
Prader-Willi Syndrome: The Disease that Opened up Epigenomic-Based Preemptive Medicine

 and
and
Abstract
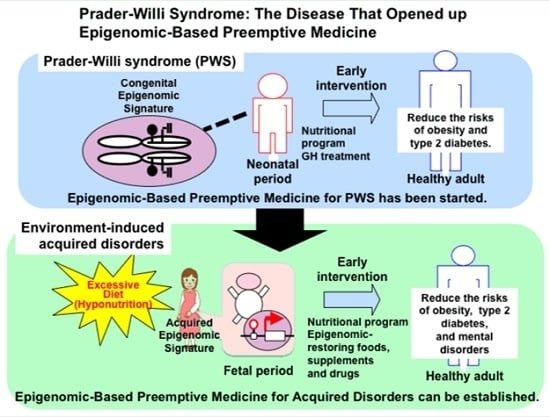
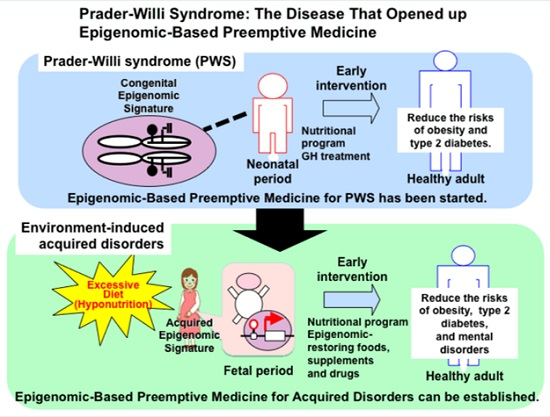
:
{kind=link}
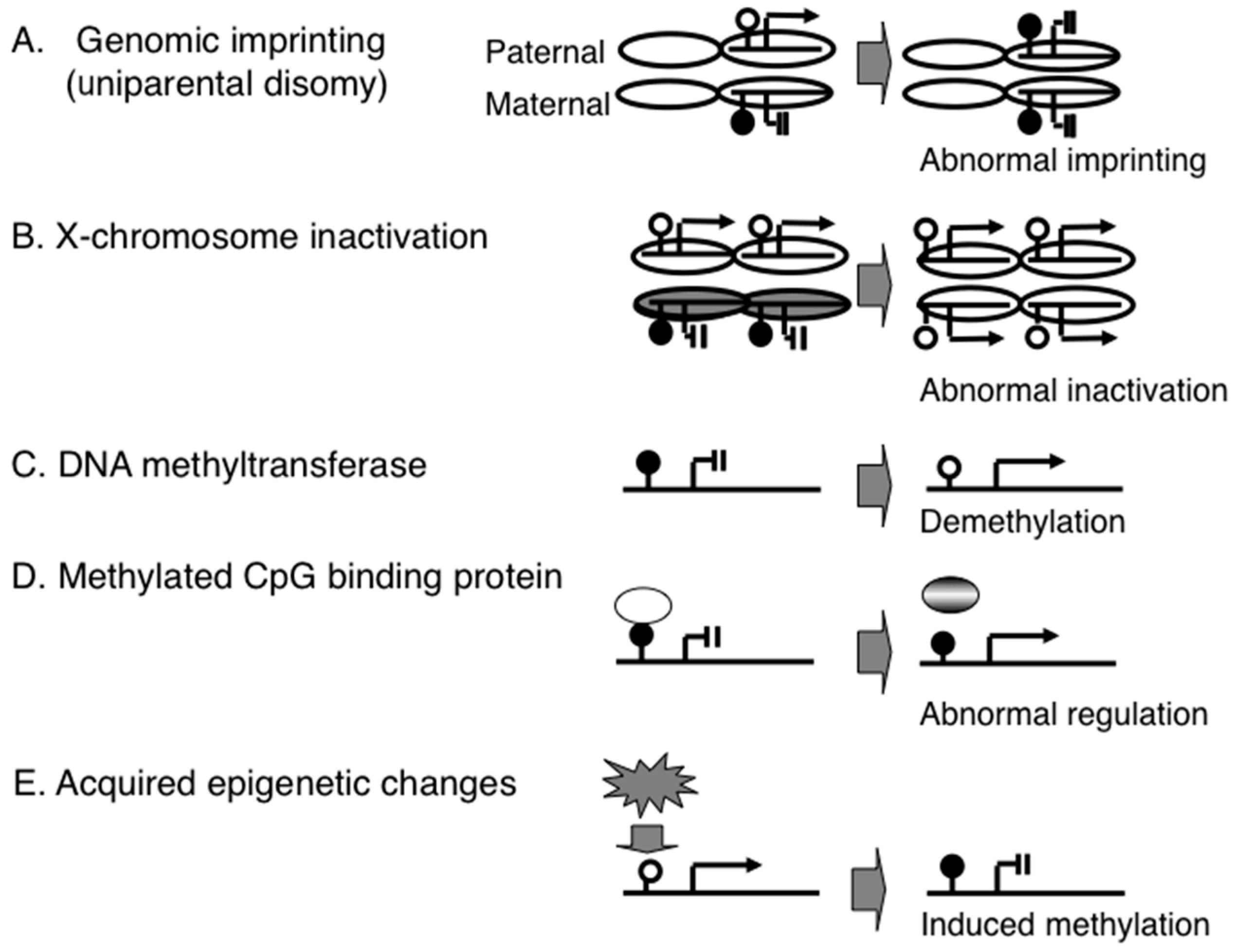{kind=link}
