Effects of Physical Activity on Brain Energy Biomarkers in Alzheimer’s Diseases


 ,
,  and
and 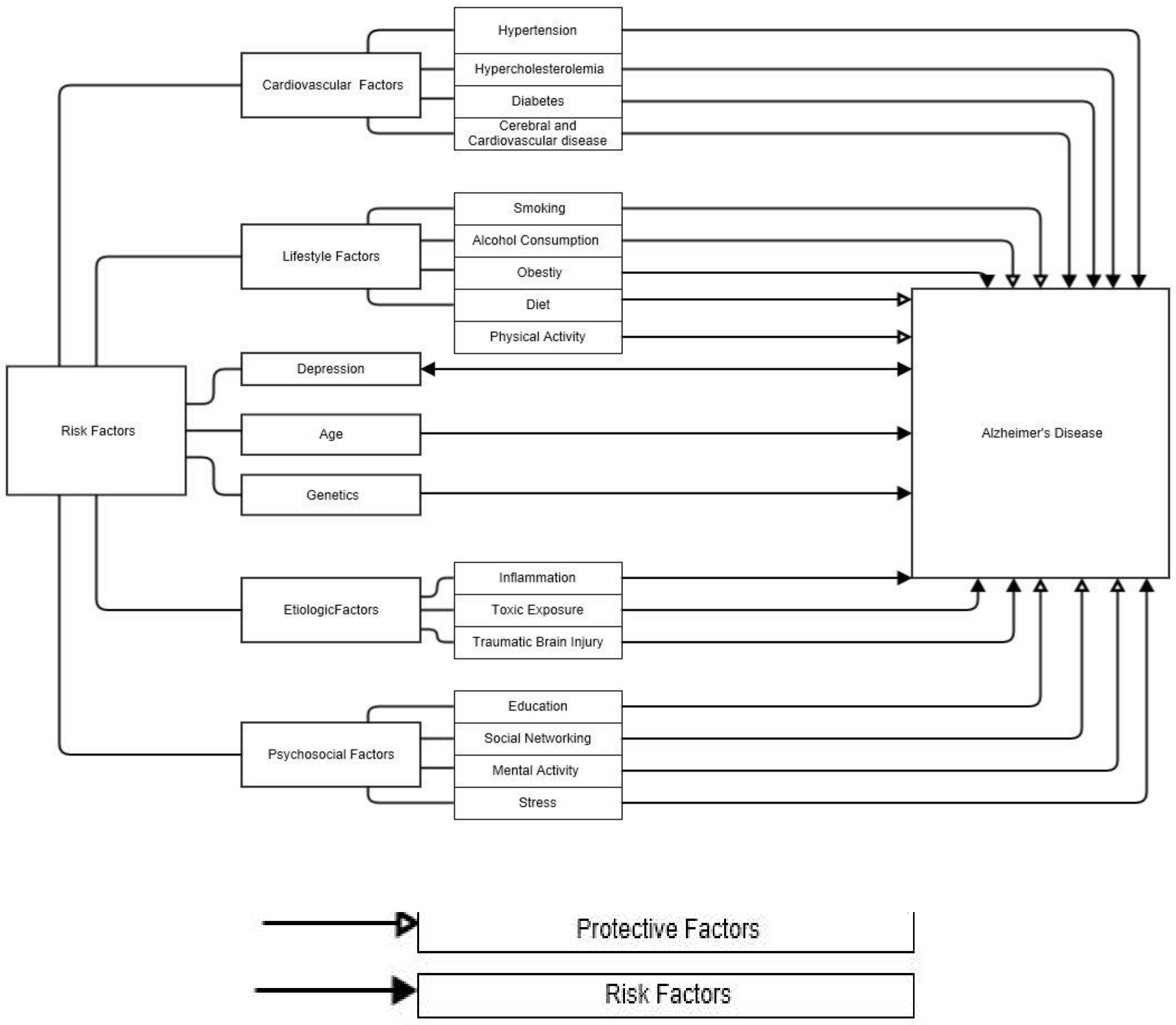{kind=link}
{kind=link}
Abstract
1. Introduction
2. Risk Factors for Alzheimer’s Disease
3. Aetology of Alzheimer’s Disease
4. Impaired Brain Creatine Kinase Activity and Cerebral Glucose Metabolism
5. The Effect of Physical Activity on Alzheimer’s Disease
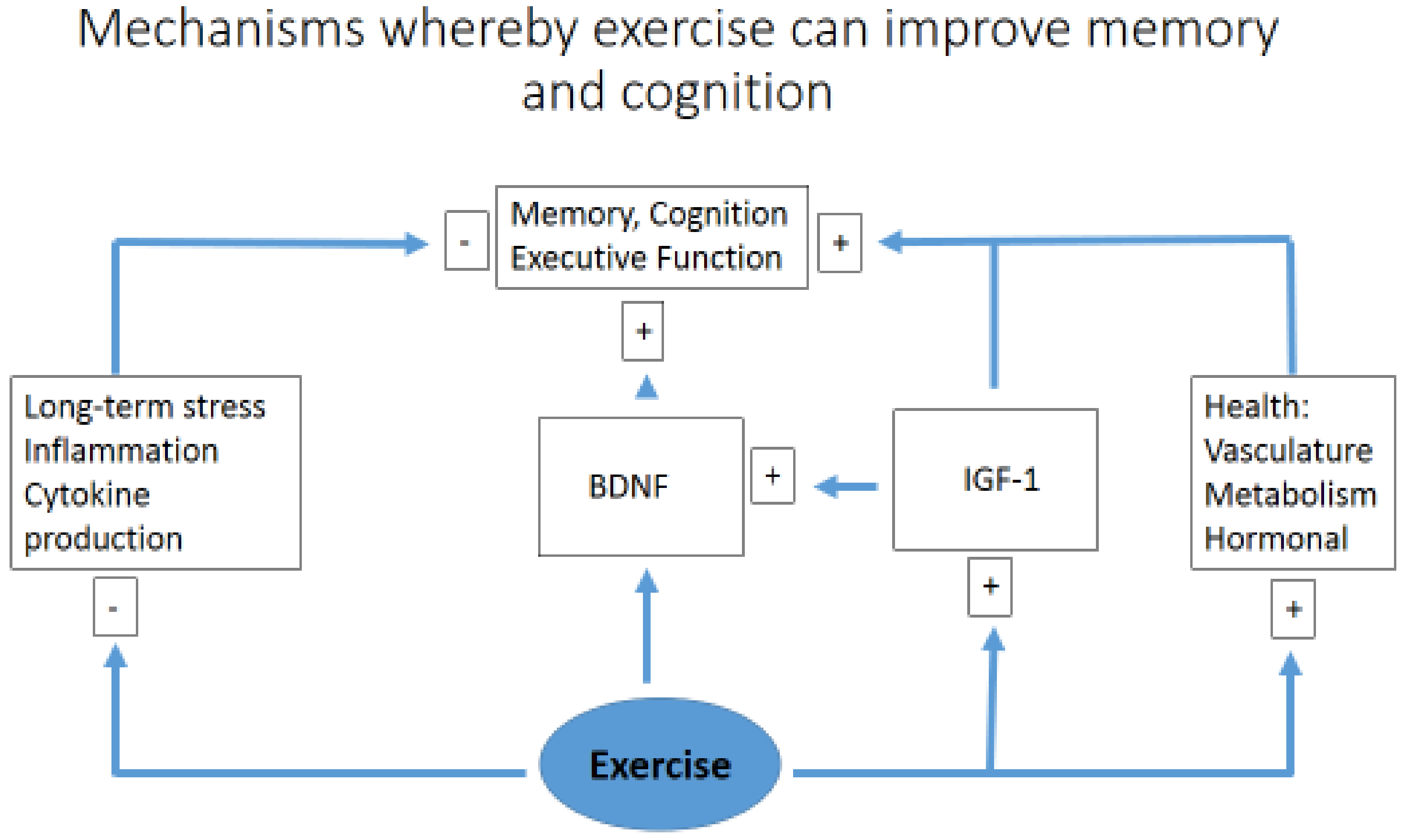
6. Exercise, Memory and Learning
7. Conclusions
Funding
Conflicts of Interest
References
- Lin, T.-W.; Shih, Y.-H.; Chen, S.-J.; Lien, C.-H.; Chang, C.-Y.; Huang, T.-Y.; Chen, S.-H.; Jen, C.J.; Kuo, Y.-M. Running exercise delays neurodegeneration in amygdala and hippocampus of Alzheimer’s disease (APP/PS1) transgenic mice. Neurobiol. Learn. Mem. 2015, 118, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 19 September 2019).
