Alqahtani, A.S.; Soliman, A.M. Synthesis of some quinazolinones inspired from the natural alkaloid L-norephedrine as EGFR inhibitors and radiosensitizers. J. Enzym. Inhib. Med. Chem. 2021, 36, 218–237. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) Chemical Computing Group. Available online: http://www.chemcomp.com (accessed on 10 May 2019).
- Zayed, M.F.; Hassan, M.H. Synthesis and biological evaluation studies of novel quinazolinone derivatives as antibacterial and anti-inflammatory agents. Saudi Pharm. J. 2014, 22, 157–162. [Google Scholar] [CrossRef]
- Swiss Institute of Bioinformatics (SwissADME). Available online: http://www.swissADME.ch (accessed on 10 November 2022).
- Zayed, M.F.; Ahmed, E.A.; Omar, A.M.; Abdelrahim, A.S.; El-Adl, K. Design, synthesis, and biological evaluation studies of novel quinazolinone derivatives as anticonvulsant agents. Med. Chem. Res. 2013, 22, 5823–5831. [Google Scholar] [CrossRef]
- Wang, D.; Gao, F. Quinazoline derivatives: Synthesis and bioactivities. Chem. Cent. J. 2013, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Connolly, D.J.; Cusack, D.; O’Sullivan, T.P.; Guiry, P.J. Synthesis of quinazolinones and quinazolines. Tetrahedron 2005, 61, 10153–10202. [Google Scholar] [CrossRef]
- Meyer, J.F.; Wagner, E.C. The Niementowski reaction. The use of methyl anthranilate or isatoic anhydride with substituted amides or amidines in the formation of 3-substituted-4-keto-3,4-dihydroquinazolines. The course of the reaction. J. Org. Chem. 1943, 8, 239–252. [Google Scholar] [CrossRef]
- Asif, M. Chemical Characteristics, Synthetic Methods, and Biological Potential of Quinazoline and Quinazolinone Derivatives. Int. J. Med. Chem. 2014, 2014, 395637. [Google Scholar] [CrossRef]
- Gupta, T.; Rohilla, A.; Pathak, A.; Akhtar, M.J.; Haider, M.R.; Yar, M.S. Current perspectives on quinazolines with potent biological activities: A review. Synth. Commun. 2018, 48, 1099–1127. [Google Scholar] [CrossRef]
- Bisht, A.S.; Negi, J.S.; Sharma, D.K. Chemistry and activity of quinazoline moiety: A systematic review study. Int. J. Pharm. Chem. Anal. 2020, 7, 61–65. [Google Scholar] [CrossRef]
- Thakur, A.; Tawa, G.J.; Henderson, M.J.; Danchik, C.; Liu, S.; Shah, P.; Wang, A.Q.; Dunn, G.; Kabir, M.; Padilha, E.C.; et al. Design, Synthesis, and Biological Evaluation of Quinazolin-4-one-Based Hydroxamic Acids as Dual PI3K/HDAC Inhibitors. J. Med. Chem. 2020, 63, 4256–4292. [Google Scholar] [CrossRef] [PubMed]
- Arachchige, K.T.P.; Yi, S.C. Synthesis of Quinazoline and Quinazolinone Derivatives via Ligand-Promoted Ruthenium-Catalyzed Dehydrogenative and Deaminative Coupling Reaction of 2-Aminophenyl Ketones and 2-Aminobenzamides with Amines. Org. Lett. 2019, 21, 3337–3341. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.M.; Sivaramakrishna, A. Remarkably flexible quinazolinones—Synthesis and biological applications. J. Heterocycl. Chem. 2019, 57, 942–954. [Google Scholar] [CrossRef]
- Zayed, M.F. Medicinal Chemistry of Quinazolines as Analgesic and Anti-Inflammatory Agents. ChemEngineering 2022, 6, 94. [Google Scholar] [CrossRef]