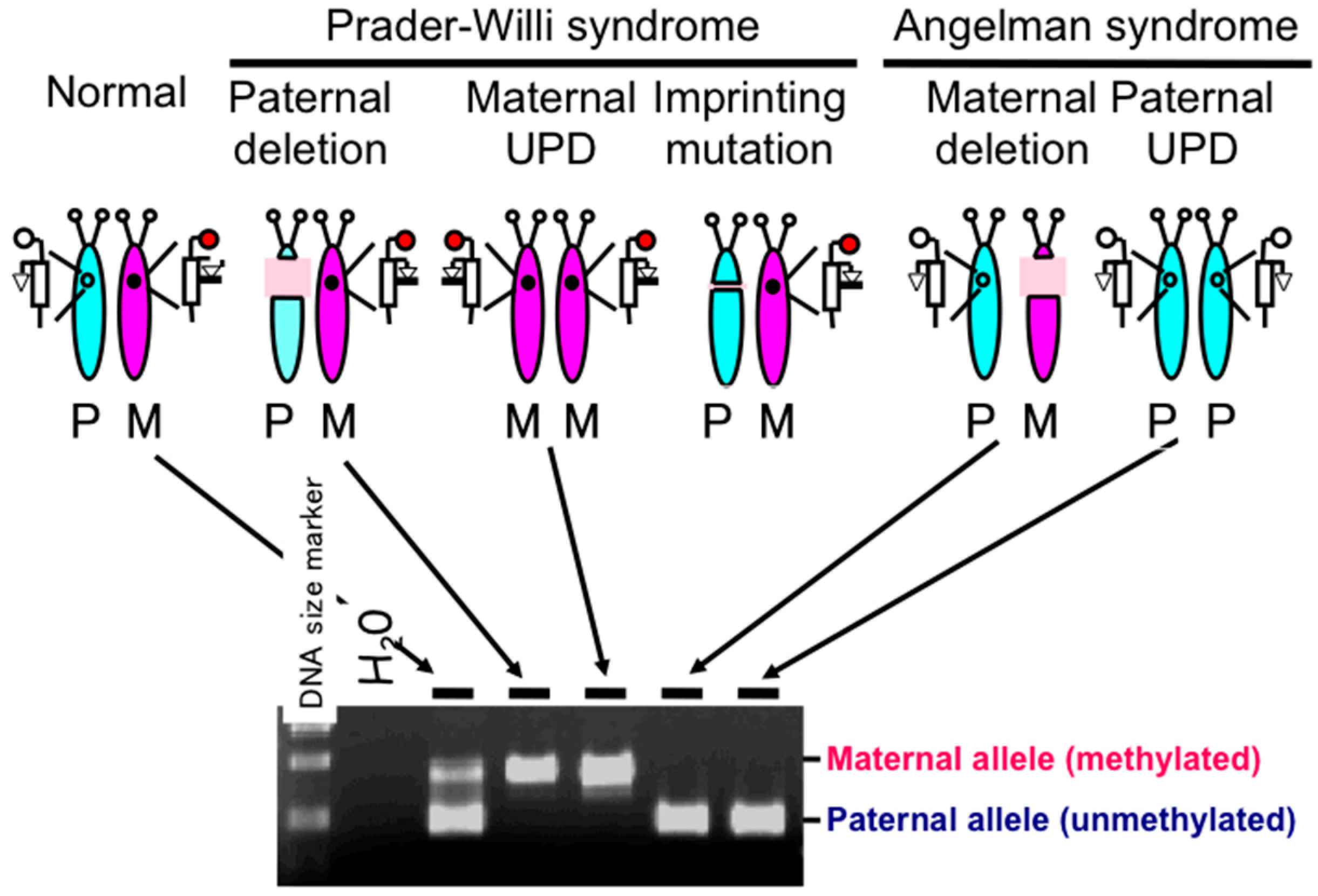{kind=link}
{kind=link}
1. Introduction
2. Development of Diagnostic Assays for PWS
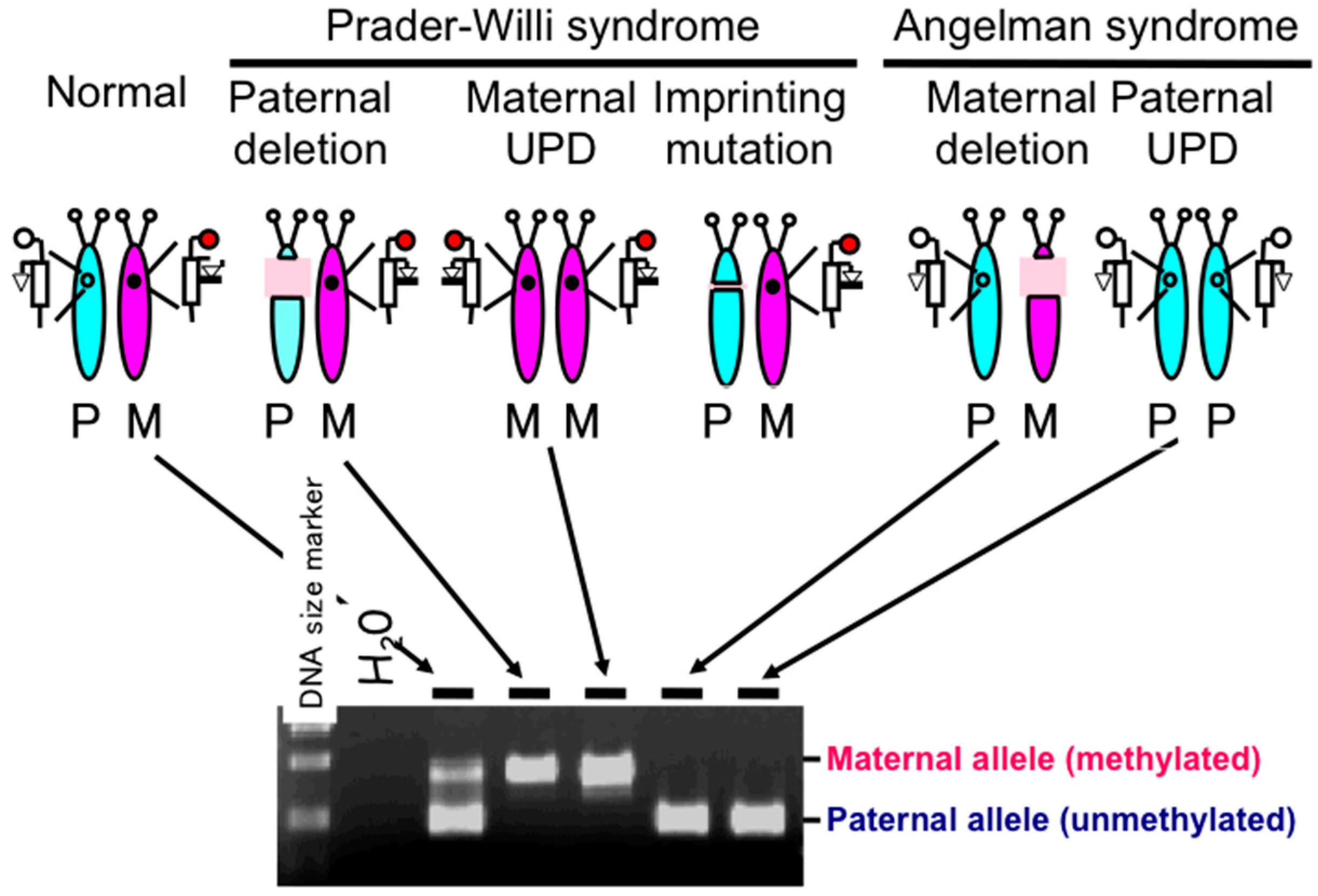
2.1. Diagnostic Assays for PWS
2.2. DNA Methylation-Based Diagnostic Assay for PWS
2.3. DNA Methylation-Based Diagnostic Assay for other Congenital Neurodevelopmental Disorders
