Modulating Effect of Diet on Alzheimer’s Disease



Abstract
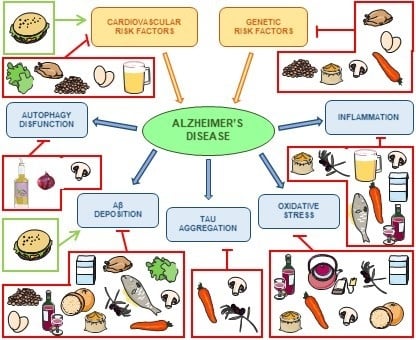
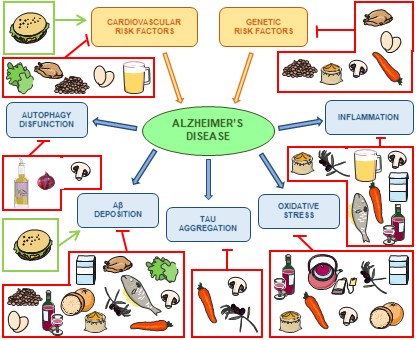
1. Introduction
2. Alzheimer’s Disease
2.1. Disease Description
2.2. Neuropathological Markers
2.3. Oxidative Stress
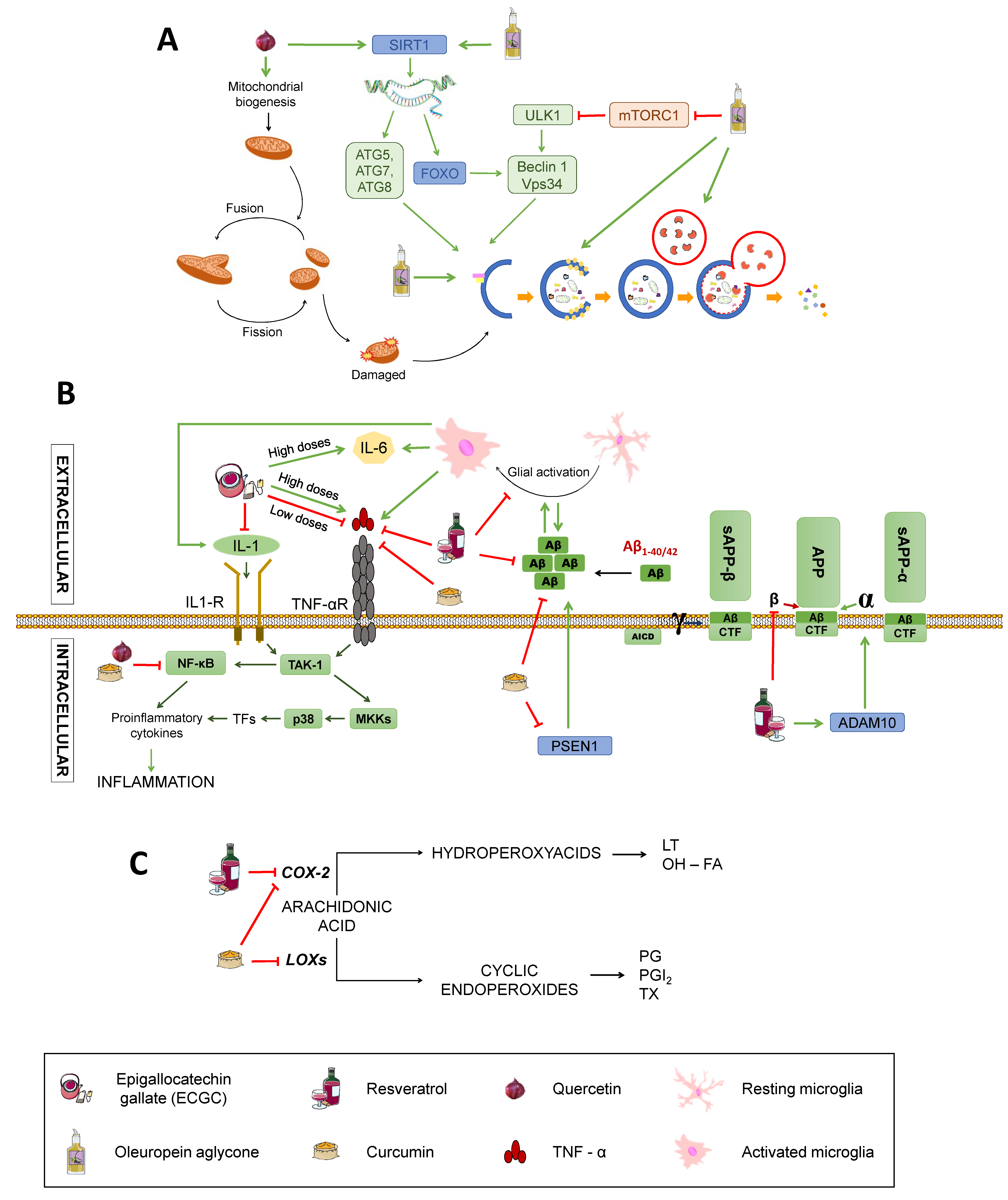
2.4. Autophagy
2.5. Inflammation
3. Relevance of Dietary Patterns
4. Nutrients That Modulate Alzheimer’s Disease
4.1. Unsaturated Fats (Monounsaturated and Polyunsaturated)
4.2. Vitamins
4.3. Polyphenols
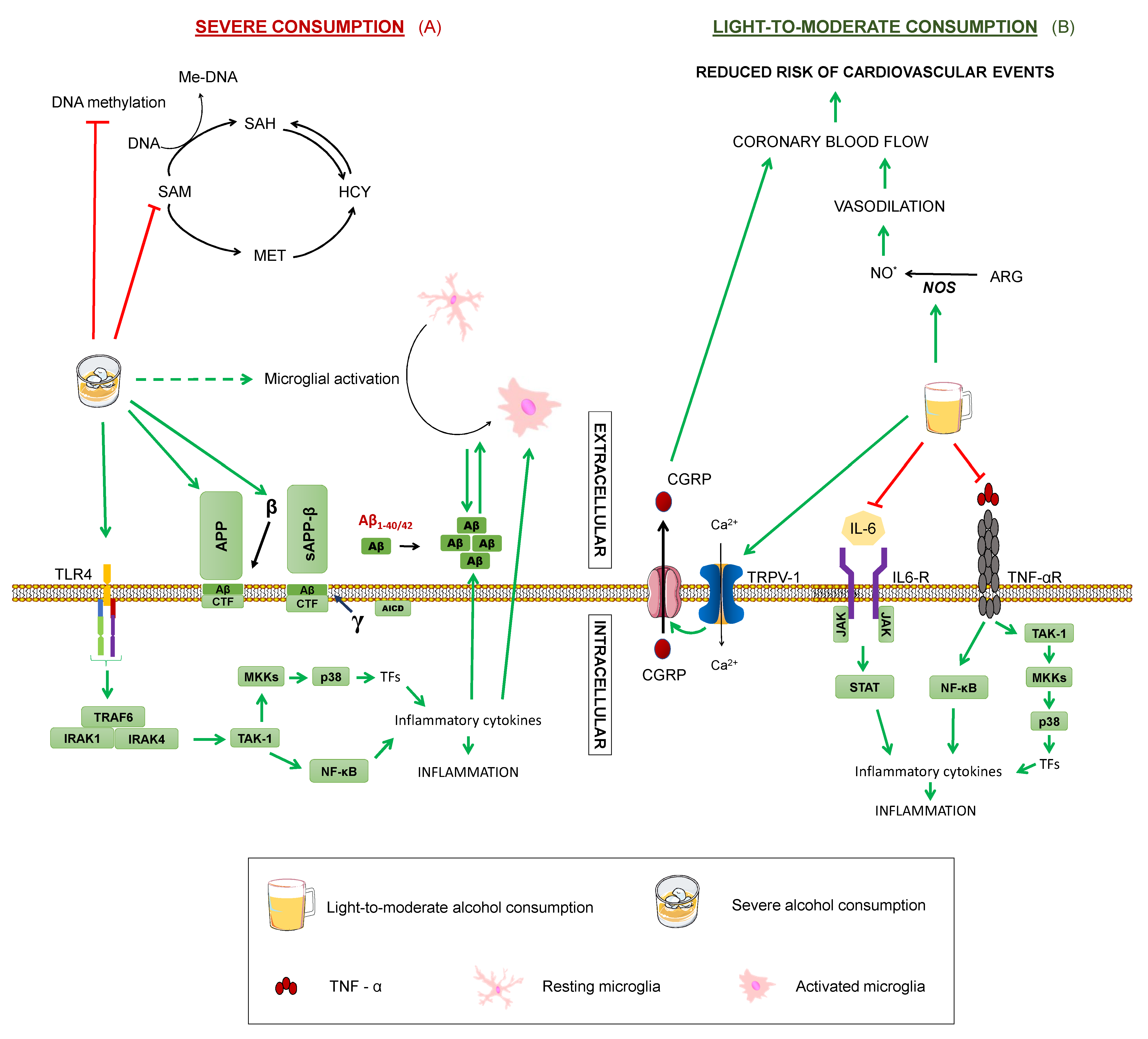
4.4. Moderate Alcohol Consumption
4.5. Trehalose
4.6. Cholesterol, Saturated Fats, Trans, or Hydrogenated Fats
4.7. Food Products Containing Alzheimer’s Disease Modulating Nutrients
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardman, R.J.; Kennedy, G.; Macpherson, H.; Scholey, A.B.; Pipingas, A. Adherence to a mediterranean-style diet and effects on cognition in adults: A qualitative evaluation and systematic review of longitudinal and prospective trials. Front. Nutr. 2016, 3, 22. [Google Scholar] [CrossRef]
- Solfrizzi, V.; Panza, F.; Frisardi, V.; Seripa, D.; Logroscino, G.; Imbimbo, B.P.; Pilotto, A. Diet and Alzheimer’s disease risk factors or prevention: The current evidence. Expert Rev. Neurother. 2011, 11, 677–708. [Google Scholar] [CrossRef] [PubMed]
- Lara, H.H.; Alanís-Garza, E.J.; Puente, M.F.E.; Mureyko, L.L.; Torres, D.A.A.; Turrent, L.I. Nutrición que previene el estrés oxidativo causante del Alzheimer. Prevención del Alzheimer. Gac. Med. Mex. 2015, 151, 245–251. [Google Scholar] [PubMed]
- Karlawish, J.; Jack, C.R., Jr.; Rocca, W.A.; Snyder, H.M.; Carrillo, M.C. Alzheimer’s disease: The next frontier—Special report 2017. Alzheimer’s Dement. 2017, 13, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Lleó, A. El Alzheimer, la enfermedad ignorada. Med. Clín. 2018, 150, 432–433. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease—Genotypes, phenotype, and treatments. Science 1997, 275, 630–631. [Google Scholar] [CrossRef]
- Burgos Peláez, R.; Virgili Casas, N. Papel de la nutrición en la prevención y evolución de las enfermedades neurodegenerativas. Nutr. Hosp. 2009, 2, 13–25. [Google Scholar]
- Arizaga, R.; Barreto, D.; Bavec, C.; Berríos, W.; Cristalli, D.; Colli, L.; Garau, M.L.; Golimstok, A.; Ollari, J.; Sarasola, D. Dieta y prevención en enfermedad de Alzheimer. Neurol. Argent. 2018, 10, 44–60. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.; Hardy, J.; Hutton, M.; Kukull, W. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and app mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864. [Google Scholar] [CrossRef]
- Sierra, L. Estrategias de investigación para el tratamiento de Alzheimer con antioxidantes polifenólicos. Rev. Acad. Colomb. Cienc. Exactas Físicas Nat. 2016, 40, 608–620. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.-C.; Crawford, F.; Houlden, H.; Warren, A.; Hughes, D.; Fidani, L.; Goate, A.; Rossor, M.; Roques, P.; Hardy, J. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature 1991, 353, 844. [Google Scholar] [CrossRef] [PubMed]
- Munoz, D.G.; Feldman, H. Causes of Alzheimer’s disease. Can. Med. Assoc. J. 2000, 162, 65–72. [Google Scholar]
- Williamson, J.; Goldman, J.; Marder, K.S. Genetic aspects of Alzheimer disease. Neurologist 2009, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2017, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- González Rodríguez, L.G.; Palmeros Exsome, C.; González Martínez, M.T.; Pérez Ávila, M.D.L.L.; Gutiérrez López, M. Factores dietéticos y nutricionales en la prevención de la enfermedad de Alzheimer. Rev. Salud Públ. Nutr. 2016, 15, 27–37. [Google Scholar]
- García-Escudero, V.; Martín-Maestro, P.; Perry, G.; Avila, J. Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid. Med. Cell. Longev. 2013, 2013, 162152. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22. [Google Scholar] [CrossRef]
- Portelius, E.; Price, E.; Brinkmalm, G.; Stiteler, M.; Olsson, M.; Persson, R.; Westman-Brinkmalm, A.; Zetterberg, H.; Simon, A.J.; Blennow, K. A novel pathway for amyloid precursor protein processing. Neurobiol. Aging 2011, 32, 1090–1098. [Google Scholar] [CrossRef]
- Portelius, E.; Mattsson, N.; Andreasson, U.; Blennow, K.; Zetterberg, H. Novel aβisoforms in Alzheimer’s disease-their role in diagnosis and treatment. Curr. Pharm. De. 2011, 17, 2594–2602. [Google Scholar] [CrossRef]
