Zebrafish Models of Rare Hereditary Pediatric Diseases

 ,
, 
Abstract
1. Introduction
2. The Zebrafish Genetic Toolkit
2.1. Transient Genetic Approaches
2.2. Stable Genetic Approaches
3. Modeling Disease with Homologs and Phenologs
4. Drug Discovery Using Zebrafish
5. Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rode, J. Rare Diseases: Understanding this Public Health Priority; EURORDIS: Paris, France, 2005. [Google Scholar]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [PubMed]
- Rath, A.; Olry, A.; Dhombres, F.; Brandt, M.M.; Urbero, B.; Ayme, S. Representation of rare diseases in health information systems: The Orphanet approach to serve a wide range of end users. Hum. Mutat. 2012, 33, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; FitzPatrick, D.R.; Firth, H.V. Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 2018, 10, e0123081. [Google Scholar]
- Strynatka, K.A.; Gurrola-Gal, M.C.; Berman, J.N.; McMaster, C.R. How Surrogate and Chemical Genetics in Model Organisms Can Suggest Therapies for Human Genetic Diseases. Genetics 2018, 208, 833–851. [Google Scholar] [CrossRef] [PubMed]
- Wangler, M.F.; Yamamoto, S.; Chao, H.-T.; Posey, J.E.; Westerfield, M.; Postlethwait, J.H.; Members of the Undiagnosed Diseases Network (UDN); Hieter, P.; Boycott, K.M.; Campeau, P.M.; et al. Model Organisms Facilitate Rare Disease Diagnosis and Therapeutic Research. Genetics 2017, 207, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Kinth, P.; Mahesh, G.; Panwar, Y. Mapping of zebrafish research: A global outlook. Zebrafish 2013, 10, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, D.J.; Eisen, J.S. Headwaters of the zebrafish—Emergence of a new model vertebrate. Nat. Rev. Genet. 2002, 3, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Varga, M. The Doctor of Delayed Publications: The Remarkable Life of George Streisinger (1927–1984). Zebrafish 2018. [Google Scholar] [CrossRef] [PubMed]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Driever, W.; Solnica-Krezel, L.; Schier, A.F.; Neuhauss, S.C.; Malicki, J.; Stemple, D.L.; Stainier, D.Y.; Zwartkruis, F.; Abdelilah, S.; Rangini, Z.; et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development 1996, 123, 37–46. [Google Scholar] [PubMed]
- Haffter, P.; Granato, M.; Brand, M.; Mullins, M.C. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development 1996, 123, 1–36. [Google Scholar] [PubMed]
- Patton, E.E.; Zon, L.I. The art and design of genetic screens: Zebrafish. Nat. Rev. Genet. 2001, 2, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Postlethwait, J.H.; Yan, Y.-L.; Gates, M.A.; Horne, S.; Amores, A.; Brownlie, A.; Donovan, A.; Egan, E.S.; Force, A.; Gong, Z.; et al. Vertebrate genome evolution and the zebrafish gene map. Nat. Genet. 1998, 18, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Knapik, E.W.; Goodman, A.; Ekker, M.; Chevrette, M.; Delgado, J.; Neuhauss, S.; Shimoda, N.; Driever, W.; Fishman, M.C.; Jacob, H.J. A microsatellite genetic linkage map for zebrafish (Danio rerio). Nat. Genet. 1998, 18, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Gates, M.A.; Kim, L.; Egan, E.S.; Cardozo, T.; Sirotkin, H.I.; Dougan, S.T.; Lashkari, D.; Abagyan, R.; Schier, A.F.; Talbot, W.S. A genetic linkage map for zebrafish: Comparative analysis and localization of genes and expressed sequences. Genome Res. 1999, 9, 334–347. [Google Scholar] [PubMed]
- Shimoda, N.; Knapik, E.W.; Ziniti, J.; Sim, C.; Yamada, E.; Kaplan, S.; Jackson, D.; de Sauvage, F.; Jacob, H.; Fishman, M.C. Zebrafish genetic map with 2000 microsatellite markers. Genomics 1999, 58, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Talbot, W.S.; Schier, A.F. Chapter 15 Positional Cloning of Mutated Zebrafish Genes. In The Zebrafish: Genetics and Genomics; Methods in Cell Biology; Elsevier: New York, NY, USA, 1998; Volume 60, pp. 259–286. [Google Scholar]
- Kettleborough, R.N.W.; Busch-Nentwich, E.M.; Harvey, S.A.; Dooley, C.M.; de Bruijn, E.; van Eeden, F.; Sealy, I.; White, R.J.; Herd, C.; Nijman, I.J.; et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature 2013, 496, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Minevich, G.; Park, D.S.; Blankenberg, D.; Poole, R.J.; Hobert, O. CloudMap: A cloud-based pipeline for analysis of mutant genome sequences. Genetics 2012, 192, 1249–1269. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, K. Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat. Rev. Genet. 2014, 15, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; Zon, L.I. Of fish and men: Using zebrafish to fight human diseases. Trends Cell Biol. 2013, 23, 584–586. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.B.; Westerfield, M. Zebrafish models in translational research: Tipping the scales toward advancements in human health. Dis. Models Mech. 2014, 7, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Baxendale, S.; van Eeden, F.; Wilkinson, R. The Power of Zebrafish in Personalised Medicine. Adv. Exp. Med. Biol. 2017, 1007, 179–197. [Google Scholar] [PubMed]
