
Effect of Dietary Bioactive Compounds on Mitochondrial and Metabolic Flexibility


Abstract
:
1. Introduction
- (1)
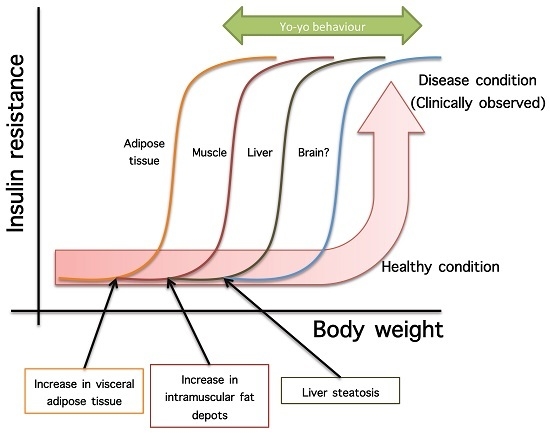
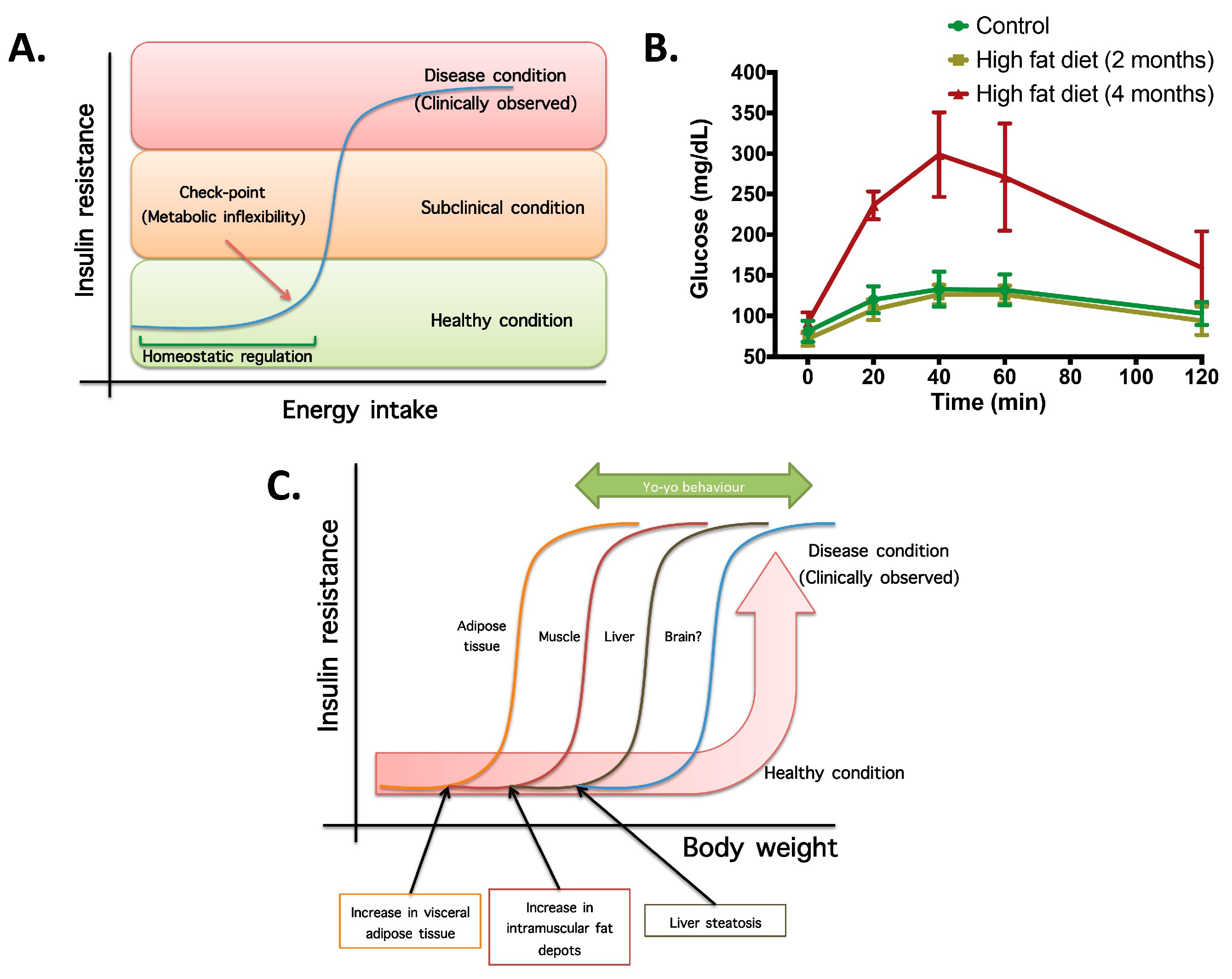
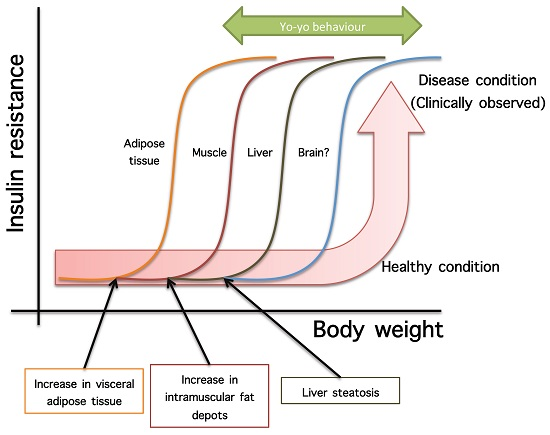
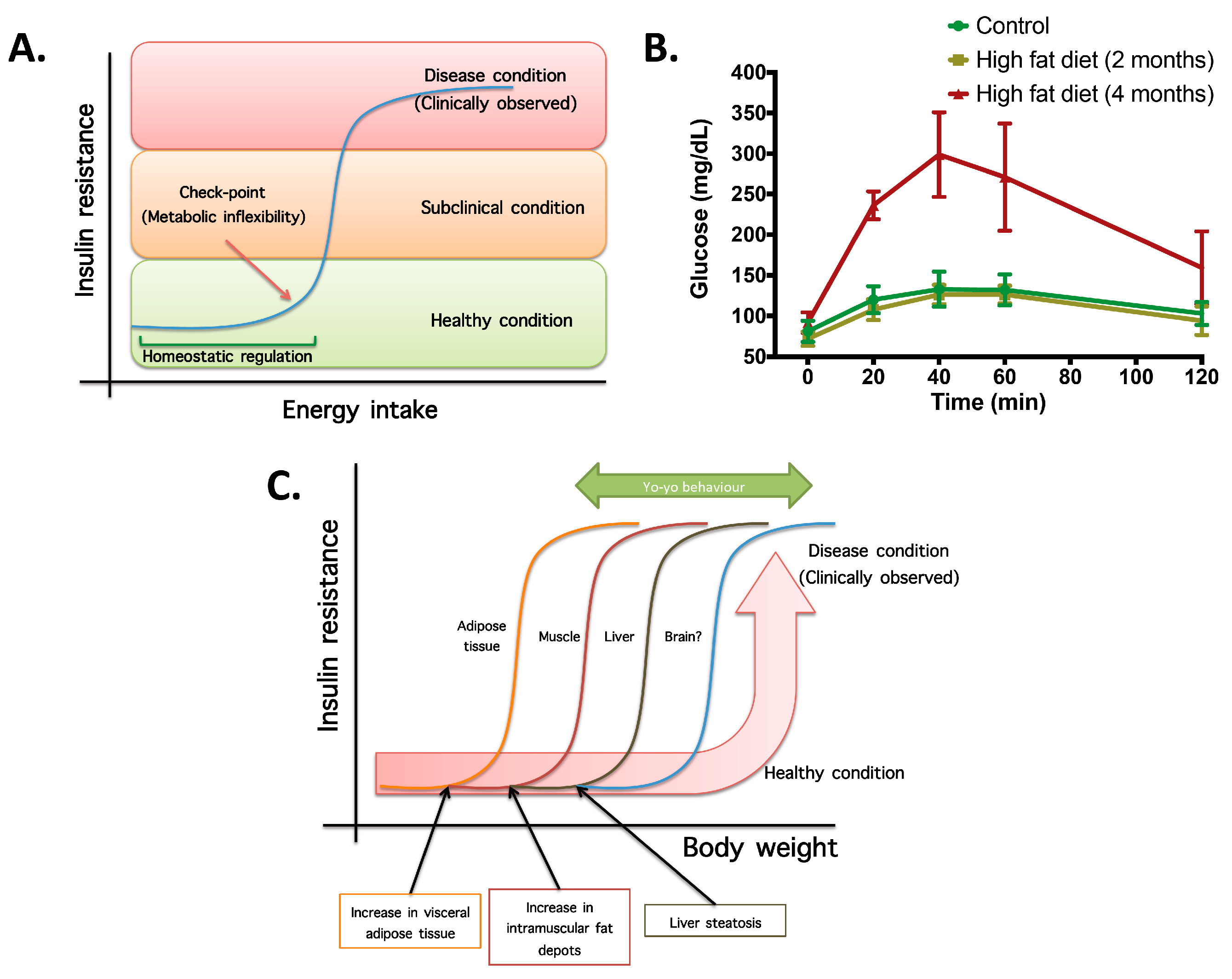
- Not all organs and systems respond in a similar way to an increase or decrease in energy availability. Differences are observed in insulin sensitivity in different organs during the development from an insulin resistance stage to a clinically type 2 diabetes stage. It seems that the onset of type 2 diabetes or other clinical complications are a consequence of the increase in the number of organs with reduced insulin sensitivity. In the clinical setting, it is common to observe a rapid shift from a non-pathological (subclinical) to a disease condition (Figure 1A) that is triggered after exceeding a metabolic checkpoint where the organism is unable to maintain homeostasis. For example, Figure 1B shows that the intake of a high-fat diet in mice is well regulated after two months, until a possible accumulation of factors that triggers a disease condition observed at Month 4 with a clinically-observed feature (unpublished observations); suggesting that, at a certain point, the homeostatic compensation is overwhelmed, and pathological conditions could be observed. Even though, it is interesting to note in the same Figure 1B that at four months of a high-fat diet, the basal glucose levels are the same, although the organism is unable to maintain normal glycemia levels; implying that, in stress conditions (glucose overload), the capacity of the system to rapidly maintain homeostasis is compromised; albeit, finally, a homeostatic condition is reached (fasting state).