- Portelius, E.; Zetterberg, H.; Andreasson, U.; Brinkmalm, G.; Andreasen, N.; Wallin, A.; Westman-Brinkmalm, A.; Blennow, K. An Alzheimer’s disease-specific β-amyloid fragment signature in cerebrospinal fluid. Neurosci. Lett. 2006, 409, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Willem, M.; Tahirovic, S.; Busche, M.A.; Ovsepian, S.V.; Chafai, M.; Kootar, S.; Hornburg, D.; Evans, L.D.; Moore, S.; Daria, A. H-secretase processing of app inhibits neuronal activity in the hippocampus. Nature 2015, 526, 443. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; Ayuso-Peralta, L.; Jabbour-Wadih, T. Estrés oxidativo y enfermedad de Alzheimer. Rev. Neurol. 2006, 42, 419–427. [Google Scholar] [PubMed]
- Mehta, V.; Desai, N.; Perwez, A.; Nemade, D.; Dawoodi, S.; Zaman, S.B. ACE Alzheimer’s: The role of vitamin A, C and E (ACE) in oxidative stress induced Alzheimer’s disease. J. Med. Res. Innov. 2018, 2, e000086. [Google Scholar] [CrossRef]
- Konigsberg Fainstein, M.; Aguilar-Maldonado, B. Radicales Libres y Estrés Oxidativo: Aplicaciones Médicas; El Manual Moderno: Bogotá, Colombia, 2008; ISBN 9707293217. [Google Scholar]
- Pupo, E.V.; Robles, L.G.; Marrero, I.R.C. Estrés oxidativo. Correo Científico Médico 2017, 21, 171–186. [Google Scholar]
- Manzano-León, N.; Mas-Oliva, J. Estrés oxidativo, péptido β-amiloide y enfermedad de Alzheimer. Gaceta Médica de México 2006, 142, 229–238. [Google Scholar] [PubMed]
- Carney, J.M.; Smith, C.D.; Carney, A.M.; Butterfield, D.A. Aging-and oxygen-induced modifications in brain biochemistry and behaviora. Ann. N. Y. Acad. Sci. 1994, 738, 44–53. [Google Scholar] [CrossRef]
- Wahlster, L.; Arimon, M.; Nasser-Ghodsi, N.; Post, K.L.; Serrano-Pozo, A.; Uemura, K.; Berezovska, O. Presenilin-1 adopts pathogenic conformation in normal aging and in sporadic Alzheimer’s disease. Acta Neuropathol. 2013, 125, 187–199. [Google Scholar] [CrossRef]
- Beydoun, M.A.; Beydoun, H.A.; Gamaldo, A.A.; Teel, A.; Zonderman, A.B.; Wang, Y. Epidemiologic studies of modifiable factors associated with cognition and dementia: Systematic review and meta-analysis. BMC Public Health 2014, 14, 643. [Google Scholar] [CrossRef]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and Alzheimer’s disease. Cell. Mol. Neurobiol. 2017, 37, 377–388. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Sagredo, A.; Aleman, L.; Villa, M.; Chávez, M.N.; García, L.; Lavandero, S. Autofagia en el sistema cardiovascular: Pasado, presente y futuro. Rev. Chil. Cardiol. 2016, 35, 228–241. [Google Scholar] [CrossRef]
- Cordero, J.G.; García-Escudero, R.; Avila, J.; Gargini, R.; García-Escudero, V. Benefit of oleuropein aglycone for Alzheimer’s disease by promoting autophagy. Oxid. Med. Cell. Longev. 2018, 2018, 5010741. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Rispoli, J.; Kaphzan, H.; Klann, E.; Chen, E.I.; Kim, J.; Komatsu, M.; Abeliovich, A. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol. Neurodegener. 2012, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, P.J.; Herman, A.M.; Hoe, H.-S.; Rebeck, G.W.; Moussa, C.E.-H. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated aβ in ad models. Hum. Mol. Genet. 2011, 20, 2091–2102. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. Park2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2015, 25, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Pedraza-Chaverri, J.; Pérez-Rojas, J.M. Role of docosahexaenoic acid in the modulation of glial cells in Alzheimer’s disease. J. Neuroinflamm. 2016, 13, 61. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Fischer, K.; Melo van Lent, D.; Wolfsgruber, S.; Weinhold, L.; Kleineidam, L.; Bickel, H.; Scherer, M.; Eisele, M.; van den Bussche, H.; Wiese, B. Prospective associations between single foods, Alzheimer’s dementia and memory decline in the elderly. Nutrients 2018, 10, 852. [Google Scholar] [CrossRef] [PubMed]