- Chiba, T. Emerging therapeutic strategies in Alzheimer’s disease. In Neurodegenerative Diseases; Kishore, U., Ed.; IntechOpon: London, UK, 2013; ISBN 978-953-51-1088-0. [Google Scholar]
- Perrin, R.J.; Fagan, A.M.; Holtzman, D.M. Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature 2009, 461, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Bird, C.M.; Burgess, N. The hippocampus and memory: Insights from spatial processing. Nat. Rev. Neurosci. 2008, 9, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.E.; Geda, Y.E.; Graff-Radford, N.R.; Petersen, R.C. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2013, 9, 208–245. [Google Scholar] [CrossRef]
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of physical exercise in Alzheimer’s disease. Biomed. Rep. 2016, 4, 403–407. [Google Scholar] [CrossRef]
- Welander, H. Alzheimer Disease: Studies on Abeta and Gamma-Secretase in Human Brain; Institutionen för Neurobiologi, Vårdvetenskap och Samhälle/Department of Neurobiology, Care Sciences and Society: Stockholm, Sweden, 2010. [Google Scholar]
- Bradbury, K.E.; Guo, W.; Cairns, B.J.; Armstrong, M.E.; Key, T.J. Association between physical activity and body fat percentage, with adjustment for BMI: A large cross-sectional analysis of UK Biobank. BMJ Open 2017, 7, e011843. [Google Scholar] [CrossRef] [PubMed]
- Daviglus, M.L.; Bell, C.C.; Berrettini, W.; Bowen, P.E.; Connolly, E.S., Jr.; Cox, N.J.; Dunbar-Jacob, J.M.; Granieri, E.C.; Hunt, G.; McGarry, K.; et al. National Institutes of Health State-of-the-Science Conference statement: Preventing Alzheimer disease and cognitive decline. Ann. Intern. Med. 2010, 153, 176–181. [Google Scholar] [CrossRef]
- Xu, W.; Yu, J.T.; Tan, M.S.; Tan, L. Cognitive reserve and Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 187–208. [Google Scholar] [CrossRef]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of Alzheimer disease: Back to the future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef]
- Schellenberg, G.D.; Montine, T.J. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 2012, 124, 305–323. [Google Scholar] [CrossRef]
- Huang, Y.; Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef]
- Poirier, J. Apolipoprotein E, cholesterol transport and synthesis in sporadic Alzheimer’s disease. Neurobiol. Aging 2005, 26, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. The pathogenesis of Alzheimer’s disease: A reevaluation of the “amyloid cascade hypothesis”. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Juniper, K.C. American Colleage; The University of Oklahama Health Sciences Center Graduate College: Oklahoma City, OK, USA, 2002. [Google Scholar]
- Rezaei-Ghaleh, N.; Giller, K.; Becker, S.; Zweckstetter, M. Effect of zinc binding on β-amyloid structure and dynamics: Implications for Aβ aggregation. Biophys. J. 2011, 101, 1202–1211. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease β-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-Beta: A crucial factor in Alzheimer’s disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef]
- Joshi, G.; Wang, Y. Golgi defects enhance APP amyloidogenic processing in Alzheimer’s disease. BioEssays 2015, 37, 240–247. [Google Scholar] [CrossRef]