- (2)
- In the same way, in the clinical setting, the effects of a mild reduction in body weight induce a rapid switch from a disease to a clinically non-pathological condition (Figure 1C). However, in this respect, frailty from this condition is also observed if an increase in body weight is observed. This implies that although most disease biomarkers are normalized during weight reduction programs, the metabolic system is unstable and liable to return to a disease condition. In other words, some organs still have metabolic inflexibility, explaining, in this way, the deleterious effects of “yo-yo” dieting.
- (3)
- The total recovery of the metabolic inflexibility condition will imply the normalization of metabolic flexibility in all organs. Thus, combined treatments with multiple mechanisms of action are required for a better handling of metabolic diseases.
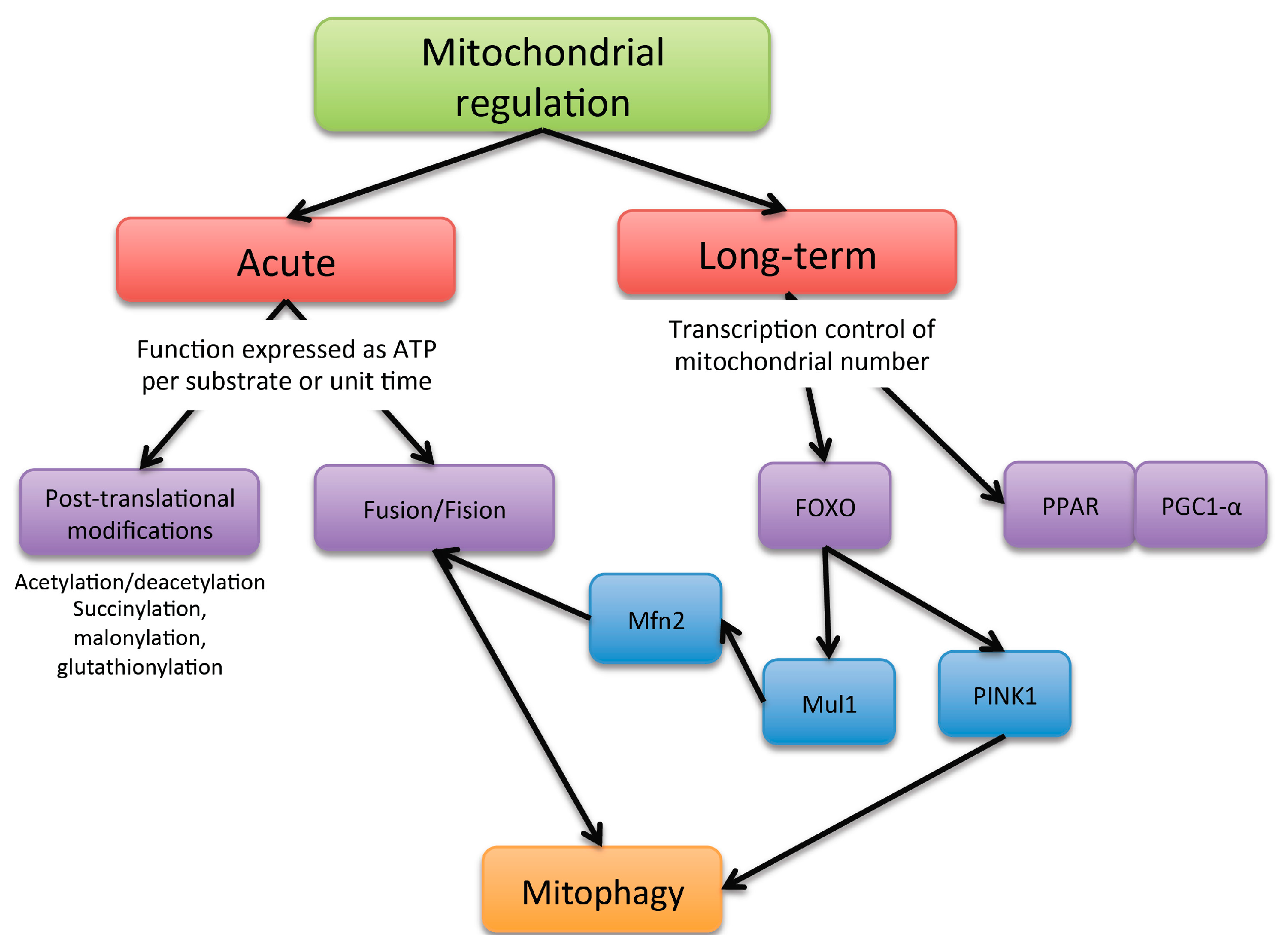
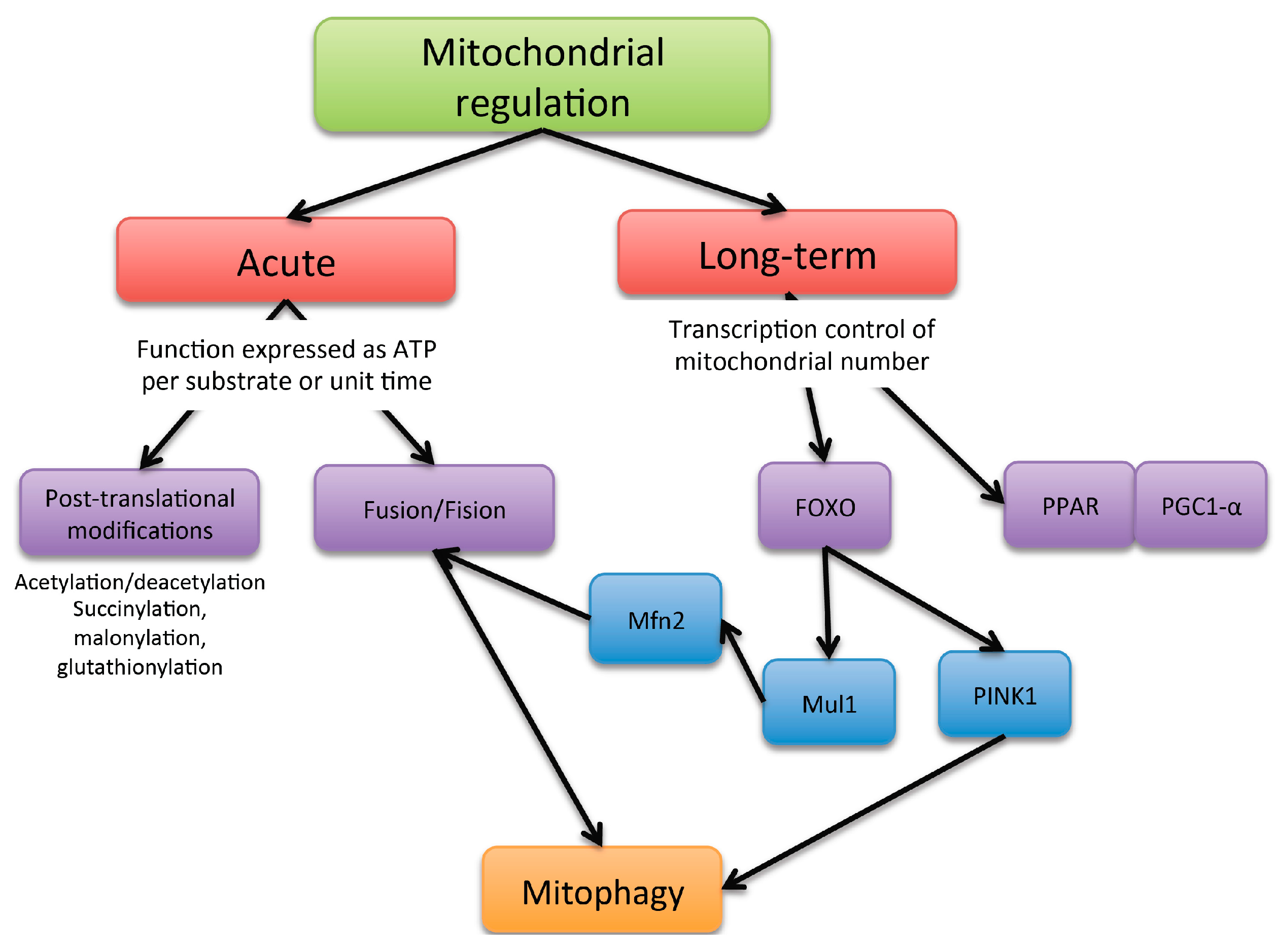
2. Mitochondria Dynamic Regulation
3. Polyphenols and Mitochondria
4. ω-3 Fatty Acids and Mitochondria
5. Dietary Fiber, Gut Microbiota and Derived Colonic Fermentation Metabolites and Mitochondria
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.W.; Cantó, C.; Houtkooper, R.H. Mitochondrial response to nutrient availability and its role in metabolic disease. EMBO Mol. Med. 2014, 6, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Corpelejin, E.; Saris, W.H.; Blaak, E.E. Metabolic flexibility in the development of insulin resistance and type 2 diabetes: Effect of lifestyle. Obes. Rev. 2009, 10, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, S.; Buskova, J.; Muniandy, M.; Kaksonen, R.; Ollikainen, M.; Ismail, K.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Vuolteenaho, K.; et al. Impaired mitochondrial biogenesis in adipose tissue in acquired obesity. Diabetes 2015, 64, 3135–3145. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.; Hurren, N.M.; Cotter, M.V.; Bhattarai, N.; Reidy, P.T.; Dillon, E.L.; Durham, W.J.; Tuvdendorj, D.; Sheffield-Moore, M.; Volpi, E.; et al. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am. J. Phsyiol. Endocrinol. MeTable 2015, 309, E224–E232. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Williams, G.R. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J. Biol. Chem. 1955, 217, 409–427. [Google Scholar] [PubMed]
- Novgorodov, S.A.; Riley, C.L.; Keffler, J.A.; Yu, J.; Kindy, M.S.; Macklin, W.B.; Lombard, D.B.; Gudz, T.I. SIRT3 deacetylates ceramide synthases: Implications for mitochondrial dysfunction and brain injury. J. Biol. Chem. 2016, 291, 1957–1973. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, R.; Staniek, K.; Kadenbach, B.; Vogt, S. Mitochondrial respiration and membrane potential are regulated by the allosteric ATP-inhibition of cytochrome c oxidase. Biochim. Biophys. Acta 2010, 1797, 1672–1680. [Google Scholar] [CrossRef] [PubMed]