- Fernández, S.S.M.; Ivanauskas, T.; Ribeiro, S.M.L. Nutritional strategies in the management of Alzheimer disease: Systematic review with network meta-analysis. J. Am. Med. Dir. Assoc. 2017, 18, 897. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Abdullah, N.; Aminudin, N. Interpretation of mushroom as a common therapeutic agent for Alzheimer’s disease and cardiovascular diseases. Crit. Rev. Biotechnol. 2016, 36, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Parsaik, A.K.; Mielke, M.M.; Erwin, P.J.; Knopman, D.S.; Petersen, R.C.; Roberts, R.O. Association of mediterranean diet with mild cognitive impairment and Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2014, 39, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Petersson, S.D.; Philippou, E. Mediterranean diet, cognitive function, and dementia: A systematic review of the evidence. Adv. Nutr. 2016, 7, 889–904. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Lapiscina, E.H.; Clavero, P.; Toledo, E.; Estruch, R.; Salas-Salvadó, J.; San Julián, B.; Sanchez-Tainta, A.; Ros, E.; Valls-Pedret, C.; Martinez-Gonzalez, M.Á. Mediterranean diet improves cognition: The predimed-navarra randomised trial. J. Neurol. Neurosurg. Psychiatry 2013. [Google Scholar] [CrossRef] [PubMed]
- Valls-Pedret, C.; Sala-Vila, A.; Serra-Mir, M.; Corella, D.; De la Torre, R.; Martínez-González, M.Á.; Martínez-Lapiscina, E.H.; Fitó, M.; Pérez-Heras, A.; Salas-Salvadó, J.; et al. Mediterranean diet and age-related cognitive decline: A randomized clinical trial. JAMA Intern. Med. 2015, 175, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Krenz, M.; Korthuis, R.J. Moderate ethanol ingestion and cardiovascular protection: From epidemiologic associations to cellular mechanisms. J. Mol. Cell. Cardiol. 2012, 52, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Berti, V.; Walters, M.; Sterling, J.; Quinn, C.G.; Logue, M.; Andrews, R.; Matthews, D.C.; Osorio, R.S.; Pupi, A.; Vallabhajosula, S.; et al. Mediterranean diet and 3-year Alzheimer brain biomarker changes in middle-aged adults. Neurology 2018, 90, e1789. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid β 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2005, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Sato, N. Bidirectional interactions between diabetes and Alzheimer’s disease. Neurochem. Int. 2017, 108, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Medina-Remón, A.; Kirwan, R.; Lamuela-Raventós, R.M.; Estruch, R. Dietary patterns and the risk of obesity, type 2 diabetes mellitus, cardiovascular diseases, asthma, and neurodegenerative diseases. Crit. Rev. Food Sci. Nutr. 2018, 58, 262–296. [Google Scholar] [CrossRef] [PubMed]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Shieh, C.-H.; Wu, Y.-S.; Kalueff, A.; Gaikwad, S.; Su, K.-P. The role of omega-3 polyunsaturated fatty acids eicosapentaenoic and docosahexaenoic acids in the treatment of major depression and Alzheimer’s disease: Acting separately or synergistically? Prog. Lipid Res. 2016, 62, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, H.; Pu, H.; Wang, G.; Li, W.; Leak, R.K.; Chen, J.; Liou, A.K.; Hu, X. N-3 pufa supplementation benefits microglial responses to myelin pathology. Sci. Rep. 2014, 4, 7458. [Google Scholar] [CrossRef]
- Corsi, L.; Momo Dongmo, B.; Avallone, R. Supplementation of omega 3 fatty acids improves oxidative stress in activated bv2 microglial cell line. Int. J. Food Sci. Nutr. 2015, 66, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Antonietta Ajmone-Cat, M.; Lavinia Salvatori, M.; De Simone, R.; Mancini, M.; Biagioni, S.; Bernardo, A.; Cacci, E.; Minghetti, L. Docosahexaenoic acid modulates inflammatory and antineurogenic functions of activated microglial cells. J. Neurosci. Res. 2012, 90, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.; Borodkin, V.S.; Albarbarawi, O.; Campbell, D.G.; Ibrahim, A.; Van Aalten, D.M. O-glcnacylation of tab1 modulates tak1-mediated cytokine release. EMBO J. 2012, 31, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Fotuhi, M.; Mohassel, P.; Yaffe, K. Fish consumption, long-chain omega-3 fatty acids and risk of cognitive decline or Alzheimer disease: A complex association. Nat. Rev. Neurol. 2009, 5, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Thomas, C.; Radcliffe, J.; Itsiopoulos, C. Omega-3 fatty acids in early prevention of inflammatory neurodegenerative disease: A focus on Alzheimer’s disease. BioMed Res. Int. 2015, 2015, 172801. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Michaelson, D.; Hartmann, T. Omega-3 fatty acids, lipids and apoe lipidation in Alzheimer’s disease: A rationale for multi-nutrient dementia prevention. J. Lipid Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.; Afonso, C.; Bandarra, N.M. Dietary dha and health: Cognitive function ageing. Nutr. Res. Rev. 2016, 29, 281–294. [Google Scholar] [CrossRef]