- Jia, J.; An, Z.; Ming, Y.; Guo, Y.; Li, W.; Li, X.; Liang, Y.; Guo, D.; Tai, J.; Chen, G.; et al. PedAM: A database for Pediatric Disease Annotation and Medicine. Nucleic Acids Res. 2018, 46, D977–D983. [Google Scholar] [CrossRef] [PubMed]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene “knockdown” in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Kleinjan, D.A.; Bancewicz, R.M.; Gautier, P.; Dahm, R.; Schonthaler, H.B.; Damante, G.; Seawright, A.; Hever, A.M.; Yeyati, P.L.; van Heyningen, V.; et al. Subfunctionalization of duplicated zebrafish pax6 genes by cis-regulatory divergence. PLoS Genet. 2008, 4, e29. [Google Scholar] [CrossRef] [PubMed]
- Force, A.; Lynch, M.; Pickett, F.B.; Amores, A.; Yan, Y.L.; Postlethwait, J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 1999, 151, 1531–1545. [Google Scholar] [PubMed]
- Lambert, M.J.; Cochran, W.O.; Wilde, B.M.; Olsen, K.G.; Cooper, C.D. Evidence for widespread subfunctionalization of splice forms in vertebrate genomes. Genome Res. 2015, 25, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.E.; Larson, J.D.; Nasevicius, A.; Beiraghi, S.; Brenner, C.; Farber, S.A.; Ekker, S.C. p53 activation by knockdown technologies. PLoS Genet. 2007, 3, e78. [Google Scholar] [CrossRef] [PubMed]
- Gentsch, G.E.; Spruce, T.; Monteiro, R.S.; Owens, N.D.L.; Martin, S.R.; Smith, J.C. Innate Immune Response and Off-Target Mis-splicing Are Common Morpholino-Induced Side Effects in Xenopus. Dev. Cell 2018, 44, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Kok, F.O.; Shin, M.; Ni, C.-W.; Gupta, A.; Grosse, A.S.; van Impel, A.; Kirchmaier, B.C.; Peterson-Maduro, J.; Kourkoulis, G.; Male, I.; et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 2015, 32, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Merker, S.; Stainier, D.Y.R. Out with the old, in with the new: Reassessing morpholino knockdowns in light of genome editing technology. Development 2014, 141, 3103–3104. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.R.; Raz, E.; Lawson, N.D.; Ekker, S.C.; Burdine, R.D.; Eisen, J.S.; Ingham, P.W.; Schulte-Merker, S.; Yelon, D.; Weinstein, B.M.; et al. Guidelines for morpholino use in zebrafish. PLoS Genet. 2017, 13, e1007000. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Hölper, S.; Krüger, M.; Stainier, D.Y.R. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Mulligan, T.S.; Shen, M.-C.; Wang, H.; Scahill, C.M.; Tan, F.J.; Du, S.J.; Busch-Nentwich, E.M.; Farber, S.A. mRNA processing in mutant zebrafish lines generated by chemical and CRISPR-mediated mutagenesis produces unexpected transcripts that escape nonsense-mediated decay. PLoS Genet. 2017, 13, e1007105. [Google Scholar] [CrossRef] [PubMed]
- Balciunas, D. Fish mutant, where is thy phenotype? PLoS Genet. 2018, 14, e1007197. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, S.; Varga, M.; Weinberg, E.S. FGF signaling is required for {beta}-catenin-mediated induction of the zebrafish organizer. Development 2006, 133, 3265–3276. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Wilkes, M.; Bibikova, E.; Youn, M.-Y.; Sakamoto, K.M.; Lin, S. Innate immune system activation in zebrafish and cellular models of Diamond Blackfan Anemia. Sci. Rep. 2018, 8, 5165. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Sakamoto, K.M.; Lin, S. Ribosomal protein L11 mutation in zebrafish leads to haematopoietic and metabolic defects. Br. J. Haematol. 2011, 152, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Heijnen, H.F.; van Wijk, R.; Pereboom, T.C.; Goos, Y.J.; Seinen, C.W.; van Oirschot, B.A.; van Dooren, R.; Gastou, M.; Giles, R.H.; van Solinge, W.; et al. Ribosomal Protein Mutations Induce Autophagy through S6 Kinase Inhibition of the Insulin Pathway. PLoS Genet. 2014, 10, e1004371. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rivera, E.; Liu, Y.P.; Verbitsky, M.; Anderson, B.R.; Capone, V.P.; Otto, E.A.; Yan, Z.; Mitrotti, A.; Martino, J.; Steers, N.J.; et al. Genetic Drivers of Kidney Defects in the DiGeorge Syndrome. N. Engl. J. Med. 2017, 376, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Pannicke, U.; Hönig, M.; Hess, I.; Friesen, C.; Holzmann, K.; Rump, E.-M.; Barth, T.F.; Rojewski, M.T.; Schulz, A.; Boehm, T.; et al. Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat. Genet. 2009, 41, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Murray, J.P.; Prykhozhij, S.V.; Dufay, J.N.; Steele, S.L.; Gaston, D.; Nasrallah, G.K.; Coombs, A.J.; Liwski, R.S.; Fernandez, C.V.; Berman, J.N.; et al. Glycine and Folate Ameliorate Models of Congenital Sideroblastic Anemia. PLoS Genet. 2016, 12, e1005783. [Google Scholar] [CrossRef] [PubMed]
- Brownlie, A.; Donovan, A.; Pratt, S.J.; Paw, B.H.; Oates, A.C.; Brugnara, C.; Witkowska, H.E.; Sassa, S.; Zon, L.I. Positional cloning of the zebrafish sauternes gene: A model for congenital sideroblastic anaemia. Nat. Genet. 1998, 20, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, F.; Fu, S.; Cooke, J.; Wilson, S.W.; Cooper, J.D.; Russell, C. A zebrafish model of CLN2 disease is deficient in tripeptidyl peptidase 1 and displays progressive neurodegeneration accompanied by a reduction in proliferation. Brain 2013, 136, 1488–1507. [Google Scholar] [CrossRef] [PubMed]
- Wager, K.; Zdebik, A.A.; Fu, S.; Cooper, J.D.; Harvey, R.J.; Russell, C. Neurodegeneration and Epilepsy in a Zebrafish Model of CLN3 Disease (Batten Disease). PLoS ONE 2016, 11, e0157365. [Google Scholar] [CrossRef] [PubMed]