2.4. Genome-Wide Epigenetic Assay
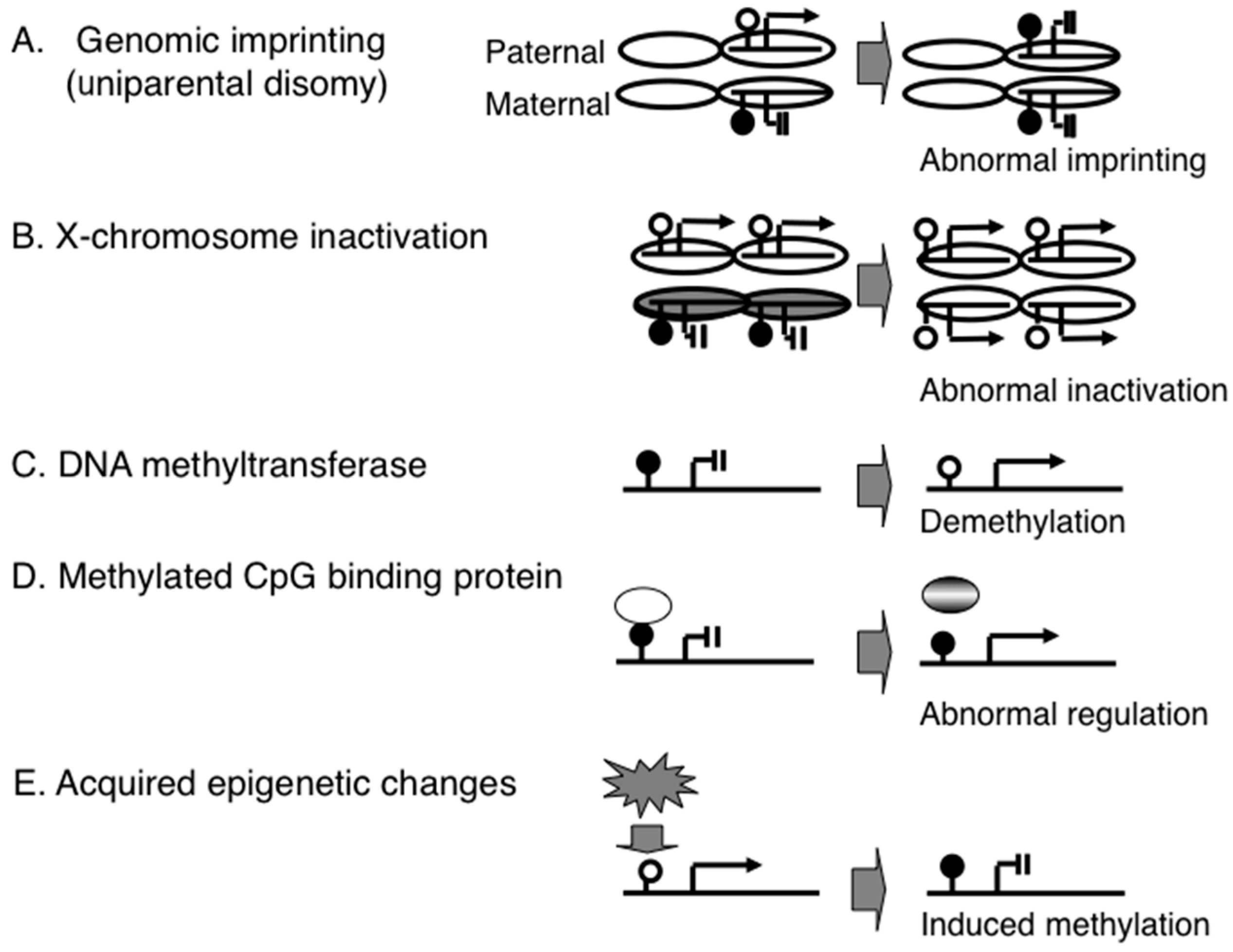
3. Acquired Epigenomic Changes Associated with Diseases
3.1. Epigenomic Changes in Cancer
3.2. Environmental Factors that Induce Epigenomic Changes
