
Advances in Nucleotide Repeat Expansion Diseases: Transcription Gets in Phase

 , and
, and 
Abstract
1. Introduction
2. A Brief Overview on DNA Transcription of Coding Genes
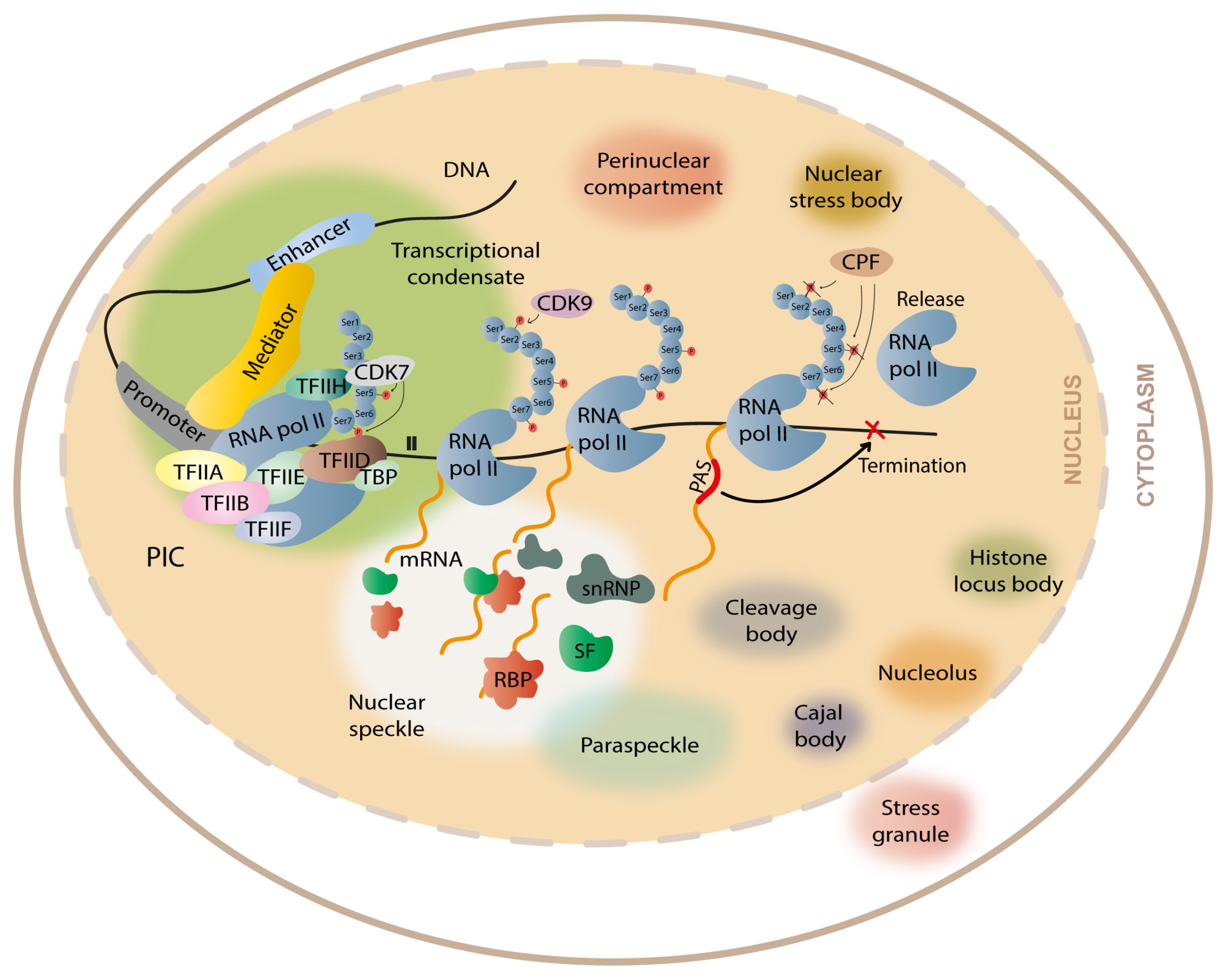
3. Formation of Membraneless Organelles during Transcription and Gene Expression
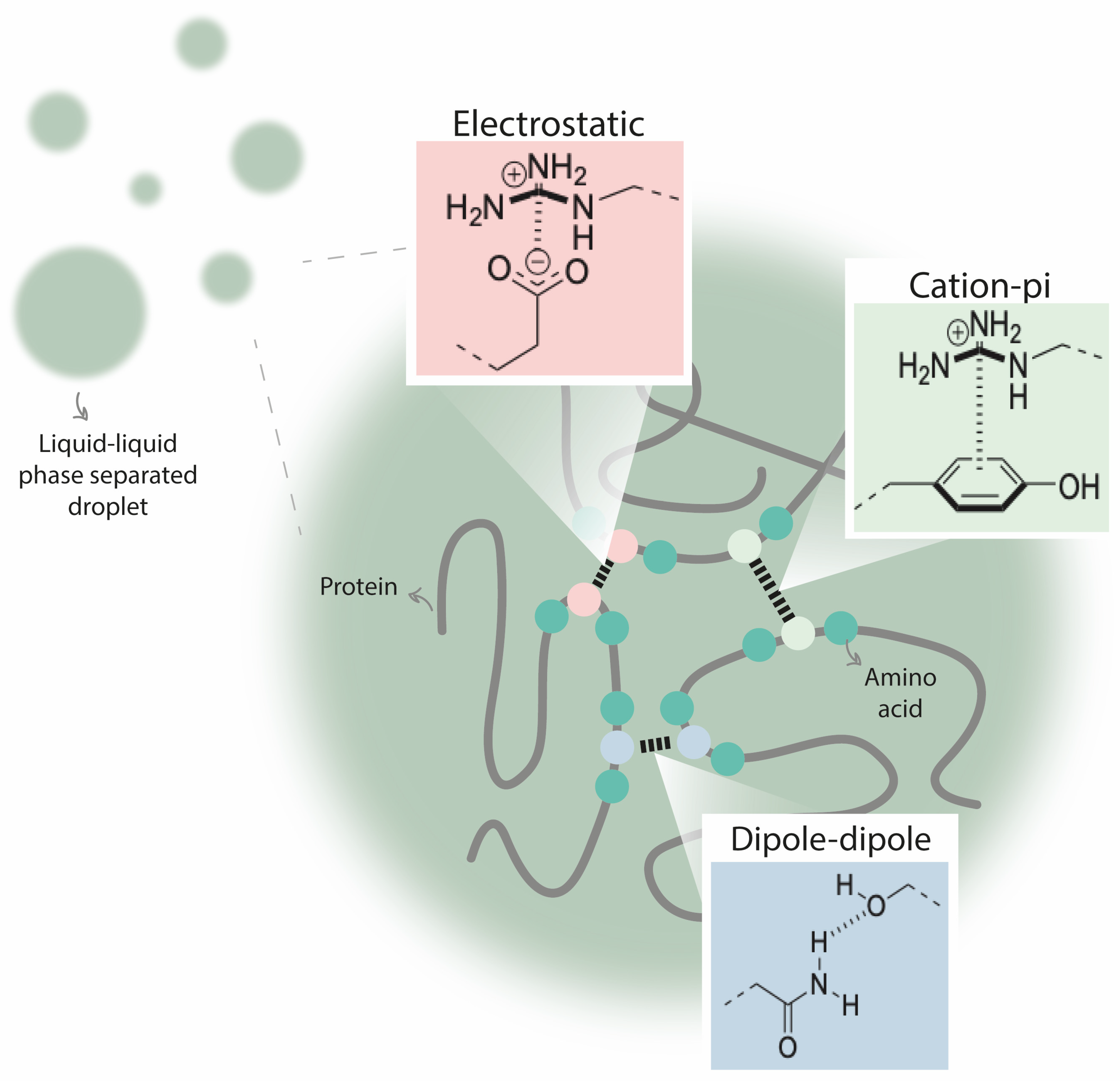
4. Repeat Expansions in Proteins Alter Their Condensation Behavior
5. Transcriptional Dysregulation in Coding Repeat Expansion Diseases
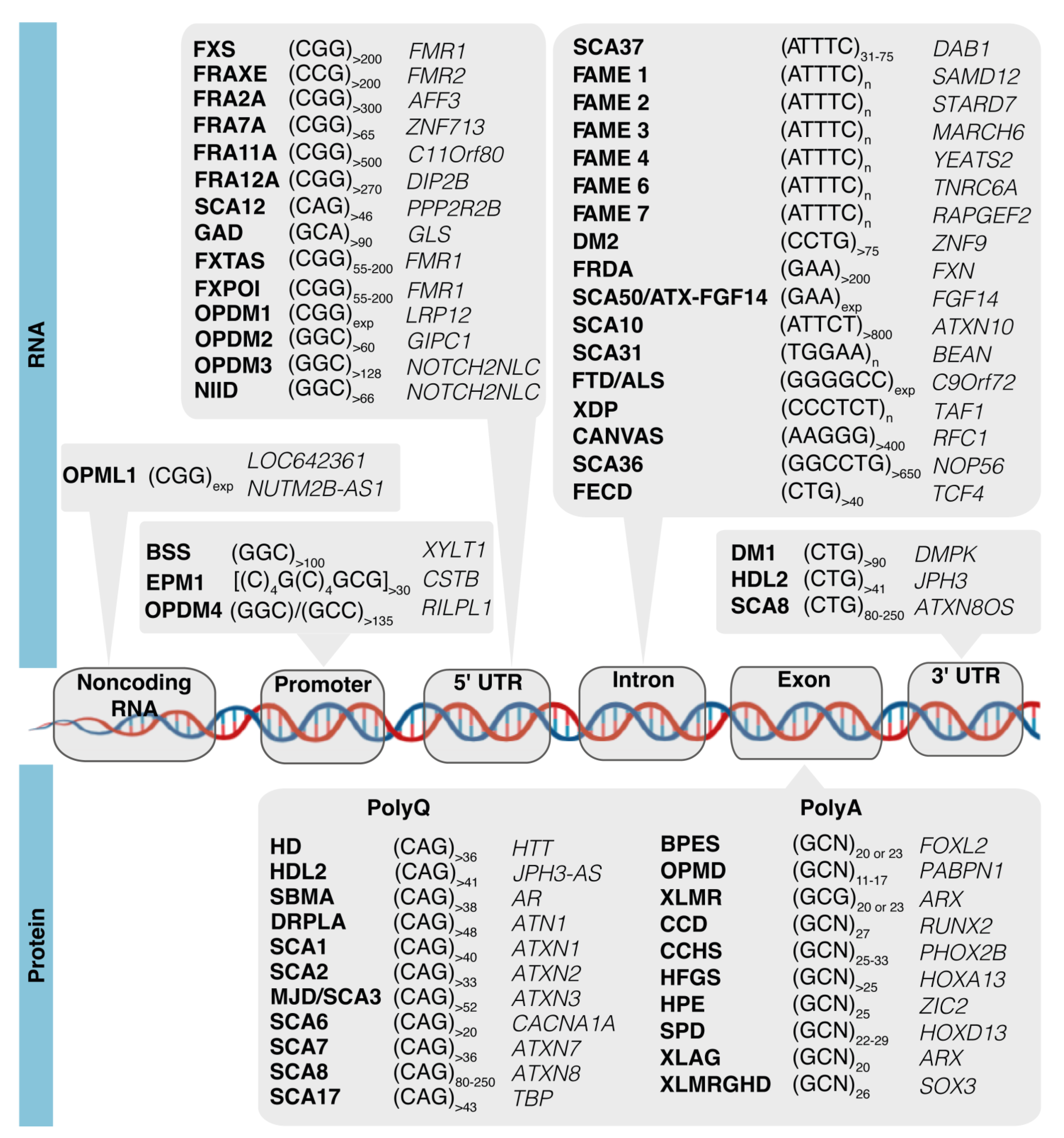
6. Aberrant Condensates and Transcriptional Dysregulation in Noncoding Repeat Expansion Diseases
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loureiro, J.R.; Castro, A.F.; Figueiredo, A.S.; Silveira, I. Molecular Mechanisms in Pentanucleotide Repeat Diseases. Cells 2022, 11, 205. [Google Scholar] [CrossRef]
- Rafehi, H.; Read, J.; Szmulewicz, D.J.; Davies, K.C.; Snell, P.; Fearnley, L.G.; Scott, L.; Thomsen, M.; Gillies, G.; Pope, K.; et al. An intronic GAA repeat expansion in FGF14 causes the autosomal-dominant adult-onset ataxia SCA50/ATX-FGF14. Am. J. Hum. Genet. 2023, 110, 105–119. [Google Scholar] [CrossRef]
- Pellerin, D.; Danzi, M.C.; Wilke, C.; Renaud, M.; Fazal, S.; Dicaire, M.J.; Scriba, C.K.; Ashton, C.; Yanick, C.; Beijer, D.; et al. Deep Intronic FGF14 GAA Repeat Expansion in Late-Onset Cerebellar Ataxia. N. Engl. J. Med. 2023, 388, 128–141. [Google Scholar] [CrossRef]
- Ishiura, H.; Shibata, S.; Yoshimura, J.; Suzuki, Y.; Qu, W.; Doi, K.; Almansour, M.A.; Kikuchi, J.K.; Taira, M.; Mitsui, J.; et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 2019, 51, 1222–1232. [Google Scholar] [CrossRef]
- Sone, J.; Mitsuhashi, S.; Fujita, A.; Mizuguchi, T.; Hamanaka, K.; Mori, K.; Koike, H.; Hashiguchi, A.; Takashima, H.; Sugiyama, H.; et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 2019, 51, 1215–1221. [Google Scholar] [CrossRef]
- Deng, J.; Yu, J.; Li, P.; Luan, X.; Cao, L.; Zhao, J.; Yu, M.; Zhang, W.; Lv, H.; Xie, Z.; et al. Expansion of GGC Repeat in GIPC1 Is Associated with Oculopharyngodistal Myopathy. Am. J. Hum. Genet. 2020, 106, 793–804. [Google Scholar] [CrossRef]
- Xi, J.; Wang, X.; Yue, D.; Dou, T.; Wu, Q.; Lu, J.; Liu, Y.; Yu, W.; Qiao, K.; Lin, J.; et al. 5′ UTR CGG repeat expansion in GIPC1 is associated with oculopharyngodistal myopathy. Brain 2021, 144, 601–614. [Google Scholar] [CrossRef]
- Yu, J.; Deng, J.; Guo, X.; Shan, J.; Luan, X.; Cao, L.; Zhao, J.; Yu, M.; Zhang, W.; Lv, H.; et al. The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3. Brain 2021, 144, 1819–1832. [Google Scholar] [CrossRef]
- Zeng, Y.H.; Yang, K.; Du, G.Q.; Chen, Y.K.; Cao, C.Y.; Qiu, Y.S.; He, J.; Lv, H.D.; Qu, Q.Q.; Chen, J.N.; et al. GGC Repeat Expansion of RILPL1 is Associated with Oculopharyngodistal Myopathy. Ann. Neurol. 2022, 92, 512–526. [Google Scholar] [CrossRef]
- Sato, N.; Amino, T.; Kobayashi, K.; Asakawa, S.; Ishiguro, T.; Tsunemi, T.; Takahashi, M.; Matsuura, T.; Flanigan, K.M.; Iwasaki, S.; et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am. J. Hum. Genet. 2009, 85, 544–557. [Google Scholar] [CrossRef]
- Seixas, A.I.; Loureiro, J.R.; Costa, C.; Ordonez-Ugalde, A.; Marcelino, H.; Oliveira, C.L.; Loureiro, J.L.; Dhingra, A.; Brandao, E.; Cruz, V.T.; et al. A Pentanucleotide ATTTC Repeat Insertion in the Non-coding Region of DAB1, Mapping to SCA37, Causes Spinocerebellar Ataxia. Am. J. Hum. Genet. 2017, 101, 87–103. [Google Scholar] [CrossRef]
- Cen, Z.; Jiang, Z.; Chen, Y.; Zheng, X.; Xie, F.; Yang, X.; Lu, X.; Ouyang, Z.; Wu, H.; Chen, S.; et al. Intronic pentanucleotide TTTCA repeat insertion in the SAMD12 gene causes familial cortical myoclonic tremor with epilepsy type 1. Brain 2018, 141, 2280–2288. [Google Scholar] [CrossRef]
- Lei, X.X.; Liu, Q.; Lu, Q.; Huang, Y.; Zhou, X.Q.; Sun, H.Y.; Wu, L.W.; Cui, L.Y.; Zhang, X. TTTCA repeat expansion causes familial cortical myoclonic tremor with epilepsy. Eur. J. Neurol. 2019, 26, 513–518. [Google Scholar] [CrossRef]
- Florian, R.T.; Kraft, F.; Leitao, E.; Kaya, S.; Klebe, S.; Magnin, E.; van Rootselaar, A.F.; Buratti, J.; Kuhnel, T.; Schroder, C.; et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with Familial Adult Myoclonic Epilepsy type 3. Nat. Commun. 2019, 10, 4919. [Google Scholar] [CrossRef]