- Dalmonte, M.E.; Forte, E.; Genova, M.L.; Giuffrè, A.; Sarti, P.; Lenaz, G. Control of respiration by cytochrome c oxidase in intact cells: Role of the membrane potential. J. Biol. Chem. 2009, 284, 32331–32335. [Google Scholar] [CrossRef] [PubMed]
- Diers, A.R.; Broniowska, K.A.; Darley-Usmar, V.M.; Hogg, N. Differential regulation of metabolism by nitric oxide and S-nitrosothiols in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H803–H812. [Google Scholar] [CrossRef] [PubMed]
- Højlund, K.; Wrzesinski, K.; Larsen, P.M.; Fey, S.J.; Roepstorff, P.; Handberg, A.; Dela, F.; Vinten, J.; McCormack, J.G.; Reynet, C.; et al. Proteome analysis reveals phosphorylation of ATP synthase β-subunit in human skeletal muscle and proteins with potential roles in type 2 diabetes. J. Biol. Chem. 2003, 278, 10436–10442. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Lee, I.; Liu, J.; Grossman, L.I. Transcription of mammalian cytochrome c oxidase subunit IV-2 is controlled by a novel conserved oxygen responsive element. FEBS J. 2007, 274, 5737–5748. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.E.; El Mohsen, M.M.A.; Rice-Evans, C. Cellular uptake and metabolism of flavonoids and their metabolites: Implications for their bioactivity. Arch. Biochem. Biophys. 2004, 423, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, M.; Guidarelli, A.; Blasa, M.; Azzolini, C.; Candiracci, M.; Piatti, E.; Cantoni, O. Mitochondria accumulate large amounts of quercetin: Prevention of mitochondrial damage and release upon oxidation of the extramitochondrial fraction of the flavonoid. J. Nutr. Biochem. 2010, 21, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, M.; Accorsi, A.; Cantoni, O. Human red blood cells as a natural flavonoid reservoir. Free Radic. Res. 2003, 37, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, E.K.; Kelsey, N.A.; Doyle, J.; Breed, E.; Bouchard, R.J.; Loucks, F.A.; Harbison, R.A.; Linseman, D.A. Green tea epigallocatechin 3-gallate accumulates in mitochondria and displays a selective antiapoptotic effect against inducers of mitochondrial oxidative stress in neurons. Antioxid. Redox Signal. 2009, 11, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Bast, F. Screening and biological evaluation of myricetin as a multiple target inhibitor insulin, epidermal growth factor, and androgen receptor; in silico and in vitro. Investig. New Drugs 2015, 33, 575–593. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H. Membrane interactions of phytochemicals as their molecular mechanism applicable to the discovery of drug leads from plants. Molecules 2015, 20, 18923–18966. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Murphy, E.A.; Carmichael, M.D.; Davis, B. Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1071–R1077. [Google Scholar] [CrossRef] [PubMed]