- Chilin, A.; Conconi, M.T.; Marzaro, G.; Guiotto, A.; Urbani, L.; Tonus, F.; Parnigotto, P. Exploring epidermal growth factor receptor (EGFR) inhibitor features: The role of fused dioxygenated rings on the quinazoline scaffold. J. Med. Chem. 2010, 53, 1862–1866. [Google Scholar] [CrossRef]
- Conconi, M.T.; Marzaro, G.; Urbani, L.; Zanusso, I.; Di, L.R.; Castagliuolo, I.; Brun, P.; Tonus, F.; Ferrarese, A.; Guiotto, A.; et al. Quinazoline-based multi-tyrosine kinase inhibitors: Synthesis, modeling, antitumor and antiangiogenic properties. Eur. J. Med. Chem. 2013, 67, 373–383. [Google Scholar] [CrossRef]
- Kumar, D.; Mariappan, G.; Husain, A.; Monga, J.; Kumar, S. Design, synthesis and cytotoxic evaluation of novel imidazolone fused quinazolinone derivatives. Arab. J. Chem. 2017, 10, 344–350. [Google Scholar] [CrossRef]
- Fitzgerald, E.C.; Patel, B.S.; Becker, W.J.; Cameron, M.P.; Zaller, D.; Pikounis, B.V.; O’Keefe, J.S.; Scapin, G. Structural basis for p38α MAP kinase quinazolinone and pyridol-pyrimidine inhibitor specificity. Nat. Struct. Mol. Biol. 2003, 10, 764–769. [Google Scholar] [CrossRef]
- Abouzid, K.; Shouman, S. Design, synthesis and in vitro antitumor activity of 4-aminoquinoline and 4-aminoquinazoline derivatives targeting EGFR tyrosine kinase. Bioorg. Med. Chem. 2008, 16, 7543–7551. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zheng, W.; Ji, L.; Luo, Q.; Hao, X.; Li, X.; Wang, F. Synthesis, characterization, screening and docking analysis of 4-anilinoquinazoline derivatives as tyrosine kinase inhibitors. Eur. J. Med. Chem. 2013, 61, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Peng, T.; Ji, X.; Li, J.; Tong, L.; Li, Z.; Yang, W.; Xu, Y.; Li, M.; Ding, J. Design, synthesis and biological evaluation of novel 4-anilinoquinazolines with C-6 urea-linked side chains as inhibitors of the epidermal growth factor receptor. Bioorg. Med. Chem. 2013, 21, 7988–7998. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yuan, Y.; Qiu, N.; Peng, P.; Sheng, R.; Hu, Y. Identification of novel 4-anilinoquinazoline derivatives as potent EGFR inhibitors both under normoxia and hypoxia. Bioorg. Med. Chem. 2014, 22, 6796–6805. [Google Scholar] [CrossRef]
- Yu, H.; Li, Y.; Ge, Y.; Song, Z.; Wang, C.; Huang, S.; Jin, Y.; Han, X.; Zhen, Y.; Liu, K.; et al. Novel 4-anilinoquinazoline derivatives featuring an 1-adamantyl moiety as potent EGFR inhibitors with enhanced activity against NSCLC cell lines. Eur. J. Med. Chem. 2016, 110, 195–203. [Google Scholar] [CrossRef]
- Wu, X.; Li, M.; Qu, Y.; Tang, W.; Zheng, Y.; Lian, J.; Ji, M.; Xu, L. Design and synthesis of novel gefitinib analogues with improved anti-tumor activity. Bioorg. Med. Chem. 2010, 18, 3812–3822. [Google Scholar] [CrossRef]
- Zhao, F.; Lin, Z.; Wang, F.; Zhao, W.; Dong, X. Four-membered heterocycles-containing 4-anilino-quinazoline derivative s as epidermal growth factor receptor (EGFR) kinase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5385–5388. [Google Scholar] [CrossRef]
- Chang, J.; Ren, H.; Zho, M.; Chong, Y.; Zhao, Y.; He, Y.; Zhao, Y.; Zhang, H.; Qi, C. Development of a se ies of novel 4-anlinoquinazoline derivatives possessing quinazoline skeleton: Design, synthesis, EGFR kinase inhibitory efficacy, and evaluation f anticancer activities in vitro. Eur. J. Med. Chem. 2017, 138, 669–688. [Google Scholar] [CrossRef]