- Yeetong, P.; Pongpanich, M.; Srichomthong, C.; Assawapitaksakul, A.; Shotelersuk, V.; Tantirukdham, N.; Chunharas, C.; Suphapeetiporn, K.; Shotelersuk, V. TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain 2019, 142, 3360–3366. [Google Scholar] [CrossRef]
- Ishiura, H.; Doi, K.; Mitsui, J.; Yoshimura, J.; Matsukawa, M.K.; Fujiyama, A.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; Suzuki, Y.; et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet. 2018, 50, 581–590. [Google Scholar] [CrossRef]
- Corbett, M.A.; Kroes, T.; Veneziano, L.; Bennett, M.F.; Florian, R.; Schneider, A.L.; Coppola, A.; Licchetta, L.; Franceschetti, S.; Suppa, A.; et al. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat. Commun. 2019, 10, 4920. [Google Scholar] [CrossRef]
- Castro, A.F.; Loureiro, J.R.; Bessa, J.; Silveira, I. Antisense Transcription across Nucleotide Repeat Expansions in Neurodegenerative and Neuromuscular Diseases: Progress and Mysteries. Genes 2020, 11, 1418. [Google Scholar] [CrossRef]
- Depienne, C.; Mandel, J.L. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am. J. Hum. Genet. 2021, 108, 764–785. [Google Scholar] [CrossRef]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [CrossRef]
- Osman, S.; Cramer, P. Structural Biology of RNA Polymerase II Transcription: 20 Years On. Annu. Rev. Cell Dev. Biol. 2020, 36, 1–34. [Google Scholar] [CrossRef]
- Soutourina, J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2018, 19, 262–274. [Google Scholar] [CrossRef]
- Poeta, L.; Drongitis, D.; Verrillo, L.; Miano, M.G. DNA Hypermethylation and Unstable Repeat Diseases: A Paradigm of Transcriptional Silencing to Decipher the Basis of Pathogenic Mechanisms. Genes 2020, 11, 684. [Google Scholar] [CrossRef]
- Kumari, D.; Biacsi, R.E.; Usdin, K. Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells. J. Biol. Chem. 2011, 286, 4209–4215. [Google Scholar] [CrossRef] [PubMed]
- Haberle, V.; Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 2018, 19, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.D.; West, S. Termination of Transcription by RNA Polymerase II: BOOM! Trends Genet 2020, 36, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Schreieck, A.; Easter, A.D.; Etzold, S.; Wiederhold, K.; Lidschreiber, M.; Cramer, P.; Passmore, L.A. RNA polymerase II termination involves C-terminal-domain tyrosine dephosphorylation by CPF subunit Glc7. Nat. Struct Mol. Biol. 2014, 21, 175–179. [Google Scholar] [CrossRef]
- Proudfoot, N.J. Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science 2016, 352, aad9926. [Google Scholar] [CrossRef]
- Mayfield, J.E.; Burkholder, N.T.; Zhang, Y.J. Dephosphorylating eukaryotic RNA polymerase II. Biochim. Biophys Acta 2016, 1864, 372–387. [Google Scholar] [CrossRef]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.; Shaw, P.G.; Kim, M.S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef]
- Goodman, L.D.; Bonini, N.M. New Roles for Canonical Transcription Factors in Repeat Expansion Diseases. Trends Genet. 2020, 36, 81–92. [Google Scholar] [CrossRef]
- Herzel, L.; Ottoz, D.S.M.; Alpert, T.; Neugebauer, K.M. Splicing and transcription touch base: Co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol. 2017, 18, 637–650. [Google Scholar] [CrossRef]
- Pinto, P.A.; Henriques, T.; Freitas, M.O.; Martins, T.; Domingues, R.G.; Wyrzykowska, P.S.; Coelho, P.A.; Carmo, A.M.; Sunkel, C.E.; Proudfoot, N.J.; et al. RNA polymerase II kinetics in polo polyadenylation signal selection. EMBO J. 2011, 30, 2431–2444. [Google Scholar] [CrossRef]
- Bentley, D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014, 15, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Dugast-Darzacq, C.; Liu, Z.; Dong, P.; Dailey, G.M.; Cattoglio, C.; Heckert, A.; Banala, S.; Lavis, L.; Darzacq, X.; et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 2018, 361, eaar2555. [Google Scholar] [CrossRef]
- Fang, X.; Wang, L.; Ishikawa, R.; Li, Y.; Fiedler, M.; Liu, F.; Calder, G.; Rowan, B.; Weigel, D.; Li, P.; et al. Arabidopsis FLL2 promotes liquid-liquid phase separation of polyadenylation complexes. Nature 2019, 569, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Peng, A.; Weber, S.C. Evidence for and against Liquid-Liquid Phase Separation in the Nucleus. Noncoding RNA 2019, 5, 50. [Google Scholar] [CrossRef]