- Taub, P.R.; Ramirez-Sanchez, I.; Ciaraldi, T.P.; Perkins, G.; Murphy, A.N.; Naviaux, R.; Hogan, M.; Maisel, A.S.; Henry, R.R.; Ceballos, G.; et al. Alterations in skeletal muscle indicators of mitochondrial structure and biogenesis in patients with type 2 diabetes and heart failure: Effects of epicatechin rich cocoa. Clin. Transl. Sci. 2012, 5, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.-B.; Jeong, H.W.; Lee, S.J.; Lee, S.J. Coumestrol induces mitochondrial biogenesis by activating Sirt1 in cultured skeletal muscle cells. J. Agric. Food Chem. 2014, 62, 4298–4305. [Google Scholar] [CrossRef] [PubMed]
- Lagoa, R.; Graziani, I.; Lopez-Sanchez, C.; Garcia-Martinez, V.; Gutierrez-Merino, C. Complex I and cytochrome c are molecular targets of flavonoids that inhibit hydrogen peroxide production by mitochondria. Biochim. Biophys. Acta 2011, 1807, 1562–1572. [Google Scholar] [CrossRef] [PubMed]
- Pajuelo, D.; Quesada, H.; Díaz, S.; Fernández-Iglesias, A.; Arona-Arnal, A.; Bladé, C.; Salvadó, J.; Arola, L. Chronic dietary supplementation of proanthocyanidins corrects the mitochondrial dysfunction of brown adipose tissue caused by diet-induced obesity in Wistar rats. Br. J. Nutr. 2012, 107, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Skemiene, K.; Liobikas, J.; Borutaite, V. Anthocyanins as substrates for mitochondrial complex I—Protective effect against heart ischemic injury. FEBS J. 2015, 282, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Dorta, D.J.; Pigoso, A.A.; Mingatto, F.E.; Rodrigues, T.; Prado, I.M.; Helena, A.F.; Uyemura, S.A.; Santos, A.C.; Curti, C. The interaction of flavonoids with mitochondria: Effects on energetic processes. Chem. Biol. Interact. 2005, 152, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Serrano, J.C.E.; Gonzalo-Benito, H.; Jové, M.; Fourcade, S.; Cassanyé, A.; Boada, J.; Delgado, M.A.; Espinel, A.E.; Pamplona, R.; Portero-Otín, M. Dietary intake of green tea polyphenols regulates insulin sensitivity with an increase in AMP-activated protein kinase α content and changes in mitochondrial respiratory complexes. Mol. Nutr. Food Res. 2013, 57, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.L.; Meyer, A.; Dal-Ros, S.; Auger, C.; Keller, N.; Ramamoorthy, T.G.; Zoll, J.; Metzger, D.; Schini-Kerth, V.; Geny, B. Polyphenols prevent ageing-related impairment in skeletal muscle mitochondrial function through decreased reactive oxygen species production. Exp. Physiol. 2013, 98, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Acuña, C.; Ferreira, J.; Speisky, H. Polyphenols and mitochondria: An update on their increasingly emerging ROS-scavenging independent actions. Arch. Biochem. Biophys. 2014, 559, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Modrianský, M.; Gabrielová, E. Uncouple my heart: The benefits of inefficiency. J. Bioenerg. Biomembr. 