3.3. Environmental Stresses May Induce Epigenetic Changes during the Neonatal Period
3.4. Environmental Stresses Induce Epigenetic Changes during Fetal Development
4. Epigenomic-Based Preemptive Medicine
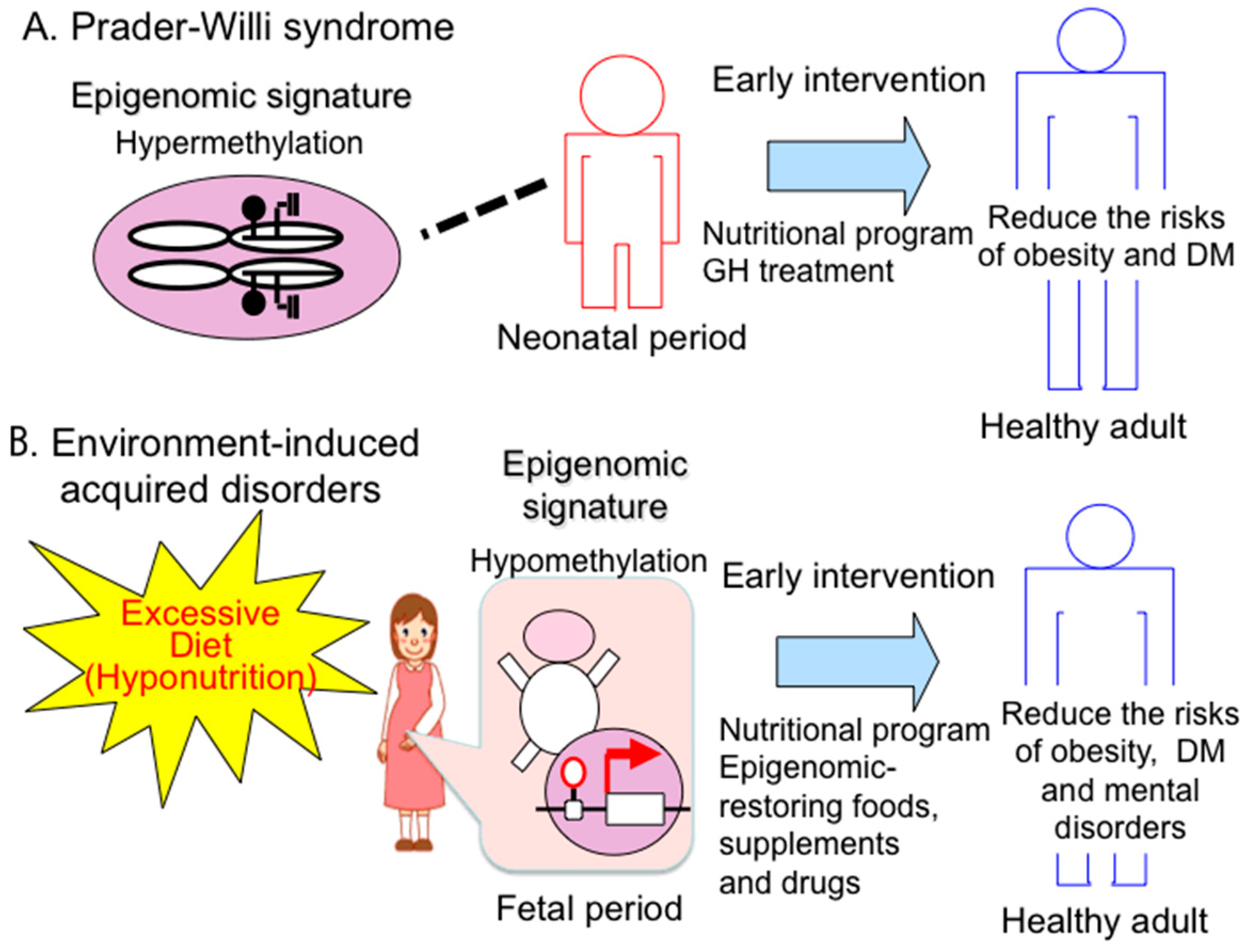
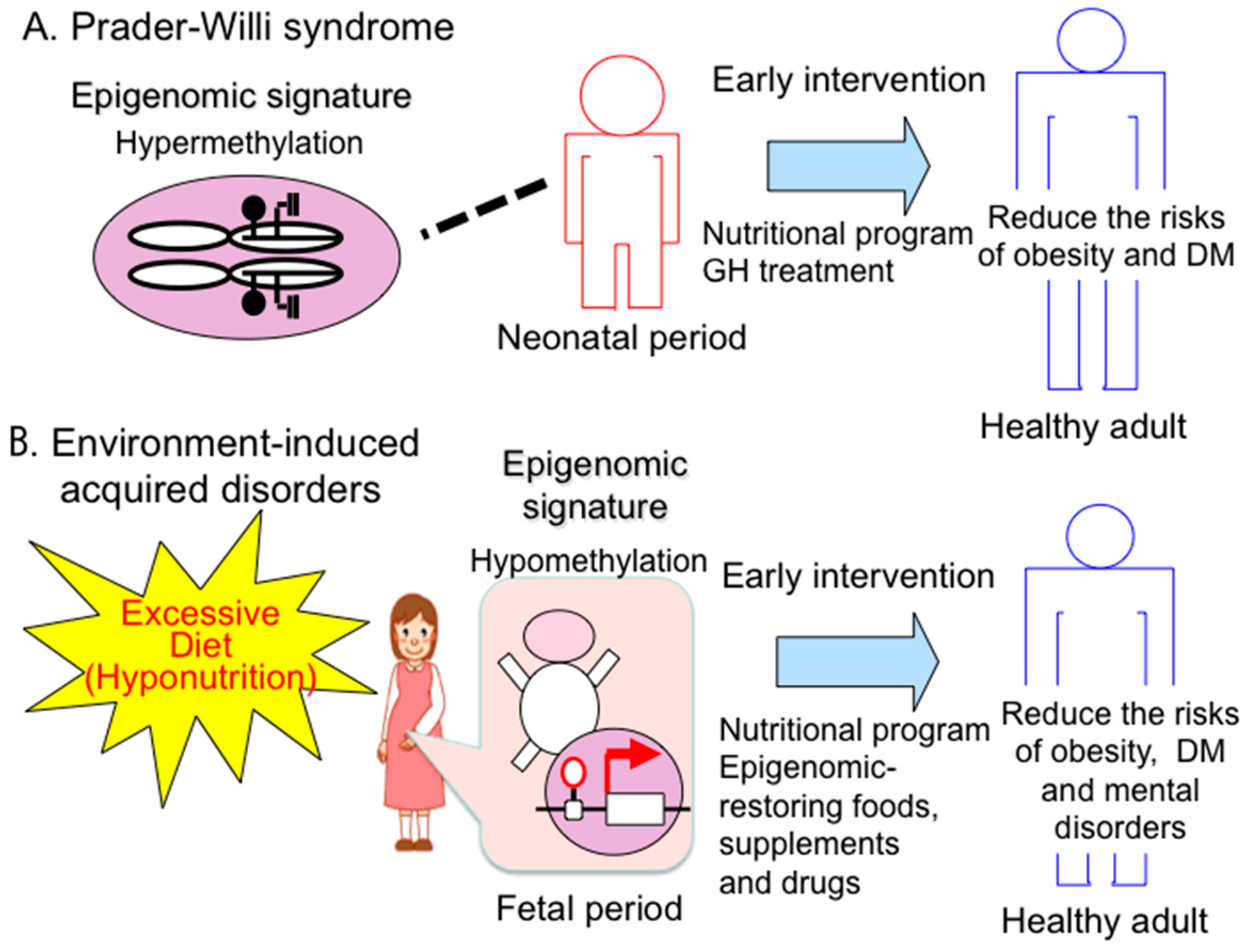
4.1. Therapeutic Strategies and Implications of Early Intervention for PWS
4.2. Epigenomic-Based Preemptive Medicine
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Online Mendelian Inheritance of Men (OMIM) #176270. Available online: http://omim.org/entry/176270 (accessed on 10 March 2016).