- Quinn, J.F.; Raman, R.; Thomas, R.G.; Yurko-Mauro, K.; Nelson, E.B.; Van Dyck, C.; Galvin, J.E.; Emond, J.; Jack, C.R.; Weiner, M. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: A randomized trial. Jama 2010, 304, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Freund-Levi, Y.; Eriksdotter-Jönhagen, M.; Cederholm, T.; Basun, H.; Faxen-Irving, G.; Garlind, A.; Vedin, I.; Vessby, B.; Wahlund, L.-O.; Palmblad, J. Ω-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: Omegad study: A randomized double-blind trial. Arch. Neurol. 2006, 63, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Fiala, M.; Halder, R.C.; Sagong, B.; Ross, O.; Sayre, J.; Porter, V.; Bredesen, D.E. Ω-3 supplementation increases amyloid-β phagocytosis and resolvin d1 in patients with minor cognitive impairment. FASEB J. 2015, 29, 2681–2689. [Google Scholar] [CrossRef] [PubMed]
- Belkouch, M.; Hachem, M.; Elgot, A.; Van, A.L.; Picq, M.; Guichardant, M.; Lagarde, M.; Bernoud-Hubac, N. The pleiotropic effects of omega-3 docosahexaenoic acid on the hallmarks of Alzheimer’s disease. J. Nutr. Biochem. 2016, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kalli, E.G. Association of nutrients with biomarkers of Alzheimer’s disease. In Genedis 2016; Springer: Cham, Switzerland, 2017; pp. 257–268. [Google Scholar]
- Volkert, D.; Chourdakis, M.; Faxen-Irving, G.; Frühwald, T.; Landi, F.; Suominen, M.H.; Vandewoude, M.; Wirth, R.; Schneider, S.M. Espen guidelines on nutrition in dementia. Clin. Nutr. 2015, 34, 1052–1073. [Google Scholar] [CrossRef]
- Huang, T.L.; Zandi, P.; Tucker, K.; Fitzpatrick, A.; Kuller, L.; Fried, L.; Burke, G.; Carlson, M. Benefits of fatty fish on dementia risk are stronger for those without apoe ε4. Neurology 2005, 65, 1409–1414. [Google Scholar] [CrossRef]
- Cao, L.; Tan, L.; Wang, H.-F.; Jiang, T.; Zhu, X.-C.; Lu, H.; Tan, M.-S.; Yu, J.-T. Dietary patterns and risk of dementia: A systematic review and meta-analysis of cohort studies. Mol. Neurobiol. 2016, 53, 6144–6154. [Google Scholar] [CrossRef]
- Yassine, H.N.; Braskie, M.N.; Mack, W.J.; Castor, K.J.; Fonteh, A.N.; Schneider, L.S.; Harrington, M.G.; Chui, H.C. Association of docosahexaenoic acid supplementation with Alzheimer disease stage in apolipoprotein e ε4 carriers: A review. JAMA Neurol. 2017, 74, 339–347. [Google Scholar] [CrossRef]
- Kosmidis, M.H.; Vlachos, G.S.; Anastasiou, C.A.; Yannakoulia, M.; Dardiotis, E.; Hadjigeorgiou, G.; Sakka, P.; Ntanasi, E.; Scarmeas, N. Dementia prevalence in greece. Alzheimer Dis. Assoc. Disord. 2018, 32, 232–239. [Google Scholar] [CrossRef]
- Cotogni, P.; Muzio, G.; Trombetta, A.; Ranieri, V.M.; Canuto, R.A. Impact of the ω-3 to ω-6 polyunsaturated fatty acid ratio on cytokine release in human alveolar cells. J. Parent. Enter. Nutr. 2011, 35, 114–121. [Google Scholar] [CrossRef]
- Lee, L.K.; Shahar, S.; Chin, A.-V.; Yusoff, N.A.M. Docosahexaenoic acid-concentrated fish oil supplementation in subjects with mild cognitive impairment (MCI): A 12-month randomised, double-blind, placebo-controlled trial. Psychopharmacology 2013, 225, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Athanasopoulos, D.; Karagiannis, G.; Tsolaki, M. Recent findings in Alzheimer disease and nutrition focusing on epigenetics. Adv. Nutr. 2016, 7, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Lillycrop, K.A. Fatty acids and epigenetics. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Mett, J.; Hartmann, T. The impact of vitamin E and other fat-soluble vitamins on Alzheimer’s disease. Int. J. Mol. Sci. 2016, 17, 1785. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Vitamin a exhibits potent antiamyloidogenic and fibril-destabilizing effects in vitro. Exp. Neurol. 2004, 189, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Presse, N.; Shatenstein, B.; Kergoat, M.-J.; Ferland, G. Low Vitamin K intakes in community-dwelling elders at an early stage of Alzheimer’s disease. J. Am. Diet. Assoc. 2008, 108, 2095–2099. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Burgos-Ramos, E. Selected micronutrients in cognitive decline prevention and therapy. Mol. Neurobiol. 2016, 53, 4083–4093. [Google Scholar] [CrossRef]
- Smith, A.D.; Refsum, H.; Bottiglieri, T.; Fenech, M.; Hooshmand, B.; McCaddon, A.; Miller, J.W.; Rosenberg, I.H.; Obeid, R. Homocysteine and dementia: An international consensus statement. J. Alzheimer’s Dis. 2018, 62, 561–570. [Google Scholar] [CrossRef]
- Fenech, M. Vitamins associated with brain aging, mild cognitive impairment, and Alzheimer disease: Biomarkers, epidemiological and experimental evidence, plausible mechanisms, and knowledge gaps. Adv. Nutr. 2017, 8, 958–970. [Google Scholar] [CrossRef]