- Cai, J.; Sun, M.; Wu, X.; Chen, P.; Wang, X.; Zong, M.J. Design and synthesis of novel 4-benzothiazole amino quinazolines dasatinib derivatives as potential anti-tumor agents. Eur. J. Med. Chem. 2013, 63, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Y.; Zhou, H.; Zheng, Q.; Li, Y.; Zheng, S.; Zhao, S.; Chen, D.; Fan, C. Structure-activity study of quinazoline derivatives leading to the discovery of potent EGFR-T790M inhibitors. Eur. J. Med. Chem. 2015, 102, 445–463. [Google Scholar] [CrossRef]
- Hou, W.; Ren, Y.; Zhang, Z.; Sun, H.; Ma, Y.; Yan, B. Novel quinazoline derivatives bearing various 6-benzamide moieties as highly selective and potent EGFR inhibitors. Bioorg. Med. Chem. 2018, 26, 1740–1750. [Google Scholar] [CrossRef]
- Zhang, Y.; Tortorella, M.D.; Liao, J.; Qin, X.; Chen, T.; Luo, J.; Guan, J.; Talley, J.J.; Tu, Z. Synthesis and evaluation of novel erlotinib-NSAID conjugates as more comprehensive anticancer agents. ACS Med. Chem. Lett. 2015, 6, 1086–1090. [Google Scholar] [CrossRef]
- Qin, X.; Lv, Y.; Liu, P.; Li, Z.; Hu, L.; Zeng, C.; Yang, L. Novel morpholin-3-one fused quinazoline derivatives as EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1571–1575. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.; Liu, J.; Wang, W.; Li, X.; Zhao, L.; Wang, W.; Li, B. Novel 4-arylaminoquinazoline derivatives with (E)-propen-1-yl moiety as potent EGFR inhibitors with enhanced antiproliferative activities against tumor cells. Eur. J. Med. Chem. 2017, 138, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, L.; Xu, H.; Li, X.; Zhao, L.; Wang, W.; Li, B.; Zhang, X. 6,7-Dimorpholinoalkoxy quinazoline derivatives as potent EGFR inhibitors with enhanced antiproliferative activities against tumor cells. Eur. J. Med. Chem. 2017, 147, 77–89. [Google Scholar] [CrossRef]
- Liu, L.T.; Yuan, T.T.; Liu, H.H.; Chen, S.F.; Wu, Y.T. Synthesis and biological evaluationJournalofsubstituted6-kynyl -4-anilinoquinazoline derivatives as potent EGFR inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6373–6377. [Google Scholar] [CrossRef]
- Li, D.D.; Fang, F.; Li, R.; Du, Q.R.; Sun, J.; Gong, H.B.; Zhu, H.L. Discovery of 6-substituted 4-anilin qinazolines with dioxygenated rings as novel EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5870–5875. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Q.; Li, D.D.; Lu, X.; Xu, Y.Y.; Zhu, H.L. Design and synthesis of 4,6-substituted-(diphenylamino)quinazolines as potent EGFR inhibitors with antitumor activity. Bioorg. Med. Chem. 2012, 20, 317–323. [Google Scholar] [CrossRef]
- Li, D.D.; Lv, P.C.; Zhang, H.; Zhang, H.J.; Hou, Y.P.; Liu, K.; Ye, Y.H.; Zhu, H.L. The combination of 4-anilinoquinazoline and cinnamic acid: A novel mode of binding to the epidermal growth factor receptor tyrosine kinase. Bioorg. Med. Chem. 2011, 19, 5012–5022. [Google Scholar] [CrossRef]