- Borcherds, W.; Bremer, A.; Borgia, M.B.; Mittag, T. How do intrinsically disordered protein regions encode a driving force for liquid-liquid phase separation? Curr. Opin. Struct. Biol. 2021, 67, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, J.B. Assembly of Mitotic Structures through Phase Separation. J. Mol. Biol. 2018, 430, 4762–4772. [Google Scholar] [CrossRef]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855.e16. [Google Scholar] [CrossRef]
- Zamudio, A.V.; Dall’Agnese, A.; Henninger, J.E.; Manteiga, J.C.; Afeyan, L.K.; Hannett, N.M.; Coffey, E.L.; Li, C.H.; Oksuz, O.; Sabari, B.R.; et al. Mediator Condensates Localize Signaling Factors to Key Cell Identity Genes. Mol. Cell 2019, 76, 753–766.e6. [Google Scholar] [CrossRef]
- Lu, Y.; Wu, T.; Gutman, O.; Lu, H.; Zhou, Q.; Henis, Y.I.; Luo, K. Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat. Cell Biol. 2020, 22, 453–464. [Google Scholar] [CrossRef]
- Cai, D.; Feliciano, D.; Dong, P.; Flores, E.; Gruebele, M.; Porat-Shliom, N.; Sukenik, S.; Liu, Z.; Lippincott-Schwartz, J. Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat. Cell Biol. 2019, 21, 1578–1589. [Google Scholar] [CrossRef]
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Ramirez Montero, D.; Ryan, V.H.; Rohatgi, R.; et al. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018, 37, e97452. [Google Scholar] [CrossRef] [PubMed]
- Franzmann, T.M.; Jahnel, M.; Pozniakovsky, A.; Mahamid, J.; Holehouse, A.S.; Nuske, E.; Richter, D.; Baumeister, W.; Grill, S.W.; Pappu, R.V.; et al. Phase separation of a yeast prion protein promotes cellular fitness. Science 2018, 359, eaao5654. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Julicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef]
- Oshidari, R.; Huang, R.; Medghalchi, M.; Tse, E.Y.W.; Ashgriz, N.; Lee, H.O.; Wyatt, H.; Mekhail, K. DNA repair by Rad52 liquid droplets. Nat. Commun. 2020, 11, 695. [Google Scholar] [CrossRef]
- Plys, A.J.; Kingston, R.E. Dynamic condensates activate transcription. Science 2018, 361, 329–330. [Google Scholar] [CrossRef]
- Wiedner, H.J.; Giudice, J. It’s not just a phase: Function and characteristics of RNA-binding proteins in phase separation. Nat. Struct. Mol. Biol. 2021, 28, 465–473. [Google Scholar] [CrossRef]
- Quinodoz, S.A.; Ollikainen, N.; Tabak, B.; Palla, A.; Schmidt, J.M.; Detmar, E.; Lai, M.M.; Shishkin, A.A.; Bhat, P.; Takei, Y.; et al. Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 2018, 174, 744–757.e24. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S. Intrinsically disordered protein regions and phase separation: Sequence determinants of assembly or lack thereof. Emerg. Top. Life Sci. 2020, 4, 307–329. [Google Scholar] [CrossRef] [PubMed]
- Dignon, G.L.; Best, R.B.; Mittal, J. Biomolecular Phase Separation: From Molecular Driving Forces to Macroscopic Properties. Annu. Rev. Phys. Chem. 2020, 71, 53–75. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, P.; Kapoor, U.; Sundaravadivelu Devarajan, D.; Phan, T.M.; Rizuan, A.; Mittal, J. Principles Governing the Phase Separation of Multidomain Proteins. Biochemistry 2022, 61, 2443–2455. [Google Scholar] [CrossRef]
- Ruff, K.M.; Choi, Y.H.; Cox, D.; Ormsby, A.R.; Myung, Y.; Ascher, D.B.; Radford, S.E.; Pappu, R.V.; Hatters, D.M. Sequence grammar underlying the unfolding and phase separation of globular proteins. Mol. Cell 2022, 82, 3193–3208.e8. [Google Scholar] [CrossRef]
- Darling, A.L.; Liu, Y.; Oldfield, C.J.; Uversky, V.N. Intrinsically Disordered Proteome of Human Membrane-Less Organelles. Proteomics 2018, 18, e1700193. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Feng, Z.; Chen, X.; Wu, X.; Zhang, M. Formation of biological condensates via phase separation: Characteristics, analytical methods, and physiological implications. J. Biol. Chem. 2019, 294, 14823–14835. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Mackowiak, S.D.; Niskanen, H.; Knezevic, D.; Asimi, V.; Grosswendt, S.; Geertsema, H.; Ali, S.; Jerkovic, I.; Ewers, H.; et al. Unblending of Transcriptional Condensates in Human Repeat Expansion Disease. Cell 2020, 181, 1062–1079.e30. [Google Scholar] [CrossRef]