- Madsen, E.C.; Morcos, P.A.; Mendelsohn, B.A.; Gitlin, J.D. In vivo correction of a Menkes disease model using antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 2008, 105, 3909–3914. [Google Scholar] [CrossRef] [PubMed]
- Elmonem, M.A.; Khalil, R.; Khodaparast, L.; Khodaparast, L.; Arcolino, F.O.; Morgan, J.; Pastore, A.; Tylzanowski, P.; Ny, A.; Lowe, M.; et al. Cystinosis (ctns) zebrafish mutant shows pronephric glomerular and tubular dysfunction. Sci. Rep. 2017, 7, 42583. [Google Scholar] [CrossRef] [PubMed]
- Strachan, L.R.; Stevenson, T.J.; Freshner, B.; Keefe, M.D.; Miranda Bowles, D.; Bonkowsky, J.L. A zebrafish model of X-linked adrenoleukodystrophy recapitulates key disease features and demonstrates a developmental requirement for abcd1 in oligodendrocyte patterning and myelination. Hum. Mol. Genet. 2017, 26, 3600–3614. [Google Scholar] [CrossRef] [PubMed]
- Gonzaga-Jauregui, C.; Harel, T.; Gambin, T.; Kousi, M.; Griffin, L.B.; Francescatto, L.; Ozes, B.; Karaca, E.; Jhangiani, S.N.; Bainbridge, M.N.; et al. Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep. 2015, 12, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Tuschl, K.; Meyer, E.; Valdivia, L.E.; Zhao, N.; Dadswell, C.; Abdul-Sada, A.; Hung, C.Y.; Simpson, M.A.; Chong, W.K.; Jacques, T.S.; et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat. Commun. 2016, 7, 11601. [Google Scholar] [CrossRef] [PubMed]
- Baraban, S.C.; Dinday, M.T.; Hortopan, G.A. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 2013, 4, 2410. [Google Scholar] [CrossRef] [PubMed]
- Bassett, D.I.; Bryson-Richardson, R.J.; Daggett, D.F.; Gautier, P.; Keenan, D.G.; Currie, P.D. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 2003, 130, 5851–5860. [Google Scholar] [CrossRef] [PubMed]
- Schubert, J.; Siekierska, A.; Langlois, M.; May, P.; Huneau, C.; Becker, F.; Muhle, H.; Suls, A.; Lemke, J.R.; de Kovel, C.G.F.; et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat. Genet. 2014, 46, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- See, K.; Yadav, P.; Giegerich, M.; Cheong, P.S.; Graf, M.; Vyas, H.; Lee, S.G.P.; Mathavan, S.; Fischer, U.; Sendtner, M.; et al. SMN deficiency alters Nrxn2 expression and splicing in zebrafish and mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2014, 23, 1754–1770. [Google Scholar] [CrossRef] [PubMed]
- Boon, K.-L.; Xiao, S.; McWhorter, M.L.; Donn, T.; Wolf-Saxon, E.; Bohnsack, M.T.; Moens, C.B.; Beattie, C.E. Zebrafish survival motor neuron mutants exhibit presynaptic neuromuscular junction defects. Hum. Mol. Genet. 2009, 18, 3615–3625. [Google Scholar] [CrossRef] [PubMed]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Züchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Ruparelia, A.A.; Oorschot, V.; Vaz, R.; Ramm, G.; Bryson-Richardson, R.J. Zebrafish models of BAG3 myofibrillar myopathy suggest a toxic gain of function leading to BAG3 insufficiency. Acta Neuropathol. 2014, 128, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, K.V.; Hennessey, J.A.; Barnett, A.S.; Yin, X.; Stadt, H.A.; Foster, E.; Shah, R.A.; Yazawa, M.; Dolmetsch, R.E.; Kirby, M.L.; et al. Calcium influx through L-type CaV1.2 Ca2+ channels regulates mandibular development. J. Clin. Investig. 2013, 123, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Little, S.C.; Xu, M.; Haupt, J.; Ast, C.; Katagiri, T.; Mundlos, S.; Seemann, P.; Kaplan, F.S.; Mullins, M.C.; et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J. Clin. Investig. 2009, 119, 3462–3472. [Google Scholar] [CrossRef] [PubMed]
- Mucha, B.E.; Hashiguchi, M.; Zinski, J.; Shore, E.M.; Mullins, M.C. Variant BMP receptor mutations causing fibrodysplasia ossificans progressiva (FOP) in humans show BMP ligand-independent receptor activation in zebrafish. Bone 2018, 109, 225–231. [Google Scholar] [CrossRef] [PubMed]
- LaBonty, M.; Pray, N.; Yelick, P.C. A Zebrafish Model of Human Fibrodysplasia Ossificans Progressiva. Zebrafish 2017, 14, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, D.; Ombrello, A.K.; Zavialov, A.V.; Toro, C.; Zavialov, A.V.; Stone, D.L.; Chae, J.J.; Rosenzweig, S.D.; Bishop, K.; et al. Early-onset stroke and vasculopathy associated with mutations in ADAN2. Engl. J. Med. 2014, 370, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Mangos, S.; Lam, P.-Y.; Zhao, A.; Liu, Y.; Mudumana, S.; Vasilyev, A.; Liu, A.; Drummond, I.A. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis. Model Mech. 2010, 3, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Bisgrove, B.W.; Snarr, B.S.; Emrazian, A.; Yost, H.J. Polaris and Polycystin-2 in dorsal forerunner cells and Kupffer's vesicle are required for specification of the zebrafish left-right axis. Dev. Biol. 2005, 287, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Schottenfeld, J.; Sullivan-Brown, J.; Burdine, R.D. Zebrafish curly up encodes a Pkd2 ortholog that restricts left-side-specific expression of southpaw. Development 2007, 134, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Golzio, C.; Willer, J.; Talkowski, M.E.; Oh, E.C.; Taniguchi, Y.; Jacquemont, S.; Reymond, A.; Sun, M.; Sawa, A.; Gusella, J.F.; et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 2012, 485, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Rooryck, C.; Diaz-Font, A.; Osborn, D.P.S.; Chabchoub, E.; Hernandez-Hernandez, V.; Shamseldin, H.; Kenny, J.; Waters, A.; Jenkins, D.; Kaissi, A.A.; et al. Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat. Genet. 2011, 43, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Galeano, M.C.R.; Ott, E.; Kaeslin, G.; Kausalya, P.J.; Kramer, C.; Ortiz-Brüchle, N.; Hilger, N.; Metzis, V.; Hiersche, M.; et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 2017, 49, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Hendee, K.E.; Sorokina, E.A.; Muheisen, S.S.; Reis, L.M.; Tyler, R.C.; Markovic, V.; Cuturilo, G.; Link, B.A.; Semina, E.V. PITX2 deficiency and associated human disease: Insights from the zebrafish model. Hum. Mol. Genet. 2018, 27, 1675–1695. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.J.; May-Simera, H.; Eichers, E.R.; Kai, M.; Hill, J.; Jagger, D.J.; Leitch, C.C.; Chapple, J.P.; Munro, P.M.; Fisher, S.; et al. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 2005, 37, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.-J.; Tayeh, M.K.; Mullins, R.F.; Stone, E.M.; Sheffield, V.C.; Slusarski, D.C. Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer’s vesicle cilia function. Hum. Mol. Genet. 2006, 15, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Badano, J.L.; Leitch, C.C.; Ansley, S.J.; May-Simera, H.; Lawson, S.; Lewis, R.A.; Beales, P.L.; Dietz, H.C.; Fisher, S.; Katsanis, N. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature 2006, 439, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Stoetzel, C.; Laurier, V.; Davis, E.E.; Muller, J.; Rix, S.; Badano, J.L.; Leitch, C.C.; Salem, N.; Chouery, E.; Corbani, S.; et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 2006, 38, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Chiang, A.P.; Beck, J.S.; Yen, H.-J.; Tayeh, M.K.; Scheetz, T.E.; Swiderski, R.E.; Nishimura, D.Y.; Braun, T.A.; Kim, K.-Y.A.; Huang, J.; et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc. Natl. Acad. Sci. USA 2006, 103, 6287–6292. [Google Scholar] [CrossRef] [PubMed]
- Stoetzel, C.; Muller, J.; Laurier, V.; Davis, E.E.; Zaghloul, N.A.; Vicaire, S.; Jacquelin, C.; Plewniak, F.; Leitch, C.C.; Sarda, P.; et al. Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am. J. Hum. Genet. 2007, 80, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jindal, G.A.; Goyal, Y.; Yamaya, K.; Futran, A.S.; Kountouridis, I.; Balgobin, C.A.; Schüpbach, T.; Burdine, R.D.; Shvartsman, S.Y. In vivo severity ranking of Ras pathway mutations associated with developmental disorders. Proc. Natl. Acad. Sci. USA 2017, 114, 510–515. [Google Scholar] [CrossRef] [PubMed]
- French, C.R.; Stach, T.R.; March, L.D.; Lehmann, O.J.; Waskiewicz, A.J. Apoptotic and proliferative defects characterize ocular development in a microphthalmic BMP model. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4636–4647. [Google Scholar] [CrossRef] [PubMed]
- Deml, B.; Kariminejad, A.; Borujerdi, R.H.R.; Muheisen, S.; Reis, L.M.; Semina, E.V. Mutations in MAB21L2 result in ocular Coloboma, microcornea and cataracts. PLoS Genet. 2015, 11, e1005002. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Willer, J.R.; Willer, G.B.; Smith, K.; Gregg, R.G.; Gross, J.M. Zebrafish blowout provides genetic evidence for Patched1-mediated negative regulation of Hedgehog signaling within the proximal optic vesicle of the vertebrate eye. Dev. Biol. 2008, 319, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Miesfeld, J.B.; Gestri, G.; Clark, B.S.; Flinn, M.A.; Poole, R.J.; Bader, J.R.; Besharse, J.C.; Wilson, S.W.; Link, B.A. Yap and Taz regulate retinal pigment epithelial cell fate. Development 2015, 142, 3021–3032. [Google Scholar] [CrossRef] [PubMed]
- Sanna-Cherchi, S.; Sampogna, R.V.; Papeta, N.; Burgess, K.E.; Nees, S.N.; Perry, B.J.; Choi, M.; Bodria, M.; Liu, Y.; Weng, P.L.; et al. Mutations in DSTYK and dominant urinary tract malformations. N. Engl. J. Med. 2013, 369, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Patten, S.A.; Jacobs-McDaniels, N.L.; Zaouter, C.; Drapeau, P.; Albertson, R.C.; Moldovan, F. Role of Chd7 in zebrafish: A model for CHARGE syndrome. PLoS ONE 2012, 7, e31650. [Google Scholar] [CrossRef] [PubMed]
- Prykhozhij, S.V.; Steele, S.L.; Razaghi, B.; Berman, J.N. A rapid and effective method for screening, sequencing and reporter verification of engineered frameshift mutations in zebrafish. Dis. Models Mech. 2017, 10, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Nam, T.-S.; Li, W.; Kim, J.H.; Yoon, W.; Choi, Y.-D.; Kim, K.-H.; Cai, H.; Kim, M.J.; Kim, C.; et al. Functional validation of novel MKS3/TMEM67 mutations in COACH syndrome. Sci. Rep. 2017, 7, 10222. [Google Scholar] [CrossRef] [PubMed]
- Leitch, C.C.; Zaghloul, N.A.; Davis, E.E.; Stoetzel, C.; Diaz-Font, A.; Rix, S.; Alfadhel, M.; Al-Fadhel, M.; Lewis, R.A.; Eyaid, W.; et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008, 40, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Edie, S.; Zaghloul, N.A.; Leitch, C.C.; Klinedinst, D.K.; Lebron, J.; Thole, J.F.; McCallion, A.S.; Katsanis, N.; Reeves, R.H. Survey of human chromosome 21 gene expression effects on early development in Danio rerio. bioRxiv 2018. [Google Scholar] [CrossRef] [PubMed]