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Ledbetter, D.H.; Riccardi, V.M.; Airhart, S.D.; Strobel, R.J.; Keenan, B.S.; Crawford, J.D. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N. Engl. J. Med. 1981, 304, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Hogart, A.; Wu, D.; LaSalle, J.M.; Schanen, N.C. The comorbidity of autism with the genomic disorders of chromosome 15q11.2–q13. Neurobiol. Dis. 2010, 38, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, T.; del Gaudio, D.; German, J.R.; Shinawi, M.; Peters, S.U.; Person, R.E.; Garnica, A.; Cheung, S.W.; Beaudet, A.L. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 2008, 40, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.P.; Gonzalez-Garay, M.L.; Xia, F.; Potocki, L.; Gripp, K.W.; Zhang, B.; Peters, B.A.; McElwain, M.A.; Drmanac, R.; Beaudet, A.L.; et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 2013, 45, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, Y.; Sagi, I.; Yanuka, O.; Eiges, R.; Benvenisty, N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat. Genet. 2014, 46, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Sutcliffe, J.S.; Aradhya, S.; Gillessen-Kaesbach, G.; Christian, S.L.; Horsthemke, B.; Beaudet, A.L.; Ledbetter, D.H. Validation studies of SNRPN methylation as a diagnostic test for Prader-Willi syndrome. Am. J. Med. Genet. 1996, 66, 77–80. [Google Scholar] [CrossRef]
- Kubota, T.; Das, S.; Christian, S.L.; Baylin, S.B.; Herman, J.G.; Ledbetter, D.H. Methylation-specific PCR simplifies imprinting analysis. Nat. Genet. 1997, 16, 16–17. [Google Scholar] [PubMed]
- Baran, Y.; Subramaniam, M.; Biton, A.; Tukiainen, T.; Tsang, E.K.; Rivas, M.A.; Pirinen, M.; Gutierrez-Arcelus, M.; Smith, K.S.; Kukurba, K.R.; et al. The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 2015, 25, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.; D’Mello, S.R.; Narayanan, V. Epigenetics, autism spectrum, and neurodevelopmental disorders. Neurotherapeutics 2013, 10, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Scoles, H.A.; Urraca, N.; Chadwick, S.W.; Reiter, L.T.; Lasalle, J.M. Increased copy number for methylated maternal 15q duplications leads to changes in gene and protein expression in human cortical samples. Mol. Autism 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Saitoh, S.; Matsumoto, T.; Narahara, K.; Fukushima, Y.; Jinno, Y.; Niikawa, N. Excess functional copy of allele at chromosomal region 11p15 may cause Wiedemann-Beckwith (EMG) syndrome. Am. J. Med. Genet. 1994, 49, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Azzi, S.; Habib, W.A.; Netchine, I. Beckwith-Wiedemann and Russell-Silver Syndromes: From new molecular insights to the comprehension of imprinting regulation. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Begemann, M.; Zirn, B.; Santen, G.; Wirthgen, E.; Soellner, L.; Büttel, H.M.; Schweizer, R.; van Workum, W.; Binder, G.; Eggermann, T. Paternally Inherited IGF2 Mutation and Growth Restriction. N. Engl. J. Med. 2015, 373, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Wakui, K.; Nakamura, T.; Ohashi, H.; Watanabe, Y.; Yoshino, M.; Kida, T.; Okamoto, N.; Matsumura, M.; Muroya, K.; et al. The proportion of cells with functional X disomy is associated with the severity of mental retardation in mosaic ring X Turner syndrome females. Cytogenet. Genome Res. 2002, 99, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Nolen, L.D.; Gao, S.; Han, Z.; Mann, M.R.; Chung, Y.G.; Otte, A.P.; Bartolomei, M.S.; Latham, K.E. X chromosome reactivation and regulation in cloned embryos. Dev. Biol. 2005, 279, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Furuumi, H.; Kamoda, T.; Iwasaki, N.; Tobita, N.; Fujiwara, N.; Goto, Y.; Matsui, A.; Sasaki, H.; Kajii, T. ICF syndrome in a girl with DNA hypomethylation but without detectable DNMT3B mutation. Am. J. Med. Genet. A 2004, 129A, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Seal, S.; Ruark, E.; Harmer, J.; Ramsay, E.; del Vecchio Duarte, S.; Zachariou, A.; Hanks, S.; O’Brien, E.; Aksglaede, L.; et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 2014, 46, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Hirasawa, T.; Soutome, M.; Itoh, M.; Goto, Y.; Endoh, K.; Takahashi, K.; Kudo, S.; Nakagawa, T.; Yokoi, S.; et al. The protocadherins, PCDHB1 and PCDH7, are regulated by MeCP2 in neuronal cells and brain tissues: Implication for pathogenesis of Rett syndrome. BMC Neurosci. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Delach, J.A.; Rosengren, S.S.; Kaplan, L.; Greenstein, R.M.; Cassidy, S.B.; Benn, P.A. Comparison of high resolution chromosome banding and fluorescence in situ hybridization (FISH) for the laboratory evaluation of Prader-Willi syndrome and Angelman syndrome. Am. J. Med. Genet. 1994, 52, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Mutirangura, A.; Greenberg, F.; Butler, M.G.; Malcolm, S.; Nicholls, R.D.; Chakravarti, A.; Ledbetter, D.H. Multiplex PCR of three dinucleotide repeats in the Prader-Willi/Angelman critical region (15q11–q13): Molecular diagnosis and mechanism of uniparental disomy. Hum. Mol. Genet. 1993, 2, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed]