- Peskett, T.R.; Rau, F.; O’Driscoll, J.; Patani, R.; Lowe, A.R.; Saibil, H.R. A Liquid to Solid Phase Transition Underlying Pathological Huntingtin Exon1 Aggregation. Mol. Cell 2018, 70, 588–601.e6. [Google Scholar] [CrossRef]
- Zhang, S.; Hinde, E.; Parkyn Schneider, M.; Jans, D.A.; Bogoyevitch, M.A. Nuclear bodies formed by polyQ-ataxin-1 protein are liquid RNA/protein droplets with tunable dynamics. Sci. Rep. 2020, 10, 1557. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kajimoto, S.; Shibata, D.; Kuroi, K.; Fujii, F.; Nakabayashi, T. Observation of liquid-liquid phase separation of ataxin-3 and quantitative evaluation of its concentration in a single droplet using Raman microscopy. Chem. Sci. 2021, 12, 7411–7418. [Google Scholar] [CrossRef] [PubMed]
- Saar, K.L.; Morgunov, A.S.; Qi, R.; Arter, W.E.; Krainer, G.; Lee, A.A.; Knowles, T.P.J. Learning the molecular grammar of protein condensates from sequence determinants and embeddings. Proc. Natl. Acad. Sci. USA 2021, 118, e2019053118. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699.e16. [Google Scholar] [CrossRef]
- Chu, X.; Sun, T.; Li, Q.; Xu, Y.; Zhang, Z.; Lai, L.; Pei, J. Prediction of liquid-liquid phase separating proteins using machine learning. BMC Bioinform. 2022, 23, 72. [Google Scholar] [CrossRef] [PubMed]
- Pancsa, R.; Vranken, W.; Mészáros, B. Computational resources for identifying and describing proteins driving liquid–liquid phase separation. Brief. Bioinform. 2021, 22, bbaa408. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Predictors, C.; DisProt, C.; Tosatto, S.C.E. Critical assessment of protein intrinsic disorder prediction. Nat. Methods 2021, 18, 472–481. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, G.; Zhang, H. Protocol for analyzing protein liquid–liquid phase separation. Biophys. Rep. 2019, 5, 1–9. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdős, G.; Szabó, B.; Schád, É.; Tantos, Á.; Abukhairan, R.; Horváth, T.; Murvai, N.; Kovács, O.P.; Kovács, M.; et al. PhaSePro: The database of proteins driving liquid–liquid phase separation. Nucleic Acids Res. 2019, 48, D360–D367. [Google Scholar] [CrossRef]
- Lu, H.; Yu, D.; Hansen, A.S.; Ganguly, S.; Liu, R.; Heckert, A.; Darzacq, X.; Zhou, Q. Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 2018, 558, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Boehning, M.; Dugast-Darzacq, C.; Rankovic, M.; Hansen, A.S.; Yu, T.; Marie-Nelly, H.; McSwiggen, D.T.; Kokic, G.; Dailey, G.M.; Cramer, P.; et al. RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol. 2018, 25, 833–840. [Google Scholar] [CrossRef]
- Cho, W.K.; Spille, J.H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Smikle, R.; Reid, M.J.; Mizielinska, S. Altered Phase Separation and Cellular Impact in C9orf72-Linked ALS/FTD. Front. Cell. Neurosci. 2021, 15, 664151. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Luo, F.; Li, Y.; Zhou, H.; Qin, Z.; Liu, Z.; Gu, J.; Xie, M.; Zhao, K.; Dai, B.; et al. Structural basis for reversible amyloids of hnRNPA1 elucidates their role in stress granule assembly. Nat. Commun. 2019, 10, 2006. [Google Scholar] [CrossRef]
- Li, H.R.; Chiang, W.C.; Chou, P.C.; Wang, W.J.; Huang, J.R. TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 2018, 293, 6090–6098. [Google Scholar] [CrossRef]
- Conicella, A.E.; Dignon, G.L.; Zerze, G.H.; Schmidt, H.B.; D’Ordine, A.M.; Kim, Y.C.; Rohatgi, R.; Ayala, Y.M.; Mittal, J.; Fawzi, N.L. TDP-43 alpha-helical structure tunes liquid-liquid phase separation and function. Proc. Natl. Acad. Sci. USA 2020, 117, 5883–5894. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Xiao, R.; Chen, J.Y.; Liang, Z.; Luo, D.; Chen, G.; Lu, Z.J.; Chen, Y.; Zhou, B.; Li, H.; Du, X.; et al. Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription. Cell 2019, 178, 107–121.e18. [Google Scholar] [CrossRef]
- Sigova, A.A.; Abraham, B.J.; Ji, X.; Molinie, B.; Hannett, N.M.; Guo, Y.E.; Jangi, M.; Giallourakis, C.C.; Sharp, P.A.; Young, R.A. Transcription factor trapping by RNA in gene regulatory elements. Science 2015, 350, 978–981. [Google Scholar] [CrossRef]