- Miras-Portugal, M.T.; Diaz-Hernandez, J.I.; Gomez-Villafuertes, R.; Diaz-Hernandez, M.; Artalejo, A.R.; Gualix, J. Role of P2X7 and P2Y 2 receptors on α-secretase-dependent APP processing: Control of amyloid plaques formation “in vivo” by P2X7 receptor. Comput. Struct. Biotechnol. J. 2015, 13, 176–181. [Google Scholar] [CrossRef]
- Marinangeli, C.; Tasiaux, B.; Opsomer, R.; Hage, S.; Sodero, A.O.; Dewachter, I.; Octave, J.N.; Smith, S.O.; Constantinescu, S.N.; Kienlen-Campard, P. Presenilin Transmembrane Domain 8 Conserved AXXXAXXXG Motifs Are Required for the Activity of the γ-Secretase Complex. J. Biol. Chem. 2015, 290, 7169–7184. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A.; Vissel, B. Therapeutic implications of how TNF links apolipoprotein E, phosphorylated tau, α-synuclein, amyloid-β and insulin resistance in neurodegenerative diseases. Br. J. Pharmacol. 2018, 175, 3859–3875. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018, 136, 663–689. [Google Scholar] [CrossRef]
- Wen, G. Alzheimer’s disease and risk factors. J. Food Drug Anal. 1998, 6, 465–476. [Google Scholar]
- Toda, N.; Ayajiki, K.; Okamura, T. Obesity-induced cerebral hypoperfusion derived from endothelial dysfunction: One of the risk factors for Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 733–744. [Google Scholar] [CrossRef]
- Radak, Z.; Hart, N.; Sarga, L.; Koltai, E.; Atalay, M.; Ohno, H.; Boldogh, I. Exercise plays a preventive role against Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, 777–783. [Google Scholar] [CrossRef]
- Um, H.S.; Kang, E.B.; Leem, Y.H.; Cho, I.H.; Yang, C.H.; Chae, K.R.; Hwang, D.Y.; Cho, J.Y. Exercise training acts as a therapeutic strategy for reduction of the pathogenic phenotypes for Alzheimer’s disease in an NSE/APPsw-transgenic model. Int. J. Mol. Med. 2008, 22, 529–534. [Google Scholar]
- Ruth, S. Association between Physical Activity and Alzheimer’s Disease. Master’s Thesis, Institute of Public Health and Clinical Nutrition, University of Eastern Finland, Kuopio, Finland, October 2014. [Google Scholar]
- Burklen, T.; Schlattner, U.; Homayouni, R.; Gough, K.; Rak, M.; Szeghalmi, A.; Wallimann, T. The creatine kinase/creatine connection to Alzheimer’s diseease: CK-inactivation, APP-CK complexes and focal creatine deposits. J. Biomed. Biotechnol. 2006, 3, 35936. [Google Scholar]
- Smith, R.N.; Agharkar, A.S.; Gonzalez, E.B. A review of creatine monohydrate supplementation in age-related diseases: More than a supplement for athletes. F1000 Res. 2014, 3, 222. [Google Scholar] [CrossRef]
- Hötting, K.; Röder, B. Beneficial effects of physical exercise on neuroplasticity and cognition. Neurosci. Biobehav. Rev. 2013, 37, 2243–2257. [Google Scholar] [CrossRef]
- Baker, L.D.; Frank, L.L.; Foster-Schubert, K.; Green, P.S.; Wilkinson, C.W.; McTiernan, A.; Plymate, S.R.; Fishel, M.A.; Watson, G.S.; Cholerton, B.A. Effects of aerobic exercise on mild cognitive impairment: A controlled trial. Arch. Neurol. 2010, 67, 71–79. [Google Scholar] [CrossRef]
- Um, H.-S.; Kang, E.-B.; Koo, J.-H.; Kim, H.-T.; Kim, E.-J.; Yang, C.-H.; An, G.-Y.; Cho, I.-H.; Cho, J.-Y. Treadmill exercise represses neuronal cell death in an aged transgenic mouse model of Alzheimer’s Disease. Neurosci. Res. 2011, 69, 161–173. [Google Scholar] [CrossRef]