- Mowafy, S.; Farag, N.A.; Abouzid, K.A.M. Design, synthesis and in vitro anti-proliferative activity of 4,6-quinazolinediamines as potent EGFR-TK inhibitors. Eur. J. Med. Chem. 2013, 61, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Chen, L.; Zhao, L.; Li, B.; Wang, W. Synthesis and in vitro biological evaluation of novel quinazoline derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 1584–1587. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Hong, J. Recent advancements of 4-aminoquinazoline derivatives as kinase inhibitors and their applications in medicinal chemistry. Eur. J. Med. Chem. 2019, 170, 55–72. [Google Scholar] [CrossRef]
- Musumeci, F.; Radi, M.; Brullo, C.; Schenone, S. Vascular endothelial growth factor (VEGF) receptors: Drugs and new inhibitors. J. Med. Chem. 2012, 55, 10797–10822. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P. The role of hypoxia-induced factorsproofintumorprogression. Oncologist 2004, 9, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Manley, P.W.; Bold, G.; Bruggen, J.; Fendrich, G.; Fuet, P.; Mestan, J.; Schnell, C.; Stolz, B.; Meyer, T.B.; Meyhack, B.; et al. Advances in the structural biology, design and clinical d v lopmentVEGF-R kinase inhibitors for the treatment of angiogenesis. Biochim. Biophys. Acta 2004, 1697, 17–27. [Google Scholar] [CrossRef] [PubMed]
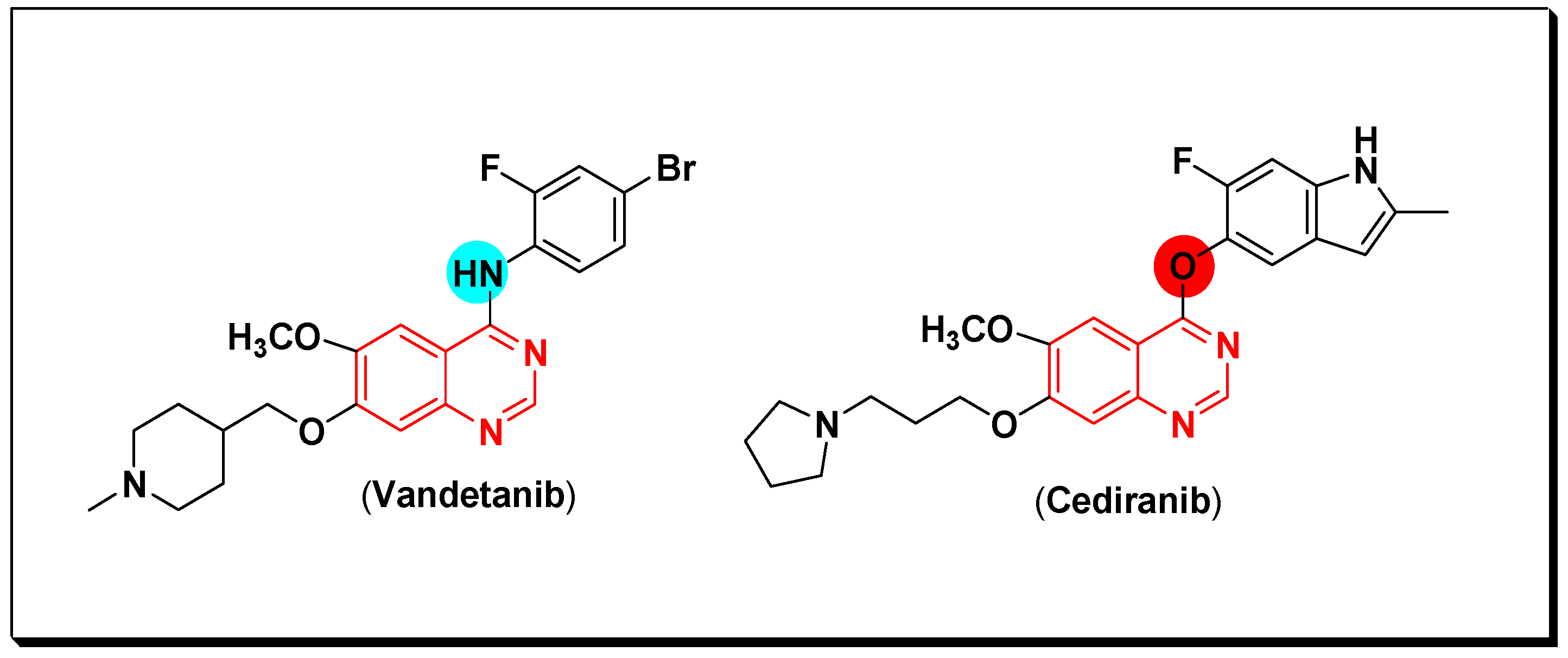
- Trimboli, P.; Castellana, M.; Virili, C.; Giorgino, F.; Giovanella, L. Efficacy of vandetanib in treating locally advanced or metastatic medullary thyroid carcinoma according to RECIST criteri: A systematic review and meta-analysis. Front. Endocrinol. 2018, 9, 224. [Google Scholar] [CrossRef]
- Batchelr, T.T.; Lholland, P.M.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef]
- Garofalo, A.; Goossens, L.; Six, P.; Lemoine, A.; Ravez, S.; Farce, A.; Depreux, P. Impact of aryloxy-linked quinazolines: A novel series of selective VEGFR-2 receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2106–2112. [Google Scholar] [CrossRef]
- Garofalo, A.; Farce, A.; Ravez, S.; Lemoine, A.; Six, P.; Chavatte, P.; Goossens, L.; Depreux, P. Synthesis and structure-activity relationships of (aryloxy)quinazoline ureas as novel, potent, and selective vascular endothelial growth factor receptor-2 inhibitors. J. Med. Chem. 2012, 55, 1189–1204. [Google Scholar] [CrossRef]
- Ravez, S.; Barczyk, A.; Six, P.; Cagnon, A.; Garofalo, A.; Goossens, L.; Depreux, P. Inhibition of tumor cell growth and angiogenesis by 7-aminoalkoxy-4-aryloxy-quinazoline ureas, a novel series of multi-tyrosine kinase inhibitors. Eur. J. Med. Chem. 2014, 79, 369–381. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, J.; Wang, N.; Kong, X.; Fu, F.; Wang, H.; Yao, J. Design and discovery of quinazoline- and thiourea-containing sorafenib analogs as EGFR and VEGFR-2 dual TK inhibitors. Molecules 2018, 23, 24. [Google Scholar] [CrossRef]
- Barbosa, M.L.; Lima, L.M.; Tesch, R.; Sant’Anna, C.M.; Totzke, F.; Kubbutat, M.H.; Schächtele, C.; Laufer, S.A.; Barreiro, E.J. Novel 2-chloro-4-anilino-quinazoline derivatives as EGFR and VEGFR-2 dual inhibitors. Eur. J. Med. Chem. 2014, 71, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Q.; Gong, F.H.; Li, C.G.; Zhang, C.; Wang, Y.J.; Xu, Y.G.; Sun, L.P. Design and discovery of 4-anilinoquinazoline-acylamino derivatives as EGFR and VEGFR-2 dual TK inhibitors. Eur. J. Med. Chem. 2016, 109, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Q.; Gong, F.H.; Ye, J.Q.; Zhang, C.; Yue, X.H.; Li, C.G.; Xu, Y.G.; Sun, L.P. Design an discovery of 4-anilinoquinazoline-urea derivatives as dual TK inhibitors of EGFR and VEGFR-2. Eur. Med. Chem. 2017, 125, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.; Zhang, J.Q.; Liu, Z.C.; Zhang, J.H.; Yan, J.F.; Jin, Y.; Lin, J. Novel 5-anilinoquinaz line-8-nitro derivatives as inhibitors of VEGFR-2 tyrosine kinase: Synthesis, biological evaluation and molecular docking. Org. Biomol. Chem. 2013, 11, 4367–4378. [Google Scholar] [CrossRef]
- Elsayed, N.M.Y.; Serya, R.A.T.; Tolba, M.F.; Ahmed, M.; Barakat, K.; Abou El Ella, D.A.; Abouzid, K.A.M. Design, synthesis, biological evaluation and dynamics simulation of indazole derivatives with antiangiogenic and antiproliferative anticancer activity. Bioorg. Chem. 2019, 82, 340–359. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 11, 1276–1312. [Google Scholar] [CrossRef]
- Shepard, D.R.; Cooney, M.M.; Elson, P.; Bukowski, R.M.; Dreicer, R.; Rini, B.J.; Garcia, J.A. A phase II study of tandutinib (MLN518), a selective inhibitor of type III tyrosine receptor kinases, in patients with metastaticrenal cell carcinoma. Invest. New Drugs 2012, 30, 364–367. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signaling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef]
- Andrews, P.D. Aurora kinases: Shining lights on the therapeutic horizon. Oncogene 2005, 24, 5005–5015. [Google Scholar] [CrossRef]
- Borisa, A.C.; Bhatt, H.G. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem. 2017, 140, 1–19. [Google Scholar] [CrossRef]
- Gadea, B.B.; Ruderman, J.V. Aurora kinase inhibit ZM447439 blocks chromosome-induced spindle assembly, the completion of ch m s me condensation, and the establishment of the spindle integrity check oint in Xenopus egg extracts. Mol. Biol. Cell. 2005, 16, 1305–1318. [Google Scholar] [CrossRef]
- Heron, N.M.; Anderson, M.; Blows, D.P.; Bred, J.; Eden, J.M.; Green, S.; Hill, G.B.; Johnson, T.; Jung, F.H.; McMiken, H.H.J.; et al. SAR and inhibitor complex structure determination of a novel class of potent specific Aurora kinase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1320–1323. [Google Scholar] [CrossRef]
- Jung, F.H.; Pasqet, G.; Brempt, C.L.; Lohmann, J.J.M.; Warin, N.; Renaud, F.; Germain, H.; De Savi, C.; Roberts, N.; Johnston, T.; et al. Discovery of novel and potent thiazoloquinazolines as selective aurora A and B kinase inhibitors. J. Med. Chem. 2006, 49, 955–970. [Google Scholar] [CrossRef] [PubMed]
- Grundy, M.; Seedhouse, C.; Shang, S.; Richardson, J.; Russell, N.; Pallis, M. The FLT3 internal tandem duplication mutation is a secondary target of the aurora B kinase inhibitor AZD1152-HQPA in acute myelogenous leukemia cells. Mol. Cancer Ther. 2010, 9, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Mortlock, A.A.; Foote, K.M.; Heron, N.M.; Jung, F.H.; Pasquet, G.; Lohmann, J.J.M.; Warin, N.; Renaud, F.; De Savi, C.; Roberts, N.J.; et al. Discovery, synthesis and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J. Med. Chem. 2007, 50, 2213–2324. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Mortlock, A.A.; Heron, N.M.; Jung, F.H.; Hill, G.B.; Pasquet, G.; Brady, M.C.; Green, S.; Heaton, S.P.; Kearney, S.; et al. Synthesis and SAR of 1-acetanilide-4-aminopyrazole-substituted quinazolines: Selective inhibitors of aurora B kinase with potent anti-tumor activity. Bioorg. Med. Chem. Lett. 2008, 18, 1904–1909. [Google Scholar] [CrossRef]