- Takasaki, J.; Ono, K.; Yoshiike, Y.; Hirohata, M.; Ikeda, T.; Morinaga, A.; Takashima, A.; Yamada, M. Vitamin a has anti-oligomerization effects on amyloid-β in vitro. J. Alzheimer’s Dis. 2011, 27, 271–280. [Google Scholar] [CrossRef]
- Goodman, A.B.; Pardee, A.B. Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2901–2905. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, C.; Goncalves, M.; Clarke, E.; Dogruel, M.; Kalindjian, S.; Thomas, S.; Maden, M.; Corcoran, J. Retinoic acid receptor-α signalling antagonizes both intracellular and extracellular amyloid-β production and prevents neuronal cell death caused by amyloid-β. Eur. J. Neurosci. 2010, 32, 1246–1255. [Google Scholar] [CrossRef]
- Ding, Y.; Qiao, A.; Wang, Z.; Goodwin, J.S.; Lee, E.-S.; Block, M.L.; Allsbrook, M.; McDonald, M.P.; Fan, G.-H. Retinoic acid attenuates β-amyloid deposition and rescues memory deficits in an Alzheimer’s disease transgenic mouse model. J. Neurosci. 2008, 28, 11622–11634. [Google Scholar] [CrossRef] [PubMed]
- Koryakina, A.; Aeberhard, J.; Kiefer, S.; Hamburger, M.; Küenzi, P. Regulation of secretases by all-trans-retinoic acid. FEBS J. 2009, 276, 2645–2655. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Quitschke, W.W.; Brewer, G.J. Upregulation of amyloid precursor protein gene promoter in rat primary hippocampal neurons by phorbol ester, IL-1 and retinoic acid, but not by reactive oxygen species. Mol. Brain Res. 1998, 60, 40–49. [Google Scholar] [CrossRef]
- Prinzen, C.; Muller, U.; Endres, K.; Fahrenholz, F.; Postina, R. Genomic structure and functional characterization of the human adam10 promoter. FASEB J. 2005, 19, 1522–1524. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Vitamin c restores behavioral deficits and amyloid-β oligomerization without affecting plaque formation in a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 26, 7–18. [Google Scholar] [CrossRef]
- Kook, S.; Lee, K.; Kim, Y.; Cha, M.; Kang, S.; Baik, S.; Lee, H.; Park, R.; Mook-Jung, I. High-dose of vitamin c supplementation reduces amyloid plaque burden and ameliorates pathological changes in the brain of 5XFAD mice. Cell Death Dis. 2014, 5, e1083. [Google Scholar] [CrossRef]
- Gu, Y.; Schupf, N.; Cosentino, S.; Luchsinger, J.; Scarmeas, N. Nutrient intake and plasma β-amyloid. Neurology 2012, 78, 1832–1840. [Google Scholar] [CrossRef]
- Jiang, Q. Natural forms of Vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Viña, J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin e. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Rottkamp, C.A.; Boux, H.; Takeda, A.; Perry, G.; Smith, M.A. Activation of p38 kinase links τ phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2000, 59, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.R.; Cominetti, C.; Cozzolino, S.M.F. Importance and management of micronutrient deficiencies in patients with Alzheimer’s disease. Clin. Interv. Aging 2013, 8, 531. [Google Scholar] [CrossRef] [PubMed]
- Allison, A. The possible role of vitamin k deficiency in the pathogenesis of Alzheimer’s disease and in augmenting brain damage associated with cardiovascular disease. Med. Hypotheses 2001, 57, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Huy, P.D.Q.; Yu, Y.-C.; Ngo, S.T.; Van Thao, T.; Chen, C.-P.; Li, M.S.; Chen, Y.-C. In silico and in vitro characterization of anti-amyloidogenic activity of Vitamin K3 analogues for Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 2960–2969. [Google Scholar] [CrossRef]
- Banerjee, A.; Khemka, V.K.; Ganguly, A.; Roy, D.; Ganguly, U.; Chakrabarti, S. Vitamin D and Alzheimer’s disease: Neurocognition to therapeutics. Int. J. Alzheimer’s Dis. 2015, 2015, 192747. [Google Scholar] [CrossRef]
- Otaegui-Arrazola, A.; Amiano, P.; Elbusto, A.; Urdaneta, E.; Martínez-Lage, P. Diet, cognition, and Alzheimer’s disease: Food for thought. Eur. J. Nutr. 2014, 53, 1–23. [Google Scholar] [CrossRef]
- Troesch, B.; Weber, P.; Mohajeri, M.H. Potential links between impaired one-carbon metabolism due to polymorphisms, inadequate B-vitamin status, and the development of Alzheimer’s disease. Nutrients 2016, 8, 803. [Google Scholar] [CrossRef]
- Clarke, R.; Smith, A.D.; Jobst, K.A.; Refsum, H.; Sutton, L.; Ueland, P.M. Folate, Vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch. Neurol. 1998, 55, 1449–1455. [Google Scholar] [CrossRef]