- Diegues, J.C.; Pauli, J.R.; Luciano, E.; Almeida Leme, J.A.C.; de Moura, L.P.; Dalia, R.A.; de Araújo, M.B.; Sibuya, C.Y.; de Mello, M.A.R.; Gomes, R.J. Spatial memory in sedentary and trained diabetic rats: Molecular mechanisms. Hippocampus 2014, 24, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Nichol, K.E.; Poon, W.W.; Parachikova, A.I.; Cribbs, D.H.; Glabe, C.G.; Cotman, C.W. Exercise alters the immune profile in Tg2576 Alzheimer mice toward a response coincident with improved cognitive performance and decreased amyloid. J. Neuroinflamm. 2008, 5, 2094–2095. [Google Scholar] [CrossRef]
- Kang, E.-B.; Kwon, I.-S.; Koo, J.-H.; Kim, E.-J.; Kim, C.-H.; Lee, J.; Yang, C.-H.; Lee, Y.-I.; Cho, I.-H.; Cho, J.-Y. Treadmill exercise represses neuronal cell death and inflammation during Aβ-induced ER stress by regulating unfolded protein response in aged presenilin 2 mutant mice. Apoptosis 2013, 18, 1332–1347. [Google Scholar] [CrossRef]
- Liu, H.L.; Zhao, G.; Zhang, H. Long-term treadmill exercise inhibits the progression of Alzheimer’s disease-like neuropathology in the hippocampus of APP/PS1 transgenic mice. Behav. Brain Res. 2013, 256, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Head, D.; Bugg, J.M.; Goate, A.M.; Fagan, A.M.; Mintun, M.A.; Benzinger, T.; Holtzman, D.M.; Morris, J.C. Exercise engagement as a moderator of the effects of APOE genotype on amyloid deposition. Arch. Neurol. 2012, 69, 636–643. [Google Scholar] [PubMed]
- Smith, J.C.; Lancaster, M.A.; Nielson, K.A.; Woodard, J.L.; Seidenberg, M.; Durgerian, S.; Sakaie, K.; Rao, S.M. Interactive effects of physical activity and APOE-ε4 on white matter tract diffusivity in healthy elders. NeuroImage 2015, 131, 102–112. [Google Scholar] [CrossRef]
- Rovio, S.; Kåreholt, I.; Helkala, E.-L.; Viitanen, M.; Winblad, B.; Tuomilehto, J.; Soininen, H.; Nissinen, A.; Kivipelto, M. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005, 4, 705–711. [Google Scholar] [CrossRef]
- Piotr, G.; Stefan, B.; Joanna, G.; Adam, Z.; Adam, M.; Roman, C.; Agnieszka, D.; Wojciech, C.; Robert, P.; Cain, C.T.C.; et al. Physical activity and Alzheimer’s disease. Aging Dis. 2019, 10, 1282–1292. [Google Scholar]
- Soto, I.; Graham, L.C.; Richter, H.J.; Simeone, S.N.; Radell, J.E.; Grabowska, W.; Funkhouser, W.K.; Howell, M.C.; Howell, G.R. APOE stabilization by exercise prevents aging neurovascular dysfunction and complement induction. PLoS Biol. 2015, 13, e1002279. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Perreau, V.M.; Pop, V.; Cotman, C.W. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 4217–4221. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Aranguiz, F.; Varela-Nallar, L.; Inestrosa, N.C. Voluntary running attenuates memory loss, decreases neuropathological changes and induces neurogenesis in a mouse model of Alzheimer’s disease. Brain Pathol. 2016, 26, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-E.; Ko, I.-G.; Kim, B.-K.; Shin, M.-S.; Cho, S.; Kim, C.-J.; Kim, S.H.; Baek, S.-S.; Lee, E.-K.; Jee, Y.-S. Treadmill exercise prevents aging-induced failure of memory through an increase in neurogenesis and suppression of apoptosis in rat hippocampus. Exp. Gerontol. 2010, 45, 357–365. [Google Scholar] [CrossRef]
- Dao, A.T.; Zagaar, M.A.; Levine, A.T.; Salim, S.; Eriksen, J.; Alkadhi, K.A. Treadmill exercise prevents learning and memory impairment in Alzheimer’s disease-like pathology. Curr. Alzheimer Res. 2013, 10, 507–515. [Google Scholar] [CrossRef]