- Long, L.; Wang, Y.H.; Zhuo, J.X.; Tu, Z.C.; Wu, R.; Yan, M.; Liu, Q.; Lu, G. Structure-based drug design: Synthesis and biological evaluation of quinazolin-4-amine derivatives as selective aurora A kinase inhibitors. Eur. J. Med. Chem. 2018, 157, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Li, L.; Hong, K.H.; Wu, X.; Chen, J.; Wang, P.; Cao, M.; Zong, X.; Ji, M. Discovery of 4-aminoquinazoline-u rea derivatives as aurora kinase inhibitors with antiproliferative activity. Bioorg. Med. Chem. 2014, 22, 5813–5823. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Coumar, M.S.; Wang, W.C.; Shiao, H.Y.; Ke, Y.Y.; Lin, W.H.; Kuo, C.C.; Chang, C.W.; Kuo, F.M.; Chen, P.Y.; et al. Discovery of BPR1K871, a quinazoline based, multi-kinase inhibit f r the treatment of AML and solid tumors: Rational design, synthesis, in vitro and in vivo evaluation. Oncotarget 2016, 7, 86239–86256. [Google Scholar]
- Malumbres, M.; Barbacid, M.; Mammalian, M. Cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cycle regulators and beyond. Dev. Cell. 2008, 14, 159–169. [Google Scholar] [CrossRef]
- Sanchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; Dios, A.D. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Shewchuk, L.; Hassell, A.; Wisely, B.; Rocque, W.; Holmes, W.; Veal, J.; Kuyper, L.F. Binding mode of the 4-anilinoquinazoline class of protein kinase inhibitor: X-ray crystallographic studies of 4-anilinoquinazolines bound to cyclin-dependent kinase 2 and p38 kinase. J. Med. Chem. 2000, 43, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Sielecki, T.M.; Johnson, T.L.; Liu, J.; Muckelbauer, J.K.; Grafstrom, R.H.; Cox, S.; Boylan, J.; Burton, C.R.; Chen, H.; Smallwood, A.; et al. Quinazolines as cyclin dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 2001, 11, 1157–1160. [Google Scholar] [CrossRef]
- Bathini, Y.; Singh, I.; Harvey, P.J.; Keller, P.R.; Singh, R.; Micetich, R.G.; Fry, D.W.; Dobrusin, E.M.; Toogood, P.L. 2-Aminoquinazoline inhibitors of cyclin-dependent kinases. Bioorg. Med. Chem. Lett. 2005, 15, 3881–3885. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Wang, S.T.; Sun, P.H.; Song, F.J. Molecular modeling studies of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives as otent CDK2/cyclin A inhibitors using 3D-QSAR and docking. Int. J. Mol. Sci. 2010, 11, 3705–3724. [Google Scholar] [CrossRef]
- Shi, H.; Li, Y.; Ren, X.; Zhang, Y.; Yang, Z.; Qi, C. A novel quinazoline-based analog induces G2/M cell cycle arrest and apoptosis in human A549 lung cancer cells via a ROS-dependentJournalmechanism. Biochem. Biophys. Res. Commun. 2017, 486, 314–320. [Google Scholar] [CrossRef]
- Marone, R.; Cmiljaovic, V.; Giese, B.; Wymann, M.P. Targeting phosphoinositide 3-kinase: Moving towards therapy. Biochim. Biophys. Acta 2008, 1784, 159–185. [Google Scholar] [CrossRef]
- Markham, A. Idelalisib: First global approval. Drugs 2014, 74, 1701–1707. [Google Scholar] [CrossRef]