- Boeynaems, S.; Dorone, Y.; Marian, A.; Shabardina, V.; Huang, G.; Kim, G.; Sanyal, A.; Şen, N.-E.; Docampo, R.; Ruiz-Trillo, I.; et al. Poly(A)-binding protein is an ataxin-2 chaperone that emulsifies biomolecular condensates. bioRxiv 2021, 1–34. [Google Scholar] [CrossRef]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808–816.e9. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef] [PubMed]
- Sprunger, M.L.; Lee, K.; Sohn, B.S.; Jackrel, M.E. Molecular determinants and modifiers of Matrin-3 toxicity, condensate dynamics, and droplet morphology. iScience 2022, 25, 103900. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Iradi, M.C.; Strunk, H.; Crown, A.M.; Davila, R.; Brown, H.; Rodriguez-Lebron, E.; Borchelt, D.R. N-terminal sequences in matrin 3 mediate phase separation into droplet-like structures that recruit TDP43 variants lacking RNA binding elements. Lab. Investig. 2019, 99, 1030–1040. [Google Scholar] [CrossRef]
- Zhang, F.; Biswas, M.; Massah, S.; Lee, J.; Lingadahalli, S.; Wong, S.; Wells, C.; Foo, J.; Khan, N.; Morin, H.; et al. Dynamic phase separation of the androgen receptor and its coactivators key to regulate gene expression. Nucleic. Acids Res. 2023, 51, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, J.; Zhu, H.; Rinaldo, L.; Lamar, K.M.; Palmenberg, A.C.; Hansel, C.; Gomez, C.M. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell 2013, 154, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Matilla, A.; Koshy, B.T.; Cummings, C.J.; Isobe, T.; Orr, H.T.; Zoghbi, H.Y. The cerebellar leucine-rich acidic nuclear protein interacts with ataxin-1. Nature 1997, 389, 974–978. [Google Scholar] [CrossRef]
- Davidson, J.D.; Riley, B.; Burright, E.N.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. Identification and characterization of an ataxin-1-interacting protein: A1Up, a ubiquitin-like nuclear protein. Hum. Mol. Genet. 2000, 9, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Okazawa, H.; Rich, T.; Chang, A.; Lin, X.; Waragai, M.; Kajikawa, M.; Enokido, Y.; Komuro, A.; Kato, S.; Shibata, M.; et al. Interaction between mutant ataxin-1 and PQBP-1 affects transcription and cell death. Neuron 2002, 34, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-K.; Fernandez-Funez, P.; Acevedo, S.F.; Lam, Y.C.; Kaytor, M.D.; Fernandez, M.H.; Aitken, A.; Skoulakis, E.M.C.; Orr, H.T.; Botas, J.; et al. Interaction of Akt-Phosphorylated Ataxin-1 with 14-3-3 Mediates Neurodegeneration in Spinocerebellar Ataxia Type 1. Cell 2003, 113, 457–468. [Google Scholar] [CrossRef]
- Tsai, C.C.; Kao, H.Y.; Mitzutani, A.; Banayo, E.; Rajan, H.; McKeown, M.; Evans, R.M. Ataxin 1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 4047–4052. [Google Scholar] [CrossRef]
- Mizutani, A.; Wang, L.; Rajan, H.; Vig, P.J.; Alaynick, W.A.; Thaler, J.P.; Tsai, C.C. Boat, an AXH domain protein, suppresses the cytotoxicity of mutant ataxin-1. EMBO J. 2005, 24, 3339–3351. [Google Scholar] [CrossRef]
- Lam, Y.C.; Bowman, A.B.; Jafar-Nejad, P.; Lim, J.; Richman, R.; Fryer, J.D.; Hyun, E.D.; Duvick, L.A.; Orr, H.T.; Botas, J.; et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 2006, 127, 1335–1347. [Google Scholar] [CrossRef]
- Serra, H.G.; Duvick, L.; Zu, T.; Carlson, K.; Stevens, S.; Jorgensen, N.; Lysholm, A.; Burright, E.; Zoghbi, H.Y.; Clark, H.B.; et al. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 2006, 127, 697–708. [Google Scholar] [CrossRef]
- Bolger, T.A.; Zhao, X.; Cohen, T.J.; Tsai, C.C.; Yao, T.P. The neurodegenerative disease protein ataxin-1 antagonizes the neuronal survival function of myocyte enhancer factor-2. J. Biol. Chem. 2007, 282, 29186–29192. [Google Scholar] [CrossRef]
- Goold, R.; Hubank, M.; Hunt, A.; Holton, J.; Menon, R.P.; Revesz, T.; Pandolfo, M.; Matilla-Duenas, A. Down-regulation of the dopamine receptor D2 in mice lacking ataxin 1. Hum. Mol. Genet. 2007, 16, 2122–2134. [Google Scholar] [CrossRef]
- Lim, J.; Crespo-Barreto, J.; Jafar-Nejad, P.; Bowman, A.B.; Richman, R.; Hill, D.E.; Orr, H.T.; Zoghbi, H.Y. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 2008, 452, 713–718. [Google Scholar] [CrossRef]