- Procter, M.; Chou, L.S.; Tang, W.; Jama, M.; Mao, R. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin. Chem. 2006, 52, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Henkhaus, R.S.; Kim, S.J.; Kimonis, V.E.; Gold, J.A.; Dykens, E.M.; Driscoll, D.J.; Butler, M.G. Methylation-specific multiplex ligation-dependent probe amplification and identification of deletion genetic subtypes in Prader-Willi syndrome. Genet. Test. Mol. Biomark. 2012, 16, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Botezatu, A.; Puiu, M.; Cucu, N.; Diaconu, C.C.; Badiu, C.; Arsene, C.; Iancu, I.V.; Plesa, A.; Anton, G. Comparative molecular approaches in Prader-Willi syndrome diagnosis. Gene 2016, 575, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Kubota, T.; Song, M.; Daniel, R.; Berry-Kravis, E.M.; Prior, T.W.; Popovich, B.; Rosser, L.; Arinami, T.; Ledbetter, D.H. Methylation analysis of the fragile X syndrome by PCR. Genet. Test. 1997, 1, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Wylie, A.A.; Coveler, K.J.; Cotter, P.D.; Papenhausen, P.R.; Sutton, V.R.; Shaffer, L.G.; Jirtle, R.L. Epigenetic detection of human chromosome 14 uniparental disomy. Hum. Mutat. 2003, 22, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Mitter, D.; Buiting, K.; von Eggeling, F.; Kuechler, A.; Liehr, T.; Mau-Holzmann, U.A.; Prott, E.C.; Wieczorek, D.; Gillessen-Kaesbach, G. Is there a higher incidence of maternal uniparental disomy 14 [upd(14)mat]? Detection of 10 new patients by methylation-specific PCR. Am. J. Med. Genet. A 2006, 140, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Sakazume, S.; Ohashi, H.; Sasaki, Y.; Harada, N.; Nakanishi, K.; Sato, H.; Emi, M.; Endoh, K.; Sohma, R.; Kido, Y.; et al. Spread of X-chromosome inactivation into chromosome 15 is associated with Prader-Willi syndrome phenotype in a boy with a t(X;15)(p21.1;q11.2) translocation. Hum. Genet. 2012, 131, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Yang, C.; Minakuchi, Y.; Ohori, K.; Soutome, M.; Hirasawa, T.; Kazuki, Y.; Adachi, N.; Suzuki, S.; Itoh, M.; et al. Comparison of genomic and epigenomic expression in monozygotic twins discordant for Rett syndrome. PLoS ONE 2013, 8, e66729. [Google Scholar] [CrossRef] [PubMed]
- Breitling, L.P.; Yang, R.; Korn, B.; Burwinkel, B.; Brenner, H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am. J. Hum. Genet. 2011, 88, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.; Stütz, A.M.; Wan, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014, 506, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A.; Jirtle, R.L. Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 2003, 23, 5293–5300. [Google Scholar] [CrossRef] [PubMed]
- Kucharski, R.; Maleszka, J.; Foret, S.; Maleszka, R. Nutritional control of reproductive status in honeybees via DNA methylation. Science 2008, 319, 1827–1830. [Google Scholar] [CrossRef] [PubMed]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Dong, E.; Nelson, M.; Grayson, D.R.; Costa, E.; Guidotti, A. Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proc. Natl. Acad. Sci. USA 2008, 105, 13614–13619. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, X.; Li, J.; Liu, J.; Gu, H.; Zhang, R.; Chen, J.; Kuang, Y.; Fei, J.; Jiang, C.; et al. Lithium, an anti-psychotic drug, greatly enhances the generation of induced pluripotent stem cells. Cell Res. 2011, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Yaoi, T.; Itoh, K.; Nakamura, K.; Ogi, H.; Fujiwara, Y.; Fushiki, S. Genome-wide analysis of epigenomic alterations in fetal mouse forebrain after exposure to low doses of bisphenol A. Biochem. Biophys. Res. Commun. 2008, 376, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.C.; Walker, D.M.; Zama, A.M.; Armenti, A.E.; Uzumcu, M. Early life exposure to endocrine-disrupting chemicals causes lifelong molecular reprogramming of the hypothalamus and premature reproductive aging. Mol. Endocrinol. 2011, 25, 2157–2168. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.H.; Larsen, A.; Nielsen, A.L. DNA methylation alterations in response to prenatal exposure of maternal cigarette smoking: A persistent epigenetic impact on health from maternal lifestyle? Arch. Toxicol. 