- Pereboom, T.C.; van Weele, L.J.; Bondt, A.; MacInnes, A.W. A zebrafish model of dyskeratosis congenita reveals hematopoietic stem cell formation failure resulting from ribosomal protein-mediated p53 stabilization. Blood 2011, 118, 5458–5465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Morimoto, K.; Danilova, N.; Zhang, B.; Lin, S. Zebrafish models for dyskeratosis congenita reveal critical roles of p53 activation contributing to hematopoietic defects through RNA processing. PLoS ONE 2012, 7, e30188. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Rao, J.; Mollet, G.; Schapiro, D.; Daugeron, M.-C.; Tan, W.; Gribouval, O.; Boyer, O.; Revy, P.; Jobst-Schwan, T.; et al. Mutations in KEOPS-complex genes cause nephrotic syndrome with primary microcephaly. Nat. Genet. 2017, 49, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Mackay, E.W.; Apschner, A.; Schulte-Merker, S. Vitamin K reduces hypermineralisation in zebrafish models of PXE and GACI. Development 2015, 142, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Apschner, A.; Huitema, L.F.A.; Ponsioen, B.; Peterson-Maduro, J.; Schulte-Merker, S. Zebrafish enpp1 mutants exhibit pathological mineralization, mimicking features of generalized arterial calcification of infancy (GACI) and pseudoxanthoma elasticum (PXE). Dis. Models Mech. 2014, 7, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sadowski, S.; Frank, M.; Chai, C.; Váradi, A.; Ho, S.-Y.; Lou, H.; Dean, M.; Thisse, C.; Thisse, B.; et al. The abcc6a gene expression is required for normal zebrafish development. J. Investig. Dermatol. 2010, 130, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Hoff, S.; Halbritter, J.; Epting, D.; Frank, V.; Nguyen, T.-M.T.; van Reeuwijk, J.; Boehlke, C.; Schell, C.; Yasunaga, T.; Helmstädter, M.; et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat. Genet. 2013, 45, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Jin, M.; Hu, R.; Wang, H.; Zhang, F.; Yuan, S.; Cao, Y. The Joubert Syndrome Protein Inpp5e Controls Ciliogenesis by Regulating Phosphoinositides at the Apical Membrane. J. Am. Soc. Nephrol. 2017, 28, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Logan, C.V.; Mougou-Zerelli, S.; Lee, J.H.; Silhavy, J.L.; Brancati, F.; Iannicelli, M.; Travaglini, L.; Romani, S.; Illi, B.; et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010, 42, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Simms, R.J.; Hynes, A.M.; Eley, L.; Inglis, D.; Chaudhry, B.; Dawe, H.R.; Sayer, J.A. Modelling a ciliopathy: Ahi1 knockdown in model systems reveals an essential role in brain, retinal, and renal development. Cell. Mol. Life Sci. 2012, 69, 993–1009. [Google Scholar] [CrossRef] [PubMed]
- Cantagrel, V.; Silhavy, J.L.; Bielas, S.L.; Swistun, D.; Marsh, S.E.; Bertrand, J.Y.; Audollent, S.; Attié-Bitach, T.; Holden, K.R.; Dobyns, W.B.; et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet. 2008, 83, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Bachmann-Gagescu, R.; Phelps, I.G.; Stearns, G.; Link, B.A.; Brockerhoff, S.E.; Moens, C.B.; Doherty, D. The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking. Hum. Mol. Genet. 2011, 20, 4041–4055. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, M.I.; Romio, L.; Castro, S.; Collins, J.E.; Goulding, D.A.; Stemple, D.L.; Woolf, A.S.; Wilson, S.W. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum. Mol. Genet. 2009, 18, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Van De Weghe, J.C.; Rusterholz, T.D.S.; Latour, B.; Grout, M.E.; Aldinger, K.A.; Shaheen, R.; Dempsey, J.C.; Maddirevula, S.; Cheng, Y.-H.H.; Phelps, I.G.; et al. Mutations in ARMC9, which Encodes a Basal Body Protein, Cause Joubert Syndrome in Humans and Ciliopathy Phenotypes in Zebrafish. Am. J. Hum. Genet. 2017, 101, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Khanna, H.; Davis, E.E.; Murga-Zamalloa, C.A.; Estrada-Cuzcano, A.; Lopez, I.; den Hollander, A.I.; Zonneveld, M.N.; Othman, M.I.; Waseem, N.; Chakarova, C.F.; et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat. Genet. 2009, 41, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Frosk, P.; Arts, H.H.; Philippe, J.; Gunn, C.S.; Brown, E.L.; Chodirker, B.; Simard, L.; Majewski, J.; Fahiminiya, S.; Russell, C.; et al. A truncating mutation in CEP55 is the likely cause of MARCH, a novel syndrome affecting neuronal mitosis. J. Med. Genet. 2017, 54, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Yourshaw, M.; Mamsa, H.; Rudnik-Schöneborn, S.; Menezes, M.P.; Hong, J.E.; Leong, D.W.; Senderek, J.; Salman, M.S.; Chitayat, D.; et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat. Genet. 2012, 44, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Hjeij, R.; Lindstrand, A.; Francis, R.; Zariwala, M.A.; Liu, X.; Li, Y.; Damerla, R.; Dougherty, G.W.; Abouhamed, M.; Olbrich, H.; et al. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet. 2013, 93, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Becker-Heck, A.; Zohn, I.E.; Okabe, N.; Pollock, A.; Lenhart, K.B.; Sullivan-Brown, J.; McSheene, J.; Loges, N.T.; Olbrich, H.; Haeffner, K.; et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011, 43, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Zariwala, M.A.; Gee, H.Y.; Kurkowiak, M.; Al-Mutairi, D.A.; Leigh, M.W.; Hurd, T.W.; Hjeij, R.; Dell, S.D.; Chaki, M.; Dougherty, G.W.; et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am. J. Hum. Genet. 2013, 93, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Person, A.D.; Beiraghi, S.; Sieben, C.M.; Hermanson, S.; Neumann, A.N.; Robu, M.E.; Schleiffarth, J.R.; Billington, C.J.; van Bokhoven, H.; Hoogeboom, J.M.; et al. WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev. Dyn. 2010, 239, 327–337. [Google Scholar] [PubMed]