- Rousseaux, M.W.C.; Tschumperlin, T.; Lu, H.C.; Lackey, E.P.; Bondar, V.V.; Wan, Y.W.; Tan, Q.; Adamski, C.J.; Friedrich, J.; Twaroski, K.; et al. ATXN1-CIC Complex Is the Primary Driver of Cerebellar Pathology in Spinocerebellar Ataxia Type 1 through a Gain-of-Function Mechanism. Neuron 2018, 97, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Ingram, M.; Wozniak, E.A.L.; Duvick, L.; Yang, R.; Bergmann, P.; Carson, R.; O’Callaghan, B.; Zoghbi, H.Y.; Henzler, C.; Orr, H.T. Cerebellar Transcriptome Profiles of ATXN1 Transgenic Mice Reveal SCA1 Disease Progression and Protection Pathways. Neuron 2016, 89, 1194–1207. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, H.; Jafar-Nejad, H.; Patel, A.J.; Sun, Y.; Chen, H.K.; Rose, M.F.; Venken, K.J.; Botas, J.; Orr, H.T.; Bellen, H.J.; et al. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell 2005, 122, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Halbach, M.V.; Gispert, S.; Stehning, T.; Damrath, E.; Walter, M.; Auburger, G. Atxn2 Knockout and CAG42-Knock-in Cerebellum Shows Similarly Dysregulated Expression in Calcium Homeostasis Pathway. Cerebellum 2017, 16, 68–81. [Google Scholar] [CrossRef]
- Li, F.; Macfarlan, T.; Pittman, R.N.; Chakravarti, D. Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J. Biol. Chem. 2002, 277, 45004–45012. [Google Scholar] [CrossRef]
- Evert, B.O.; Araujo, J.; Vieira-Saecker, A.M.; de Vos, R.A.; Harendza, S.; Klockgether, T.; Wullner, U. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J. Neurosci. 2006, 26, 11474–11486. [Google Scholar] [CrossRef]
- Chai, Y.; Wu, L.; Griffin, J.D.; Paulson, H.L. The role of protein composition in specifying nuclear inclusion formation in polyglutamine disease. J. Biol. Chem. 2001, 276, 44889–44897. [Google Scholar] [CrossRef]
- Araujo, J.; Breuer, P.; Dieringer, S.; Krauss, S.; Dorn, S.; Zimmermann, K.; Pfeifer, A.; Klockgether, T.; Wuellner, U.; Evert, B.O. FOXO4-dependent upregulation of superoxide dismutase-2 in response to oxidative stress is impaired in spinocerebellar ataxia type 3. Hum. Mol. Genet. 2011, 20, 2928–2941. [Google Scholar] [CrossRef]
- Chou, A.H.; Chen, S.Y.; Yeh, T.H.; Weng, Y.H.; Wang, H.L. HDAC inhibitor sodium butyrate reverses transcriptional downregulation and ameliorates ataxic symptoms in a transgenic mouse model of SCA3. Neurobiol. Dis. 2011, 41, 481–488. [Google Scholar] [CrossRef]
- Zeng, L.; Zhang, D.; McLoughlin, H.S.; Zalon, A.J.; Aravind, L.; Paulson, H.L. Loss of the Spinocerebellar Ataxia type 3 disease protein ATXN3 alters transcription of multiple signal transduction pathways. PLoS ONE 2018, 13, e0204438. [Google Scholar] [CrossRef]
- Toonen, L.J.A.; Overzier, M.; Evers, M.M.; Leon, L.G.; van der Zeeuw, S.A.J.; Mei, H.; Kielbasa, S.M.; Goeman, J.J.; Hettne, K.M.; Magnusson, O.T.; et al. Transcriptional profiling and biomarker identification reveal tissue specific effects of expanded ataxin-3 in a spinocerebellar ataxia type 3 mouse model. Mol. Neurodegener. 2018, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, E.; Balbo, I.; Miniaci, M.C.; Tempia, F. Purkinje Cell Signaling Deficits in Animal Models of Ataxia. Front. Synaptic. Neurosci. 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomska-Cimicka, A.; Hache, A.; Le Gras, S.; Keime, C.; Ye, T.; Eisenmann, A.; Harichane, I.; Roux, M.J.; Messaddeq, N.; Clerin, E.; et al. Polyglutamine-expanded ATXN7 alters a specific epigenetic signature underlying photoreceptor identity gene expression in SCA7 mouse retinopathy. J. Biomed. Sci. 2022, 29, 107. [Google Scholar] [CrossRef]
- Shah, A.G.; Friedman, M.J.; Huang, S.; Roberts, M.; Li, X.J.; Li, S. Transcriptional dysregulation of TrkA associates with neurodegeneration in spinocerebellar ataxia type 17. Hum. Mol. Genet. 2009, 18, 4141–4152. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M.J.; Wang, C.E.; Li, X.J.; Li, S. Polyglutamine expansion reduces the association of TATA-binding protein with DNA and induces DNA binding-independent neurotoxicity. J. Biol. Chem. 2008, 283, 8283–8290. [Google Scholar] [CrossRef]
- Friedman, M.J.; Shah, A.G.; Fang, Z.H.; Ward, E.G.; Warren, S.T.; Li, S.; Li, X.J. Polyglutamine domain modulates the TBP-TFIIB interaction: Implications for its normal function and neurodegeneration. Nat. Neurosci. 2007, 10, 1519–1528. [Google Scholar] [CrossRef]