2009, 41, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Duckles, S.P.; Krause, D.N. Mechanisms of cerebrovascular protection: Oestrogen, inflammation and mitochondria. Acta Physiol. (Oxf.) 2011, 203, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, E.; Karthik, D.; Anuradha, C.V. Genistein sensitizes hepatic insulin signaling and modulates lipid regulatory genes through p70 ribosomal S6 kinase-1 inhibition in high-fat-high-fructose diet-fed mice. Pharm. Biol. 2013, 51, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Nettleton, J.A.; Katz, R. N-3 long-chain polyunsaturated fatty acids in type 2 diabetes: A review. J. Am. Diet. Assoc. 2005, 105, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, Y.B.; Hein, G.; Chicco, A. Metabolic syndrome: Effects of n-3 PUFAs on a model of dyslipidemia, insulin resistance and adiposity. Lipids 2007, 42, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Peoples, G.E.; McLennan, P.L. Dietary fish oil reduces skeletal muscle oxygen consumption, provides fatigue resistance and improves contractile recovery in the rat in vivo hindlimb. Br. J. Nutr. 2010, 104, 1771–1779. [Google Scholar] [CrossRef] [PubMed]
- Couet, C.; Delarue, J.; Ritz, P.; Antoine, J.M.; Lamisse, F. Effect of dietary fish oil on body fat mass and basal fat oxidation in healthy adults. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Delarue, J.; Couet, C.; Cohen, R.; Bréchot, J.F.; Antoine, J.M.; Lamisse, F. Effects of fish oil on metabolic responses to oral fructose and glucose loads in healthy humans. Am. J. Physiol. 1996, 270, E353–E362. [Google Scholar] [PubMed]
- Flachs, P.; Rossmeisl, M.; Bryhn, M.; Kopecky, J. Cellular and molecular effects of n-3 polyunsaturated fatty acids on adipose tissue biology and metabolism. Clin. Sci. 2009, 116, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sparagna, G.C.; Lesnefsky, E.J. Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol. 2009, 53, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Herbst, E.A.F.; Paglialunga, S.; Gerling, C.; Whitfield, J.; Mukai, K.; Chabowski, A.; Heigenhauser, G.J.; Spriet, L.L.; Holloway, G.P. ω-3 Supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J. Physiol. 2014, 592, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.A.; Dabkowski, E.R.; de Fatima Galvao, T.; Xu, W.; Daneault, C.; de Rosiers, C.; Stanley, W.C. Dietary saturated fat and docosahexaenoic acid differentially effect cardiac mitochondrial phospholipid fatty acyl composition and Ca(2+) uptake, without altering permeability transition or left ventricular function. Physiol. Rep. 2013, 1, e00009. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, K.M.; Khairallah, R.J.; Sparagna, G.C.; Xu, W.; Hecker, P.A.; Robillard-Frayne, I.; Des Rosiers, C.; Kristian, T.; Murphy, R.C.; Fiskum, G.; et al. Dietary ω-3 fatty acids alter cardiac mitochondrial phospholipid composition and delay Ca2+-induced permeability transition. J. Mol. Cell. Cardiol. 2009, 47, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Pepe, S.; Tsuchiya, N.; Lakatta, E.G.; Hansford, R.G. PUFA and aging modulate cardiac mitochondrial membrane lipid composition and Ca2+ activation of PDH. Am. J. Physiol. 1999, 276, H149–H158. [Google Scholar] [PubMed]