- Douaud, G.; Refsum, H.; de Jager, C.A.; Jacoby, R.; Nichols, T.E.; Smith, S.M.; Smith, A.D. Preventing Alzheimer’s disease-related gray matter atrophy by b-vitamin treatment. Proc. Natl. Acad. Sci. 2013, 110, 9523–9528. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.-M.; Praticò, D. Acceleration of brain amyloidosis in an Alzheimer’s disease mouse model by a folate, Vitamin B6 and B12-deficient diet. Exp. Gerontol. 2010, 45, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Caruana, M.; Cauchi, R.; Vassallo, N. Putative role of red wine polyphenols against brain pathology in Alzheimer’s and parkinson’s disease. Front. Nutr. 2016, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Li, X.; Che, X.; Wei, C.; He, S.; Lu, J.; Jia, Z.; Pang, K.; Fan, L. SIRT1 is a regulator of autophagy: Implications in gastric cancer progression and treatment. FEBS Lett. 2015, 589, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Li, X.; Wei, C.; Che, X.; He, S.; Lu, J.; Wang, S.; Pang, K.; Fan, L. The prognostic role of SIRT1-autophagy axis in gastric cancer. Dis. Markers 2016, 2016, 6869415. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, B.; Rathinasamy, B.; Lohanathan, B.; Thiyagarajan, V.; Weng, C.-F. Neuroprotective role of phytochemicals. Molecules 2018, 23, 2485. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Tan, M.-S.; Yu, J.-T.; Tan, L. Resveratrol as a therapeutic agent for Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 350516. [Google Scholar] [CrossRef] [PubMed]
- Menard, C.; Bastianetto, S.; Quirion, R. Neuroprotective effects of resveratrol and epigallocatechin gallate polyphenols are mediated by the activation of protein kinase C gamma. Front. Cell. Neurosci. 2013, 7, 281. [Google Scholar] [CrossRef]
- Huang, T.-C.; Lu, K.-T.; Wo, Y.-Y.P.; Wu, Y.-J.; Yang, Y.-L. Resveratrol protects rats from aβ-induced neurotoxicity by the reduction of inos expression and lipid peroxidation. PLoS ONE 2011, 6, e29102. [Google Scholar] [CrossRef]
- Liu, T.F.; McCall, C.E. Deacetylation by sirt1 reprograms inflammation and cancer. Genes Cancer 2013, 4, 135–147. [Google Scholar] [CrossRef]
- Reed, S.; Quelle, D. P53 acetylation: Regulation and consequences. Cancers 2014, 7, 30–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Cao, N.; Li, Z.; Han, J.; Li, L. Resveratrol, an activator of SIRT1, induces protective autophagy in non-small-cell lung cancer via inhibiting Akt/mTOR and activating p38-MAPK. OncoTargets Ther. 2018, 11, 7777. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; Mithieux, G.; Vega, F.V. Resveratrol protects primary rat hepatocytes against oxidative stress damage::Activation of the NRF2 transcription factor and augmented activities of antioxidant enzymes. Eur. J. Pharmacol. 2008, 591, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Lange, K.W.; Li, S. Resveratrol, pterostilbene, and dementia. BioFactors 2018, 44, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Thapa, A.; Carroll, N.J. Dietary modulation of oxidative stress in Alzheimer’s disease. Int. J. Mol. Sci. 2017, 18, 1583. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef]
- Ramesh, B.N.; Rao, T.; Prakasam, A.; Sambamurti, K.; Rao, K. Neuronutrition and Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 19, 1123–1139. [Google Scholar] [CrossRef]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef]
- Davinelli, S.; Sapere, N.; Zella, D.; Bracale, R.; Intrieri, M.; Scapagnini, G. Pleiotropic protective effects of phytochemicals in Alzheimer’s disease. Oxid. Med. Cell. Longev. 2012, 2012, 386527. [Google Scholar] [CrossRef]
- Noguchi-Shinohara, M.; Yuki, S.; Dohmoto, C.; Ikeda, Y.; Samuraki, M.; Iwasa, K.; Yokogawa, M.; Asai, K.; Komai, K.; Nakamura, H.; et al. Consumption of green tea, but not black tea or coffee, is associated with reduced risk of cognitive decline. PLoS ONE 2014, 9, e96013. [Google Scholar] [CrossRef]
- Sawikr, Y.; Yarla, N.S.; Peluso, I.; Kamal, M.A.; Aliev, G.; Bishayee, A. Neuroinflammation in Alzheimer’s disease: The preventive and therapeutic potential of polyphenolic nutraceuticals. Adv. Protein Chem. Struct. Biol. 2017, 108, 33–57. [Google Scholar] [PubMed]
- Venkataraman, A.; Kalk, N.; Sewell, G.; Ritchie, C.W.; Lingford-Hughes, A. Alcohol and Alzheimer’s disease—Does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s disease? Alcohol Alcohol. 2017, 52, 151–158. [Google Scholar] [PubMed]