- Yang, S.; Huang, S.; Gaertig, M.A.; Li, X.J.; Li, S. Age-dependent decrease in chaperone activity impairs MANF expression, leading to Purkinje cell degeneration in inducible SCA17 mice. Neuron 2014, 81, 349–365. [Google Scholar] [CrossRef]
- Hao, F.; Yang, C.; Chen, S.-S.; Wang, Y.-Y.; Zhou, W.; Hao, Q.; Lu, T.; Hoffer, B.; Zhao, L.-R.; Duan, W.-M.; et al. Long-term protective effects of AAV9-mesencephalic astrocyte-derived neurotrophic factor gene transfer in parkinsonian rats. Exp. Neurol. 2017, 291, 120–133. [Google Scholar] [CrossRef]
- Guo, J.; Cui, Y.; Liu, Q.; Yang, Y.; Li, Y.; Weng, L.; Tang, B.; Jin, P.; Li, X.J.; Yang, S.; et al. Piperine ameliorates SCA17 neuropathology by reducing ER stress. Mol. Neurodegener. 2018, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Yang, S.; Guo, J.; Yan, S.; Gaertig, M.A.; Li, S.; Li, X.J. Large Polyglutamine Repeats Cause Muscle Degeneration in SCA17 Mice. Cell Rep. 2015, 13, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.T.; Chung, M.Y.; Banfi, S.; Kwiatkowski, T.J., Jr.; Servadio, A.; Beaudet, A.L.; McCall, A.E.; Duvick, L.A.; Ranum, L.P.; Zoghbi, H.Y. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993, 4, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; DeFranco, D.B.; Orr, H.T.; Zoghbi, H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148–154. [Google Scholar] [CrossRef]
- Waragai, M.; Lammers, C.H.; Takeuchi, S.; Imafuku, I.; Udagawa, Y.; Kanazawa, I.; Kawabata, M.; Mouradian, M.M.; Okazawa, H. PQBP-1, a novel polyglutamine tract-binding protein, inhibits transcription activation by Brn-2 and affects cell survival. Hum. Mol. Genet. 1999, 8, 977–987. [Google Scholar] [CrossRef]
- Okazawa, H. PQBP1, an intrinsically disordered/denatured protein at the crossroad of intellectual disability and neurodegenerative diseases. Neurochem. Int. 2018, 119, 17–25. [Google Scholar] [CrossRef]
- Lu, H.C.; Tan, Q.; Rousseaux, M.W.; Wang, W.; Kim, J.Y.; Richman, R.; Wan, Y.W.; Yeh, S.Y.; Patel, J.M.; Liu, X.; et al. Disruption of the ATXN1-CIC complex causes a spectrum of neurobehavioral phenotypes in mice and humans. Nat. Genet. 2017, 49, 527–536. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Young, R.A. Biomolecular Condensates in the Nucleus. Trends Biochem. Sci. 2020, 45, 961–977. [Google Scholar] [CrossRef]
- Drobot, B.; Iglesias-Artola, J.M.; Le Vay, K.; Mayr, V.; Kar, M.; Kreysing, M.; Mutschler, H.; Tang, T.D. Compartmentalised RNA catalysis in membrane-free coacervate protocells. Nat. Commun. 2018, 9, 3643. [Google Scholar] [CrossRef]
- Lin, Y.; McCarty, J.; Rauch, J.N.; Delaney, K.T.; Kosik, K.S.; Fredrickson, G.H.; Shea, J.E.; Han, S. Narrow equilibrium window for complex coacervation of tau and RNA under cellular conditions. Elife 2019, 8, e42571. [Google Scholar] [CrossRef]
- Sharp, P.A.; Chakraborty, A.K.; Henninger, J.E.; Young, R.A. RNA in formation and regulation of transcriptional condensates. RNA 2022, 28, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225.e24. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Gao, R.; Xu, W.; Mandal, S.M.; Lim, J.G.; Hazra, T.K.; Wakamiya, M.; Edwards, S.F.; Raskin, S.; Teive, H.A.; et al. Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 2010, 6, e1000984. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Robin Highley, J.; Dickman, M.J.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.A.; Kirby, J.; et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef]
- Jain, A.; Vale, R.D. RNA phase transitions in repeat expansion disorders. Nature 2017, 546, 243–247. [Google Scholar] [CrossRef]
- Fay, M.M.; Anderson, P.J.; Ivanov, P. ALS/FTD-Associated C9ORF72 Repeat RNA Promotes Phase Transitions In Vitro and in Cells. Cell Rep. 2017, 21, 3573–3584. [Google Scholar] [CrossRef]
- Berry, J.; Weber, S.C.; Vaidya, N.; Haataja, M.; Brangwynne, C.P. RNA transcription modulates phase transition-driven nuclear body assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E5237–E5245. [Google Scholar] [CrossRef]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef]
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct stages in stress granule assembly and disassembly. eLife 2016, 5, e18413. [Google Scholar] [CrossRef]