- Liu, X.; Shibata, T.; Hisaka, S.; Kawai, Y.; Osawa, T. DHA hydroperoxides as a potential inducer of neuronal cell death: A mitochondrial dysfunction-mediated pathway. J. Clin. Biochem. Nutr. 2008, 43, 26–33. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Afshordel, S.; Hagl, S.; Werner, D.; Röhner, N.; Kögel, D.; Bazan, N.G.; Eckert, G.P. ω-3 Polyunsaturated fatty acids improve mitochondrial dysfunction in brain aging—Impact of Bcl-2 and NPD-1 like metabolites. Prostaglandins. Leukot. Essent. Fatty Acids 2015, 92, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, L.; Hu, W.; Zheng, Q.; Xiang, W. Mitochondrial dysfunction during in vitro hepatocyte steatosis is reversed by ω-3 fatty acid-induced up-regulation of mitofusin 2. Metabolism 2011, 60, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Lalia, A.Z.; Dasari, S.; Pallauf, M.; Fitch, M.; Hellerstein, M.K.; Lanza, I.R. Eicosapentaenoic acid but not docosahexaenoic acid restores skeletal muscle mitochondrial oxidative capacity in old mice. Aging Cell 2015, 14, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Horakova, O.; Medrikova, D.; van Schothorst, E.M.; Bunschoten, A.; Flachs, P.; Kus, V.; Kuda, O.; Bardova, K.; Janovska, P.; Hensler, M.; et al. Preservation of metabolic flexibility in skeletal muscle by a combined use of n-3 PUFA and rosiglitazone in dietary obese mice. PLoS ONE 2012, 7, e43764. [Google Scholar] [CrossRef] [PubMed]
- Sleeth, M.L.; Thompson, E.L.; Ford, H.E.; Zac-Varghese, S.E.K.; Frost, G. Free fatty acid receptor 2 and nutrient sensing: A proposed role for fibre, fermentable carbohydrates and short-chain fatty acids in appetite regulation. Nutr. Res. Rev. 2010, 23, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Kovatcheva-Datchary, P.; Nilsson, A.; Akrami, R.; Lee, Y.S.; de Vadder, F.; Arora, T.; Hallen, A.; Martens, E.; Björk, I.; Bäckhed, F. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell MeTable 2015, 22, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Inoue, D.; Hirano, K.; Tsujimoto, G. The SCFA Receptor GPR43 and Energy Metabolism. Front. Endocrinol. (Lausanne) 2014, 5, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Compound | Effect | Mechanism | Type of Study | Reference |
|---|---|---|---|---|
| Resveratrol | Increased number of mitochondria in liver and muscle | SIRT1 and PGC-1α activation | Animal model | [22] |
| Quercetin | Increased mtDNA and cytochrome c content in muscle and brain | SIRT1 and PGC-1α activation | Animal model | [23] |
| Epicatechin-rich cocoa | Mitochondrial biogenesis stimulation in muscle | SIRT1 and PGC-1α activation | Human study | [24] |
| Coumestrol | Increased mitochondrial content in muscle cells | SIRT1 activation | Cell culture | [25] |