- Alfonso-Loeches, S.; Pascual-Lucas, M.; Blanco, A.M.; Sanchez-Vera, I.; Guerri, C. Pivotal role of tlr4 receptors in alcohol-induced neuroinflammation and brain damage. J. Neurosci. 2010, 30, 8285–8295. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Araiz, A.; Porcu, F.; Pérez-Hernández, M.; García-Gutiérrez, M.S.; Aracil-Fernández, M.A.; Gutierrez-López, M.D.; Guerri, C.; Manzanares, J.; O’shea, E.; Colado, M.I. Disruption of blood–brain barrier integrity in postmortem alcoholic brain: Preclinical evidence of TLR4 involvement from a binge-like drinking model. Addict. Biol. 2017, 22, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hazell, A.S. Microglial activation is a major contributor to neurologic dysfunction in thiamine deficiency. Biochem. Biophys. Res. Commun. 2010, 402, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-R.; Jeong, H.-Y.; Yang, S.; Choi, S.-P.; Seo, M.Y.; Yun, Y.-K.; Choi, Y.; Baik, S.-H.; Park, J.-S.; Gwon, A.-R.; et al. Effects of chronic alcohol consumption on expression levels of app and abeta-producing enzymes. BMB Rep. 2011, 44, 135–139. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Xu, H.; Shi, Q.; Chen, L.H.; Pedrini, S.; Pechman, D.; Baker, H.; Beal, M.F.; Gandy, S.E.; Gibson, G.E. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer’s mouse model. Neurobiol. Aging 2009, 30, 1587–1600. [Google Scholar] [CrossRef]
- Poli, A.; Marangoni, F.; Avogaro, A.; Barba, G.; Bellentani, S.; Bucci, M.; Cambieri, R.; Catapano, A.; Costanzo, S.; Cricelli, C. Moderate alcohol use and health: A consensus document. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 487–504. [Google Scholar] [CrossRef]
- Bate, C.; Williams, A. Ethanol protects cultured neurons against amyloid-β and α-synuclein-induced synapse damage. Neuropharmacology 2011, 61, 1406–1412. [Google Scholar] [CrossRef]
- Liu, R.; Barkhordarian, H.; Emadi, S.; Park, C.B.; Sierks, M.R. Trehalose differentially inhibits aggregation and neurotoxicity of β-amyloid 40 and 42. Neurobiol. Dis. 2005, 20, 74–81. [Google Scholar] [CrossRef]
- Du, J.; Liang, Y.; Xu, F.; Sun, B.; Wang, Z. Trehalose rescues a lzheimer’s disease phenotypes in APP/PS 1 transgenic mice. J. Pharm. Pharmacol. 2013, 65, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Holler, C.J.; Taylor, G.; McEachin, Z.T.; Deng, Q.; Watkins, W.J.; Hudson, K.; Easley, C.A.; Hu, W.T.; Hales, C.M.; Rossoll, W.; et al. Trehalose upregulates progranulin expression in human and mouse models of grn haploinsufficiency: A novel therapeutic lead to treat frontotemporal dementia. Mol. Neurodegener. 2016, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.S.; Min, S.-W.; Krabbe, G.; Wang, C.; Zhou, Y.; Asgarov, R.; Li, Y.; Martens, L.H.; Elia, L.P.; Ward, M.E.; et al. Progranulin protects against amyloid β deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 2014, 20, 1157. [Google Scholar] [CrossRef] [PubMed]
- Tien, N.T.; Karaca, I.; Tamboli, I.Y.; Walter, J. Trehalose alters subcellular trafficking and the metabolism of the Alzheimer-associated amyloid precursor protein. J. Biol. Chem. 2016. [Google Scholar] [CrossRef]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Krüger, U.; Wang, Y.; Kumar, S.; Mandelkow, E.-M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol. Aging 2012, 33, 2291–2305. [Google Scholar] [CrossRef]
- Minutoli, L.; Altavilla, D.; Bitto, A.; Polito, F.; Bellocco, E.; Laganà, G.; Giuliani, D.; Fiumara, T.; Magazù, S.; Ruggeri, P.; et al. The disaccharide trehalose inhibits proinflammatory phenotype activation in macrophages and prevents mortality in experimental septic shock. Shock 2007, 27, 91–96. [Google Scholar] [CrossRef]
- Echigo, R.; Shimohata, N.; Karatsu, K.; Yano, F.; Kayasuga-Kariya, Y.; Fujisawa, A.; Ohto, T.; Kita, Y.; Nakamura, M.; Suzuki, S.; et al. Trehalose treatment suppresses inflammation, oxidative stress, and vasospasm induced by experimental subarachnoid hemorrhage. J. Transl. Med. 2012, 10, 80. [Google Scholar] [CrossRef]
- Crowe, J.H. Trehalose as a “chemical chaperone”. In Molecular Aspects of the Stress Response: Chaperones, Membranes and Networks; Springer: New York, NY, USA, 2007; pp. 143–158. [Google Scholar]
- De Backer, G.; Ambrosioni, E.; Borch-Johnsen, K.; Brotons, C.; Cifkova, R.; Dallongeville, J.; Ebrahim, S.; Faergeman, O.; Graham, I.; Mancia, G. European guidelines on cardiovascular disease prevention in clinical practice: Third joint task force of european and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of eight societies and by invited experts). Eur. Heart J. 2003, 24, 1601–1610. [Google Scholar]