- Otto, E.A.; Hurd, T.W.; Airik, R.; Chaki, M.; Zhou, W.; Stoetzel, C.; Patil, S.B.; Levy, S.; Ghosh, A.K.; Murga-Zamalloa, C.A.; et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 2010, 42, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Van Karnebeek, C.D.M.; Bonafé, L.; Wen, X.-Y.; Tarailo-Graovac, M.; Balzano, S.; Royer-Bertrand, B.; Ashikov, A.; Garavelli, L.; Mammi, I.; Turolla, L.; et al. NANS-mediated synthesis of sialic acid is required for brain and skeletal development. Nat. Genet. 2016, 48, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Cade, L.; Khayter, C.; Reyon, D.; Peterson, R.T.; Joung, J.K.; Yeh, J.-R.J. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat. Biotechnol. 2011, 29, 697–698. [Google Scholar] [CrossRef] [PubMed]
- Bedell, V.M.; Wang, Y.; Campbell, J.M.; Poshusta, T.L.; Starker, C.G.; Krug, R.G.; Tan, W.; Penheiter, S.G.; Ma, A.C.; Leung, A.Y.H.; et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012, 491, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Reyon, D.; Tsai, S.Q.; Khayter, C.; Foden, J.A.; Sander, J.D.; Joung, J.K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012, 30, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Jao, L.-E.; Wente, S.R.; Chen, W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. USA 2013, 110, 13904–13909. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Ahkmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, e98186. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Kaini, P.; Sander, J.D.; Joung, J.K.; Peterson, R.T.; Yeh, J.-R.J. Heritable and Precise Zebrafish Genome Editing Using a CRISPR-Cas System. PLoS ONE 2013, 8, e68708. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.C.; Amacher, S.L. A Streamlined CRISPR Pipeline to Reliably Generate Zebrafish Frameshifting Alleles. Zebrafish 2014, 11, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Hruscha, A.; Krawitz, P.; Rechenberg, A.; Heinrich, V.; Hecht, J.; Haass, C.; Schmid, B. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 2013, 140, 4982–4987. [Google Scholar] [CrossRef] [PubMed]
- Varshney, G.K.; Sood, R.; Burgess, S.M. Understanding and Editing the Zebrafish Genome. Adv. Genet. 2015, 92, 1–52. [Google Scholar] [PubMed]
- Irion, U.; Krauss, J.; Nüsslein-Volhard, C. Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development 2014, 141, 4827–4830. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Sakuma, T.; Nakade, S.; Ohga, R.; Ota, S.; Okamoto, H.; Yamamoto, T.; Kawahara, A. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Sci. Rep. 2015, 5, 8841. [Google Scholar] [CrossRef] [PubMed]
- Hoshijima, K.; Jurynec, M.J.; Grunwald, D.J. Precise Editing of the Zebrafish Genome Made Simple and Efficient. Dev. Cell 2016, 36, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Lee, J.; Kim, J.-S. Measuring and Reducing Off-Target Activities of Programmable Nucleases Including CRISPR-Cas9. Mol. Cells 2015, 38, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Kulcsár, P.I.; Tálas, A.; Huszár, K.; Ligeti, Z.; Tóth, E.; Weinhardt, N.; Fodor, E.; Welker, E. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biol. 2017, 18, 190. [Google Scholar] [CrossRef] [PubMed]
- Varshney, G.K.; Zhang, S.; Pei, W.; Adomako-Ankomah, A.; Fohtung, J.; Schaffer, K.; Carrington, B.; Maskeri, A.; Slevin, C.; Wolfsberg, T.; et al. CRISPRz: A database of zebrafish validated sgRNAs. Nucleic Acids Res. 2016, 44, D822–D826. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.T.; Shaw, S.Y.; Peterson, T.A.; Milan, D.J.; Zhong, T.P.; Schreiber, S.L.; Macrae, C.A.; Fishman, M.C. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nat. Biotechnol. 2004, 22, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Kwan, K.M.; Fujimoto, E.; Grabher, C.; Mangum, B.D.; Hardy, M.E.; Campbell, D.S.; Parant, J.M.; Yost, H.J.; Kanki, J.P.; Chien, C.-B. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 2007, 236, 3088–3099. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K. Tol2: A versatile gene transfer vector in vertebrates. Genome Biol. 2007, 8, S7. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, H.; Kawakami, K. Chapter 5—Transient and Stable Transgenesis Using Tol2 Transposon Vectors. In Zebrafish; Humana Press: New York, NY, USA, 2009. [Google Scholar]
- Urasaki, A.; Kawakami, K. Chapter 6—Analysis of Genes and Genome by the Tol2-Mediated Gene and Enhancer Trap Methods. In Zebrafish; Humana Press: New York, NY, USA, 2009. [Google Scholar]
- Kawakami, K.; Abe, G.; Asada, T.; Asakawa, K.; Fukuda, R.; Ito, A.; Lal, P.; Mouri, N.; Muto, A.; Suster, M.L.; et al. zTrap: Zebrafish gene trap and enhancer trap database. BMC Dev. Biol. 2010, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Jungke, P.; Hans, S.; Brand, M. The Zebrafish CreZoo: An Easy-to-Handle Database for Novel CreER T2-Driver Lines. Zebrafish 2013, 10, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; Durand, E.M.; Yang, S.; Zhou, Y.; Zon, L.I. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev. Cell 2015, 32, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, V.; De Santis, F.; Auer, T.O.; Testa, N.; Sánchez-Iranzo, H.; Mercader, N.; Concordet, J.-P.; Del Bene, F. 2C-Cas9: A versatile tool for clonal analysis of gene function. Genome Res. 2016, 26, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Bill, B.R.; Petzold, A.M.; Clark, K.J.; Schimmenti, L.A.; Ekker, S.C. A primer for morpholino use in zebrafish. Zebrafish 2009, 6, 69–77. [Google Scholar] [CrossRef] [PubMed]