- Dreyling, M.; Santoro, A.; Mollica, L.; Leppa, S.; Follows, G.A.; Lenz, G.; Kim, W.S.; Nagler, A.; Panayiotidis, P.; Demeter, P.; et al. Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J. Clin. Oncol. 2017, 35, 3898–3905. [Google Scholar] [CrossRef] [PubMed]
- Shao, T.; Wang, J.; Chen, J.G.; Wang, X.M.; Li, H.; Li, Y.P.; Li, Y.; Yang, G.D.; Mei, Q.B.; Zhang, S.Q. Discovery of 2-methoxy-3-phenylsulfonamido-5-(quinazolin-6-yl or quinolin-6-yl)benzamides as novel PI3K inhibitors and anticancer agents by bioisostere. Eur. J. Med. Chem. 2014, 75, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Wani, Z.A.; Guru, S.K.; Rao, A.V.; Sharma, S.; Mahajan, G.; Behl, A.; Kumar, A.; Sharma, P.R.; Kamal, A.; Bhushan, S.; et al. A novel quinazolinone chalcone derivative induces mitochondrial dependent apoptosis and inhibits PI3K/Akt/mTOR signaling pathway in human colon cancer HCT-116 cells. Food Chem. Toxicol. 2016, 87, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Dai, W.; Chen, X.; Geng, A.; Chen, Y.; Lu, T.; Zhu, Y. Design, synthesis and biological evaluation of 2,3-dihydroimidazo[1,2-c]quinazoline derivatives as novel phosphatidylinositol 3-kinase and histone deacetylase dual inhibitors. RSC Adv. 2017, 7, 52180–52186. [Google Scholar] [CrossRef]
- Xin, M.; Hei, Y.Y.; Zhang, H.; Xie, X.X.; Mao, S.; Zu, S.J.; Zhang, S.Q. Discovery of 6-benzamide containing 4-phenylquinazoline de ivatives as novel PI3Kδ inhibitors. Lett. Drug Des. Dis. 2017, 14, 167–174. [Google Scholar] [CrossRef]
- Xin, M.; Hei, Y.Y.; Zhang, H.; Shen, Y.; Zhang, S.Q. Design and synthesis of novel 6-aryl substituted 4-anilinequinazoline de ivatives as potential PI3Kδ inhibitors. Bioorg. Med. Chem. Lett. 2017, 14, 1972–1977. [Google Scholar] [CrossRef]
- Xin, M.; Duan, W.; Feng, Y.; Hei, Y.; Zhang, H.; Shen, Y.; Zhao, H.Y.; Mao, S.; Zhang, S.Q. Novel 6- yl substituted 4-pyrrolidineaminoquinazoline derivatives as potent phosphoinositide 3-kinase delta (PI3Kδ) inhibitors. Bioorg. Med. Chem. 2018, 26, 2028–2040. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 25 January 2023).






































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Character | 4-Aminoquinazoline |
|---|---|
| Molecular formula | C8H9N3 |
| Molecular weight | 147.18 g/mol |
| Number of heavy atoms | 11 |
| Number of aromatic heavy atoms | 6 |
| Fraction Csp3 | 0.12 |
| Number of rotatable bonds | 0 |
| Number of H-bond acceptors | 2 |
| Number of H-bond donors | 2 |
| Molar refractivity | 51.25 |
| Tropological polar surface area | 50.41 A2 |
| Lipophilicity | 0.66 |
| Water solubility | Soluble |
| GI absorption | High |
| BBB permeation | No |
| Bioavailability score | 0.55 |
| Lipinski | Yes |
| Synthetic accessibility | Easy |
| Molecular Structure | Generic Name | Chemical Name | Biological Target |
|---|---|---|---|
 | Gefitinib Iressa Irressat NSC 759856 UNII-S65743JHBS ZD 1839 CCRIS 9011 | 4-(3’-Chloro-4’-fluoroanilino)-7-methoxy-6-(3-morpholinopropoxy)quinazoline | Tyrosine kinase (EGFR) IC50 = 33 nM |
 | Erlotinib HSDB 8082 UNII-J4T82NDH7E | 4-Quinazolinamine, N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)- | Tyrosine kinase (EGFR) IC50 = 2 nM |