- Refolo, L.M.; Pappolla, M.A.; LaFrancois, J.; Malester, B.; Schmidt, S.D.; Thomas-Bryant, T.; Tint, G.S.; Wang, R.; Mercken, M.; Petanceska, S.S.; et al. A cholesterol-lowering drug reduces β-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2001, 8, 890–899. [Google Scholar] [CrossRef]
- Wolozin, B.; Kellman, W.; Ruosseau, P.; Celesia, G.G.; Siegel, G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme a reductase inhibitors. Arch. Neurol. 2000, 57, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Anstey, K.J.; Ashby-Mitchell, K.; Peters, R. Updating the evidence on the association between serum cholesterol and risk of late-life dementia: Review and meta-analysis. J. Alzheimer’s Dis. 2017, 56, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-L.; Wang, Y.-Y.; Liu, X.-G.; Kuo, S.-H.; Liu, N.; Song, Q.-Y.; Wang, M.-W. Cholesterol, 24-hydroxycholesterol, and 27-hydroxycholesterol as surrogate biomarkers in cerebrospinal fluid in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. J. Alzheimer’s Dis. 2016, 51, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Jensen, M.K. Hdl-cholesterol and apolipoproteins in relation to dementia. Curr. Opin. Lipidol. 2016, 27, 76. [Google Scholar] [CrossRef] [PubMed]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B 2010, 86, 484–493. [Google Scholar] [CrossRef]
- Tudorache, I.F.; Trusca, V.G.; Gafencu, A.V. Apolipoprotein e-a multifunctional protein with implications in various pathologies as a result of its structural features. Comput. Struct. Biotechnol. J. 2017, 15, 359–365. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.-C.; Qiao, W.; Bu, G. Apolipoprotein e, receptors, and modulation of Alzheimer’s disease. Biol. Psychiatry 2017, 83, 347–357. [Google Scholar] [CrossRef]
- Leoni, V.; Caccia, C. Potential diagnostic applications of side chain oxysterols analysis in plasma and cerebrospinal fluid. Biochem. Pharmacol. 2013, 86, 26–36. [Google Scholar] [CrossRef]
- Xu, Q.; Cao, S.; Rajapakse, S.; Matsubara, J.A. Understanding amd by analogy: Systematic review of lipid-related common pathogenic mechanisms in amd, ad, as and gn. Lipids Health Dis. 2018, 17, 3. [Google Scholar] [CrossRef]
- Barnard, N.D.; Bunner, A.E.; Agarwal, U. Saturated and trans fats and dementia: A systematic review. Neurobiol. Aging 2014, 35, S65–S73. [Google Scholar] [CrossRef] [PubMed]
- Alimentos. Available online: https://alimentos.org.es/ (accessed on 25 July 2018).
- BotanicalOnline. Available online: https://www.botanical-online.com/ (accessed on 25 July 2018).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nutrients | Food Products | |||
|---|---|---|---|---|
| DHA | Oily Fish Salmon | Nuts Walnuts, Almonds | egg yolk | |
| Vitamin A | Meat Chicken, turkey, beef | Vegetables Carrot, broccoli, sweet potato, pumpkin, spinach | Dairy Milk, cheese | Fruit Melon, apricot, papaya |
| Vitamins C and E | Swiss chard | Cranberries and currants | Black olives | |
| Vitamin B12 | Meat Pork, beef liver | Fish Salmon, sardines, mackerel, clams | Egg | |
| Vitamin B9 | Meat Chicken, beef | Vegetables and legumes Spinach, lentils, soy | Egg yolk | |
| Vitamin B6 | Meat Chicken, turkey, beef, pork | Vegetables and fruits Spinach, green peas, broccoli, asparagus, banana | Fish Salmon | Nuts Sunflower seeds, hazelnut, cashew nut |
| EGCG | Green tea | Blackberry | Apple | |
| Quercetin | Capers | Dark chocolate | Red onion | Clove |
| Resveratrol | Red wine | Berries Blackberry, raspberry, currant | Dark chocolate | Nuts Walnuts, peanuts |
| Oleuropein aglycone | Extra-virgin olive oil | |||
| Trehalose | Mushrooms | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Sanz, P.; Ruiz-Gabarre, D.; García-Escudero, V. Modulating Effect of Diet on Alzheimer’s Disease. Diseases 2019, 7, 12. https://doi.org/10.3390/diseases7010012
Fernández-Sanz P, Ruiz-Gabarre D, García-Escudero V. Modulating Effect of Diet on Alzheimer’s Disease. Diseases. 2019; 7(1):12. https://doi.org/10.3390/diseases7010012
Chicago/Turabian StyleFernández-Sanz, Paloma, Daniel Ruiz-Gabarre, and Vega García-Escudero. 2019. "Modulating Effect of Diet on Alzheimer’s Disease" Diseases 7, no. 1: 12. https://doi.org/10.3390/diseases7010012
APA StyleFernández-Sanz, P., Ruiz-Gabarre, D., & García-Escudero, V. (2019). Modulating Effect of Diet on Alzheimer’s Disease. Diseases, 7(1), 12. https://doi.org/10.3390/diseases7010012