- McGary, K.L.; Park, T.J.; Woods, J.O.; Cha, H.J.; Wallingford, J.B.; Marcotte, E.M. Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proc. Natl. Acad. Sci. USA 2010, 107, 6544–6549. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhang, X.; Jia, S.; Yelick, P.C.; Zhao, C. Zebrafish as a Model for Human Ciliopathies. J. Genet. Genom. 2016, 43, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, C.K.; White, R.M.; Zon, L. Chemical genetic screening in the zebrafish embryo. Nat. Protoc. 2009, 4, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Griffin, A.; Hamling, K.R.; Knupp, K.; Hong, S.; Lee, L.P.; Baraban, S.C. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 2017, 140, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S. Industrializing rare disease therapy discovery and development. Nat. Biotechnol. 2017, 35, 117–118. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Bibikova, E.; Covey, T.M.; Nathanson, D.; Dimitrova, E.; Konto, Y.; Lindgren, A.; Glader, B.; Radu, C.G.; Sakamoto, K.M.; et al. The role of the DNA damage response in zebrafish and cellular models of Diamond Blackfan anemia. Dis. Model Mech. 2014, 7, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Andersson-Lendahl, M.; Sejersen, T.; Arner, A. Muscle dysfunction and structural defects of dystrophin-null sapje mutant zebrafish larvae are rescued by ataluren treatment. FASEB J. 2014, 28, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Rasighaemi, P.; Basheer, F.; Liongue, C.; Ward, A.C. Zebrafish as a model for leukemia and other hematopoietic disorders. J. Hematol. Oncol. 2015, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Deveau, A.P.; Bentley, V.L.; Berman, J.N. Using zebrafish models of leukemia to streamline drug screening and discovery. Exp. Hematol. 2017, 45, 1–9. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Jing, C.-B.; Look, A.T. Zebrafish models of leukemia. Methods Cell Biol. 2017, 138, 563–592. [Google Scholar] [PubMed]
- Fior, R.; Póvoa, V.; Mendes, R.V.; Carvalho, T.; Gomes, A.; Figueiredo, N.; Ferreira, M.G. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc. Natl. Acad. Sci. USA 2017, 114, E8234–E8243. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhang, W.; Zhao, J.-J.; Kwart, A.H.; Yang, C.; Ma, D.; Ren, X.; Tai, Y.-T.; Anderson, K.C.; Handin, R.I.; et al. A clinically relevant in vivo zebrafish model of human multiple myeloma to study preclinical therapeutic efficacy. Blood 2016, 128, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Gaudenzi, G.; Albertelli, M.; Dicitore, A.; Würth, R.; Gatto, F.; Barbieri, F.; Cotelli, F.; Florio, T.; Ferone, D.; Persani, L.; et al. Patient-derived xenograft in zebrafish embryos: A new platform for translational research in neuroendocrine tumors. Endocrine 2017, 57, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Liu, Y.; Yang, B.; Wang, X.; Wei, J.; Lu, Z.; Zhang, Y.; Wu, J.; Huang, X.; et al. Base editing with a Cpf1-cytidine deaminase fusion. Nat. Biotechnol. 2018, 36, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Poon, K.L.; Wang, X.; Ng, A.S.; Goh, W.H.; McGinnis, C.; Fowler, S.; Carney, T.J.; Wang, H.; Ingham, P.W. Humanizing the zebrafish liver shifts drug metabolic profiles and improves pharmacokinetics of CYP3A4 substrates. Arch. Toxicol. 2017, 91, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Bastarache, L.; Hughey, J.J.; Hebbring, S.; Marlo, J.; Zhao, W.; Ho, W.T.; Van Driest, S.L.; McGregor, T.L.; Mosley, J.D.; Wells, Q.S.; et al. Phenotype risk scores identify patients with unrecognized Mendelian disease patterns. Science 2018, 359, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Sulem, P.; Helgason, H.; Oddson, A.; Stefansson, H.; Gudjonsson, S.A.; Zink, F.; Hjartarson, E.; Sigurdsson, G.T.; Jonasdottir, A.; Jonasdottir, A.; et al. Identification of a large set of rare complete human knockouts. Nat. Genet. 2015, 47, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, S.; Bradley, R.K. Translational plasticity facilitates the accumulation of nonsense genetic variants in the human population. Genome Res. 2016, 26, 1639–1650. [Google Scholar] [CrossRef] [PubMed]


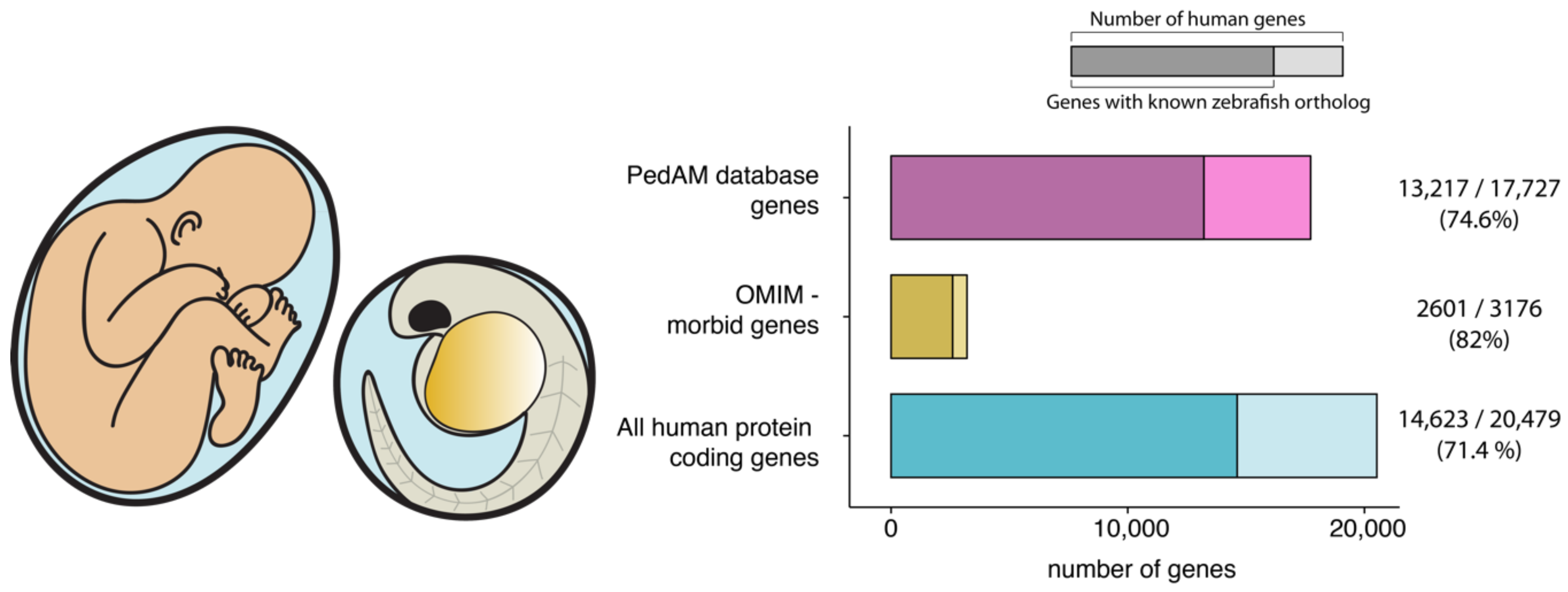{kind=link}
| Disease Name | ICD-10 | Genes Targeted in Models | OMIM IDs | Model Type | References |
|---|---|---|---|---|---|
| Diseases of the blood and blood forming organs | |||||
| Blackfan-Diamond anemia | D61.0 | RPS19, RPL11, RPS7 | 105650, 612562, 603658 | MO, mutant | [41,42,43] |
| DiGeorge syndrome | D82.1 | SNAP29, AIFM3, CRKL | 604202, 617298, 602007 | MO, crispant | [44] |
| Reticular dysgenesis | D81.0 | AK2 | 267500 | MO | [45] |
| Sideroblastic anemia (AR) | D64.0 | SLC25A38 | 205950 | MO | [46] |
| X-linked sideroblastic anemia | D64.0 | ALAS2 | 300751 | mutant | [47] |
| Endocrine and metabolic diseases | |||||