- Loureiro, J.R.; Oliveira, C.L.; Silveira, I. Unstable repeat expansions in neurodegenerative diseases: Nucleocytoplasmic transport emerges on the scene. Neurobiol. Aging 2016, 39, 174–183. [Google Scholar] [CrossRef]
- Castelo-Branco, P.; Furger, A.; Wollerton, M.; Smith, C.; Moreira, A.; Proudfoot, N. Polypyrimidine tract binding protein modulates efficiency of polyadenylation. Mol. Cell Biol. 2004, 24, 4174–4183. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Nussbacher, J.K.; Tabet, R.; Yeo, G.W.; Lagier-Tourenne, C. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 2019, 102, 294–320. [Google Scholar] [CrossRef] [PubMed]
- Prudencio, M.; Belzil, V.V.; Batra, R.; Ross, C.A.; Gendron, T.F.; Pregent, L.J.; Murray, M.E.; Overstreet, K.K.; Piazza-Johnston, A.E.; Desaro, P.; et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 2015, 18, 1175–1182. [Google Scholar] [CrossRef]
- Curinha, A.; Oliveira Braz, S.; Pereira-Castro, I.; Cruz, A.; Moreira, A. Implications of polyadenylation in health and disease. Nucleus 2014, 5, 508–519. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Proteins with LLPS Capacity | UniProt ID | IDRs (n) a | RRMs (n) | DBMs (n) a | Disease | Experimental Model | References |
|---|---|---|---|---|---|---|---|
| FUS | P35637 | 1 | 1 | 1 | ALS, FTD, SCA31 | Human SK-ES-1 cells | [36,40,85] |
| EWS | Q01844 | 3 | 1 | 1 | ALS, FTD, cancer | ||
| TAF15 | Q92804 | 3 | 1 | 1 | ALS, FTD, cancer | ||
| hnRNPA1 | P09651 | 2 | 2 | ND | ALS | Human HeLa and U2OS | [77,80] |
| TDP-43 | Q13148 | 2 | 2 | ND | ALS, SCA31 | In vitro | [78,80,84] |
| MATRIN-3 | P43243 | 4 | 2 | 1 | ALS, FTD, distal myopathy | Yeast, mouse C2C12 myoblast cells | [88,89] |
| ATXN1 | P54253 | 5 | ND | ND | SCA1 | Mouse Neuro 2a cells | [64] |
| ATXN2 | Q99700 | 3 | ND | ND | SCA2, ALS | Human U2OS cells | [83] |
| ATXN3 | P54252 | 1 | ND | ND | MJD/SCA3 | In vitro | [65] |
| CACNA1A/α-1ACT | O00555 | 3 | ND | b | SCA6 | ND | |
| ATXN7 | O15265 | 6 | ND | ND | SCA7 | ND | |
| TBP | P20226 | 3 | ND | ND | SCA17 | Human HEK293T cells | [62] |
| AR | P10275 | 2 | ND | ND | SBMA | In vitro and in human LNCaP and LAPC4 cell lines | [90] |
| Disease | Protein | Transcriptional Interactors | Dysregulated Genes | References |
|---|---|---|---|---|
| SCA1 | ATXN1 | HDAC3, RORα/TIP60, CIC, LANP, SMRT/NCOR2, HDAC4/MEF2, ATXNL1 (BOAT), GFI-1, 14-3-3, SP1, A1Up, PQBP1, RBM17 | Cerebellar PC-specific | [92,93,94,95,96,97,98,99,100,101,102,103,104,105] |
| SCA2 | ATXN2 | PABP-1, A2BP1/Fox1, DDX6, TDP-43 | Cerebellar GC- and PC-specific (e.g., RORα, Itpr1, Atp2a2, Inpp5a) | [106] |
| MJD/SCA3 | ATXN3 | CBP, PCAF, P300, HDAC3/NCOR, FOXO4, TBP, PML, TAF130, MAML1, SC35, NCoR | Involved in glutamatergic neurotransmission; intracellular calcium signaling; MAP Kinase signaling | [107,108,109,110,111,112,113] |
| SCA6 | CACNA1A | NA | Involved in neurite outgrowth (e.g., TAF, BTG1, PMCA2, GRN) | [91] |
| SCA7 | ATXN7 | GCN5, USP22, CRX, RORα, SIRT1, TBP, KAT2A/GCN5/ATXN7L3 (components of SAGA complex) | Cerebellar PC-specific; involved in morphological and physiological identities of mature photoreceptors | [114,115] |
| SCA17 | TBP | TFIIB, XBP1, NFY, MYOD, SP1 | Hspb1, Manf, Trka, Mhc4, Mck | [116,117,118,119,120,121,122] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueiredo, A.S.; Loureiro, J.R.; Macedo-Ribeiro, S.; Silveira, I. Advances in Nucleotide Repeat Expansion Diseases: Transcription Gets in Phase. Cells 2023, 12, 826. https://doi.org/10.3390/cells12060826
Figueiredo AS, Loureiro JR, Macedo-Ribeiro S, Silveira I. Advances in Nucleotide Repeat Expansion Diseases: Transcription Gets in Phase. Cells. 2023; 12(6):826. https://doi.org/10.3390/cells12060826
Chicago/Turabian StyleFigueiredo, Ana S., Joana R. Loureiro, Sandra Macedo-Ribeiro, and Isabel Silveira. 2023. "Advances in Nucleotide Repeat Expansion Diseases: Transcription Gets in Phase" Cells 12, no. 6: 826. https://doi.org/10.3390/cells12060826
APA StyleFigueiredo, A. S., Loureiro, J. R., Macedo-Ribeiro, S., & Silveira, I. (2023). Advances in Nucleotide Repeat Expansion Diseases: Transcription Gets in Phase. Cells, 12(6), 826. https://doi.org/10.3390/cells12060826