| Quercetin, kaempferol, epicatechin | Inhibitors of H2O2 production by mitochondria | Inhibition of complex I activity | Cell culture | [26] |
| Grape seed proanthocyanidin extract | Enhanced thermogenic capacity and improvement in mitochondrial function in brown and adipose tissue | Not described | Animal model | [27] |
| Anthocyanins | Complex I activity recovery and increase in the rate of ATP synthesis | Functioning as electron carriers in a similar way as coenzyme Q1 | Isolated mitochondria | [28] |
| Galangin | Modulation of the mitochondrial permeability transition pore | Decreased fluidity of the mitochondrial membrane | Isolated mitochondria | [29] |
| Epigallocatechin | Modification in mitochondrial architecture | AMPKα activation | Animal model | [30] |
| Product | Effect | Mechanism | Type of Study | Reference |
|---|---|---|---|---|
| Fish oil | Improvement in mitochondrial efficiency | Increased content or enhanced kinetics of ETC | Animal model | [38] |
| Fish oil | Reduced body fat mass | Stimulation of lipid oxidation | Human study | [39] |
| Fish oil | Decrease in insulinemia | Increased lipid oxidation | Human study | [40] |
| DHA + EPA | Improve in mitochondrial ADP kinetics | Incorporation in mitochondrial membranes, displacing ω-6 species in several phospholipids population | Human study | [44] |
| DHA + EPA | Decrease in H2O2 production | Increased tolerance to Ca2+-induced MPTP opening | Isolated mitochondria | [47] |
| Fish oil | Improvement in ATP production in brain | Improvement in membrane fluidity | Animal model | [49] |
| EPA and DHA | Increase in ATP and reduction in ROS levels in hepatocytes | Increase in the length of mitochondrial tubes by an increase in Mfn2 mRNA levels | Cell culture | [50] |
| EPA | Restoration of skeletal muscle mitochondrial capacity | Increase in coupling efficiency of the ETC | Animal model | [51] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serrano, J.C.E.; Cassanye, A.; Martín-Gari, M.; Granado-Serrano, A.B.; Portero-Otín, M. Effect of Dietary Bioactive Compounds on Mitochondrial and Metabolic Flexibility. Diseases 2016, 4, 14. https://doi.org/10.3390/diseases4010014
Serrano JCE, Cassanye A, Martín-Gari M, Granado-Serrano AB, Portero-Otín M. Effect of Dietary Bioactive Compounds on Mitochondrial and Metabolic Flexibility. Diseases. 2016; 4(1):14. https://doi.org/10.3390/diseases4010014
Chicago/Turabian StyleSerrano, Jose C. E., Anna Cassanye, Meritxell Martín-Gari, Ana Belen Granado-Serrano, and Manuel Portero-Otín. 2016. "Effect of Dietary Bioactive Compounds on Mitochondrial and Metabolic Flexibility" Diseases 4, no. 1: 14. https://doi.org/10.3390/diseases4010014
APA StyleSerrano, J. C. E., Cassanye, A., Martín-Gari, M., Granado-Serrano, A. B., & Portero-Otín, M. (2016). Effect of Dietary Bioactive Compounds on Mitochondrial and Metabolic Flexibility. Diseases, 4(1), 14. https://doi.org/10.3390/diseases4010014