| Batten disease (Juvenile neuronal ceroid lipofuscinosis) | E75.4 | CLN3, TPP1 | 204200, 204500 | mutant, MO | [48,49] |
| Menkes disease | E83.0 | ATP7A | 309400 | mutant | [50] |
| Nephropatic infantile cystinosis | E72.0 | CTNS | 219800 | mutant | [51] |
| X-linked adrenoleukodystrophy (ALD) | E71.3 | ABCD1 | 300100 | mutant | [52] |
| Diseases of the nervous system | |||||
| Charcot-Marie-Tooth syndrome | G60.0 | MFN2, GDAP1, ABHD12, MED25, HSPB1, WNK1 | 608507, 606598, 613599, 610197, 602195, 605232 | MO | [53] |
| Childhood-onset parkinsonism-dystonia | SLC39A14 | 617013 | mutant | [54] | |
| Dravet syndrome | G40.4 | SCN1A | 182389 | mutant | [55] |
| Duchenne muscular dystrophy | G71.0 | DMD | 310200 | mutant | [56] |
| Generalized epilepsy with febrile seizures-plus | G40.3 | STX1B | 616172 | MO | [57] |
| Spinal muscular atrophy | G12 | SMN1 | 600354 | MO, mutant | [58,59] |
| Diseases of the circulatory system | |||||
| Dilated cardiomyopathy | I42.0 | BAG3 | 603883 | MO, transgenic | [60,61] |
| Timothy syndrome | I45.8 | CACNA1C | 601005 | MO | [62] |
| Diseases of the musculoskeletal system | |||||
| Fibrodysplasia ossificans progressiva (FOP) | M61.1 | ACVR1 | 135100 | mRNA, transgenic | [63,64,65] |
| Vasculitis due to ADA2 deficiency | M30.8 | ADA2 | 615688 | MO | [66] |
| Diseases of the genitourinary system | |||||
| Polycystic kidney disease (PKD) | PKD1, PKD2 | 173900, 613095 | MO, mutant | [67,68,69] | |
| Congenital malformations | |||||
| 16p11.2 microdeletion/microduplication syndrome | KCTD13 | 608947 | MO, mRNA | [70] | |
| 3MC syndrome | Q87.8 | COLEC11, MASP1 | 265050, 257920 | MO | [71] |
| Autosomal recessive polycistic kidney disease | Q61.1 | DZIP1L | 617610 | MO, mutant | [72] |
| Axenfeld-Rieger syndrome | Q13.8 | PITX2 | 180500 | mutant | [73] |
| Bardet-Biedl syndrome (BBS) | Q87.8 | BBS1, BBS2, BBS4, BBS5, BBS6, BBS7, BBS8, BBS10, BBS11, BBS12, CCDC28B | 20991, 600374, 605231, 615981, 615983, 615984, 615985, 615987, 615988, 615989, 610162 | MO, | [74,75,76,77,78,79] |
| Cardiofaciocutaneous syndrome | Q87.8 | MEK1 | 615279 | mRNA | [80] |
| Coloboma | GDF6, MAB21L2, PTCH1, YAP1 | 601147, 615877, 601309, 120433 | mutant | [81,82,83,84] | |
| Congenital anomalies of kidney and urinary tract (CAKUT) | DSTYK | 612666 | MO | [85] | |
| CHARGE syndrome | Q87.8 | CHD7 | 608892 | MO, mutant | [86,87] |
| COACH syndrome | Q04.3 | MKS3/TMEM67 | 216360 | MO | [88,89] |
| Down syndrome | Q90 | 21q22.3 | 190685 | mRNA | [90] |
| Dyskeratosis congenita | Q82.8 | DKC1, NOLA3/NOP10 | 305000, 224230 | MO, mutant | [91,92] |
| Galloway-Mowat syndrome | Q04.3 | OSGEP, TPRKB | 617729, 617731 | crispant | [93] |
| Generalized arterial calcification in infancy (GACI) | Q28.8 | ABCC6, ENPP1 | 614473, 208000 | MO, mutants | [94,95,96] |
| Infantile nephronophthisis | Q61.5 | ANKS6 | 615382 | MO | [97] |
| Joubert syndrome | Q04.3 | JBTS1/INPP5E, JBTS2/TMEM216, JBTS3/AHI1, JBTS5/CEP290, JBTS7/RPGRIP1L, JBTS8/ARL13B, JBTS9/CC2D2A, JBTS10/OFD1, ARMC9 | 213300, 608091, 608629, 610188, 611560, 610688, 612291, 612285, 300804, 617612 | MO, mutant | [89,98,99,100,101,102,103,104,105] |
| MARCH syndrome | CEP55 | 610000 | MO, crispant | [106] | |
| Pontocerebellar hypoplasia (1B) | Q04.3 | EXOSC3 | 614678 | MO | [107] |
| Primary ciliary dyskenesia | Q34.8 | ARMC4, CCDC40, ZMYND10 | 615451, 613799, 615444 | MO, mutant | [108,109,110] |
| Robinow syndrome (AD) | Q87.1 | WNT5A | 180700 | mRNA | [111] |
| Senior-Løken syndrome | Q61.5 | SDCCAG8 | 613615 | MO | [112] |
| Spondyloepimetaphyseal dysplasia | Q77.7 | NANS | 610442 | MO | [113] |
| Syndrome | Drug/Small Molecule Used | Target/Function | References |
|---|---|---|---|
| Aortic coarctation | GS4012 | VEGF inducer | [130] |
| Blackfan-Diamond anemia | PF477736 | CHK1 inhibitor | [146] |
| Childhood-onset parkinsonism-dystonia | Na2CaEDTA | Mn chelator | [54] |
| Dravet syndrome | clemizole | Serotonin modulators | [55,144] |
| lorcaserin | |||
| Duchenne muscular dystrophy | Ataluren (PTC124) | Translational readthrough agonist | [147] |
| Fibrodysplasia ossificans progressiva (FOP) | Dorsomorphin (and derivatives) | BMP Type 1 Receptor inhibitor | [142] |
| Generalized arterial calcification in infancy (GACI) | Etidronate | PPi analog | [95] |
| Sideroblastic anemia (AR) | Glycine and folate | supplement | [46] |
| Spondyloepimetaphyseal dysplasia | Sialic acid | supplement | [113] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varga, M.; Ralbovszki, D.; Balogh, E.; Hamar, R.; Keszthelyi, M.; Tory, K. Zebrafish Models of Rare Hereditary Pediatric Diseases. Diseases 2018, 6, 43. https://doi.org/10.3390/diseases6020043
Varga M, Ralbovszki D, Balogh E, Hamar R, Keszthelyi M, Tory K. Zebrafish Models of Rare Hereditary Pediatric Diseases. Diseases. 2018; 6(2):43. https://doi.org/10.3390/diseases6020043
Chicago/Turabian StyleVarga, Máté, Dorottya Ralbovszki, Eszter Balogh, Renáta Hamar, Magdolna Keszthelyi, and Kálmán Tory. 2018. "Zebrafish Models of Rare Hereditary Pediatric Diseases" Diseases 6, no. 2: 43. https://doi.org/10.3390/diseases6020043
APA StyleVarga, M., Ralbovszki, D., Balogh, E., Hamar, R., Keszthelyi, M., & Tory, K. (2018). Zebrafish Models of Rare Hereditary Pediatric Diseases. Diseases, 6(2), 43. https://doi.org/10.3390/diseases6020043