- Aguiar, A.S.; Castro, A.A.; Moreira, E.L.; Glaser, V.; Santos, A.R.S.; Tasca, C.I.; Latini, A.; Prediger, R.D.S. Short bouts of mild-intensity physical exercise improve spatial learning and memory in aging rats: Involvement of hippocampal plasticity via AKT, CREB and BDNF signaling. Mech. Ageing Dev. 2011, 132, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Okamoto, M.; Liu, Y.F.; Inoue, K.; Matsui, T.; Nogami, H.; Soya, H. Voluntary resistance running with short distance enhances spatial memory related to hippocampal BDNF signaling. J. Appl. Physiol. 2012, 113, 1260–1266. [Google Scholar] [CrossRef]
- Van Praag, H.; Shubert, T.; Zhao, C.; Gage, F.H. Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 2005, 25, 8680–8685. [Google Scholar] [CrossRef]
- Liu-Ambrose, T.; Nagamatsu, L.S.; Graf, P.; Beattie, B.L.; Ashe, M.C.; Handy, T.C. Resistance training and executive functions: A 12-month randomized controlled trial. Arch. Intern. Med. 2010, 170, 170–178. [Google Scholar] [CrossRef]
- Cassilhas, R.C.; Viana, V.A.; Grassmann, V.; Santos, R.T.; Santos, R.F.; Tufik, S.; Mello, M.T. The impact of resistance exercise on the cognitive function of the elderly. Med. Sci. Sport. Exerc. 2007, 39, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Peig-Chiello, P.; Perrig, W.J.; Ehrsam, R.; Staehelin, H.B.; Krings, F. The effects of resistance training on well-being and memory in elderly volunteers. Age Ageing 1998, 27, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Liu-Ambrose, T.; Nagamatsu, L.S.; Voss, M.W.; Khan, K.M.; Handy, T.C. Resistance training and functional plasticity of the aging brain: A 12-month randomized controlled trial. Neurobiol. Aging 2012, 33, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Özkaya, G.Y.; Aydin, H.; Toraman, F.N.; Kizilay, F.; Özdemir, Ö.; Cetinkaya, V. Effect of strength and endurance training on cognition in older people. J. Sports Sci. Med. 2005, 4, 300–313. [Google Scholar] [PubMed]
- Cassilhas, R.; Lee, K.; Fernandes, J.; Oliveira, M.; Tufik, S.; Meeusen, R.; De Mello, M.T. Spatial memory is improved by aerobic and resistance exercise through divergent molecular mechanisms. Neuroscience 2012, 202, 309–317. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebrahimi, K.; Jourkesh, M.; Sadigh-Eteghad, S.; Stannard, S.R.; Earnest, C.P.; Ramsbottom, R.; Antonio, J.; Navin, K.H. Effects of Physical Activity on Brain Energy Biomarkers in Alzheimer’s Diseases. Diseases 2020, 8, 18. https://doi.org/10.3390/diseases8020018
Ebrahimi K, Jourkesh M, Sadigh-Eteghad S, Stannard SR, Earnest CP, Ramsbottom R, Antonio J, Navin KH. Effects of Physical Activity on Brain Energy Biomarkers in Alzheimer’s Diseases. Diseases. 2020; 8(2):18. https://doi.org/10.3390/diseases8020018
Chicago/Turabian StyleEbrahimi, Khadijeh, Morteza Jourkesh, Saeed Sadigh-Eteghad, Stephen R Stannard, Conrad P. Earnest, Roger Ramsbottom, Jose Antonio, and Khan H. Navin. 2020. "Effects of Physical Activity on Brain Energy Biomarkers in Alzheimer’s Diseases" Diseases 8, no. 2: 18. https://doi.org/10.3390/diseases8020018
APA StyleEbrahimi, K., Jourkesh, M., Sadigh-Eteghad, S., Stannard, S. R., Earnest, C. P., Ramsbottom, R., Antonio, J., & Navin, K. H. (2020). Effects of Physical Activity on Brain Energy Biomarkers in Alzheimer’s Diseases. Diseases, 8(2), 18. https://doi.org/10.3390/diseases8020018