 | Vandetanib Caprelsa HSDB 8198 UNII-YO460OQ37K Zactima ZD 6474 | 4-Quninazolinamine, N-(4-bromo-2-fluorophenyl)-6-methoxy-7-((1-methyl-4-piperidinyl)methoxy)- | Tyrosine kinase (VEGFR2) IC50 = 40 nM |
 | Dacomitinib Vizimpro UNII-5092U85G58 PF-00299804 | 2-Butenamide, N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxy-6-quinazolinyl)-4-(1-piperidinyl)-, hydrate (1:1), (2E)- | Tyrosine kinase (EGFR) IC50 = 50 nM |
 | Afatinib BIBW 2992 Tomtovok Tovok UNII-41UD74L59M | 2-Butenamide, N-(4-((3-chloro-4-fluorophenyl)amino)-7-(((3S)-tetrahydro-3-furanyl)oxy)-6-quinazolinyl)-4-(dimethylamino)-, (2E)- | Tyrosine kinase (EGFR) IC50 = 0.5 nM |
 | Canertinib UNII-C78W1K5ASF | N-(4-((3-Chloro-4-fluorophenyl)amino)-7-(3-(morpholin-4-yl)propoxy)quinazolin-6-yl)prop-2-enamide | Tyrosine kinase (EGFR) IC50 = 0.8 nM |
 | Trimetrexate Trimetrexatum HSDB 6545 JB 11 NSC 249008 TMQ UNII-UPN4ITI8T4 | 2,4-Quinazolinediamine, 5-methyl-6-(((3,4,5-trimethoxyphenyl)amino)methyl) | Dihydrofolate reductase (DHFR) IC50 = 0.04 nM |
 | Lapatinib GSK 572016 HSDB 8209 Tykerb UNII-0VUA21238F | 4-Quinazolinamine, N-(3-chloro-4-((3-fluorophenyl)methoxy)phenyl)-6-(5-(((2-(methylsulfonyl)ethyl)amino)methyl)-2-furanyl) | Tyrosine Kinase (EGFR) IC50 = 10.8 nM; (HER) IC50 = 29.2 nM |
 | Raltitrexed D 1694 UNII-FCB9EGG971 ZD1694 | L-Glutamic acid, N-((5-(((1,4-dihydro-2-methyl-4-oxo-6-quinazolinyl)methyl)methylamino)-2-thienyl)carbonyl) | Thymidylate synthase inhibitor IC50 = 9 nM |
 | Cediranib USAN AZD-2171 Recentin UNII-NQU9IPY4K9 | Quinazoline, 4-((4-fluoro-2-methyl-1H-indol-5-yl)oxy)-6-methoxy-7-(3-(1-pyrrolidinyl)propoxy) | Tyrosine Kinase (VEGFR) IC50 = 0.4 nM |
 | Tandutinib CT 53518 MLN 518 NSC 759851 NII-E1IO3ICJ9A | 1-Piperazinecarboxamide, 4-(6-methoxy-7-(3-(1-piperidinyl)propoxy)-4-quinazolinyl)-N-(4-(1-methylethoxy)phenyl) | Tyrosine Kinase (PDGFR) IC50 = 0.20 μM |
 | Barasertib AZD 1152 UNII-16XC2U7W8N | 1H-Pyrazole-3-acetamide, 5-((7-(3-(ethyl(2- (phosphonooxy)ethyl)amino)propoxy)-4-quinazolinyl)amino)-N-(3-fluorophenyl) | Tyrosine Kinase (Aurora B) IC50 = 0.37 nM |
 | Idelalisib GS 1101 UNII-YG57I8T5M0 Zydelig HSDB 8408 CAL-101 | 5-Fluoro-3-phenyl-2-((S)-1-(9H-purin-6-ylamino)-propyl)-3H-quinazolin-4-one | Tyrosine Kinase (PI3Kδ) IC50 = 2.5 nM |
 | Copanlisib BAY 80-6946 UNII-WI6V529FZ9 Aliqopa | 2-amino-N-(7-methoxy-8-(3-morpholinopropoxy)-2,3-dihydroimidazo(1,2-c)quinazolin-4-yl)pyrimidine-5-carboxamide | Tyrosine Kinase (PI3Kα) (PI3Kϒ) IC50 = 19 nM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zayed, M.F. Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases. Sci. Pharm. 2023, 91, 18. https://doi.org/10.3390/scipharm91020018
Zayed MF. Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases. Scientia Pharmaceutica. 2023; 91(2):18. https://doi.org/10.3390/scipharm91020018
Chicago/Turabian StyleZayed, Mohamed F. 2023. "Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases" Scientia Pharmaceutica 91, no. 2: 18. https://doi.org/10.3390/scipharm91020018
APA StyleZayed, M. F. (2023). Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases. Scientia Pharmaceutica, 91(2), 18. https://doi.org/10.3390/scipharm91020018