2016, 90, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.K.; Jang, M.H.; Guo, J.U.; Kitabatake, Y.; Chang, M.L.; Pow-Anpongkul, N.; Flavell, R.A.; Lu, B.; Ming, G.L.; Song, H. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 2009, 323, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Bowdin, S.C.; Tee, L. Clinical and molecular genetic features of Beckwith-Wiedemann syndrome associated with assisted reproductive technologies. Hum. Reprod. 2009, 24, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Loke, Y.J.; Galati, J.C.; Saffery, R.; Craig, J.M. Association of in vitro fertilization with global and IGF2/H19 methylation variation in newborn twins. J. Dev. Orig. Health Dis. 2015, 6, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Amor, D.J.; Halliday, J. A review of known imprinting syndromes and their association with assisted reproduction technologies. Hum. Reprod. 2008, 23, 2826–2834. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Murakami, N.; Fukami, M.; Kagami, M.; Nagai, T.; Ogata, T. Risk assessment of medically assisted reproduction and advanced maternal ages in the development of Prader-Willi syndrome due to UPD(15)mat. Clin. Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 9, 847–854. [Google Scholar] [CrossRef] [PubMed]
- McGowan, P.O.; Sasaki, A.; D’Alessio, A.C.; Dymov, S.; Labonté, B.; Szyf, M.; Turecki, G.; Meaney, M.J. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 2009, 12, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Bockmühl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Almeida, O.F.; Spengler, D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Phillips, E.S.; Torrens, C.; Hanson, M.A.; Jackson, A.A.; Burdge, G.C. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR α promoter of the offspring. Br. J. Nutr. 2008, 100, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, C.A.; van Veldhoven, K.; Relton, C.; Stringhini, S.; Kyriacou, K.; Vineis, P. Biological embedding of early-life exposures and disease risk in humans: A role for DNA methylation. Eur. J. Clin. Investig. 2015, 45, 303–332. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Seng, C.Y.; Fukuoka, H.; Beedle, A.S.; Hanson, M.A. Low birthweight and subsequent obesity in Japan. Lancet 2007, 369, 1081–1082. [Google Scholar] [CrossRef]
- St Clair, D.; Xu, M.; Wang, P.; Yu, Y.; Fang, Y.; Zhang, F.; Zheng, X.; Gu, N.; Feng, G.; Sham, P.; et al. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959–1961. JAMA 2005, 294, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Silveira, P.P.; Manfro, G.G. Retrospective studies. Adv. Neurobiol. 2015, 10, 251–267. [Google Scholar] [PubMed]
- Benyshek, D.C. The “early life” origins of obesity-related health disorders: New discoveries regarding the intergenerational transmission of developmentally programmed traits in the global cardiometabolic health crisis. Am. J. Phys. Anthropol. 2013, 152 (Suppl. S57), 79–93. [Google Scholar] [CrossRef] [PubMed]
- Youngson, N.A.; Lecomte, V.; Maloney, C.A.; Leung, P.; Liu, J.; Hesson, L.B.; Luciani, F.; Krause, L.; Morris, M.J. Obesity-induced sperm DNA methylation changes at satellite repeats are reprogrammed in rat offspring. Asian J. Androl. 2015. [Google Scholar] [CrossRef]
- Soubry, A.; Murphy, S.K.; Wang, F.; Huang, Z.; Vidal, A.C.; Fuemmeler, B.F.; Kurtzberg, J.; Murtha, A.; Jirtle, R.L.; Schildkraut, J.M.; et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int. J. Obes. 2015, 39, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Soubry, A.; Schildkraut, J.M.; Murtha, A.; Wang, F.; Huang, Z.; Bernal, A.; Kurtzberg, J.; Jirtle, R.L.; Murphy, S.K.; Hoyo, C. Paternal obesity is associated with IGF2 hypomethylation in newborns: Results from a Newborn Epigenetics Study (NEST) cohort. BMC Med. 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Donkin, I.; Versteyhe, S.; Ingerslev, L.R.; Qian, K.; Mechta, M.; Nordkap, L.; Mortensen, B.; Appel, E.V.; Jørgensen, N.; Kristiansen, V.B.; et al. Obesity and Bariatric Surgery Drive Epigenetic Variation of Spermatozoa in Humans. Cell Metab. 2016, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Fullston, T.; Ohlsson Teague, E.M.; Palmer, N.O.; DeBlasio, M.J.; Mitchell, M.; Corbett, M.; Print, C.G.; Owens, J.A.; Lane, M. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB J. 2013, 27, 4226–4243. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Lynn, C.H.; Shuster, J.; Driscoll, D.J. A reduced-energy intake, well-balanced diet improves weight control in children with Prader-Willi syndrome. J. Hum. Nutr. Diet. 2013, 26, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Schlumpf, M.; Eiholzer, U.; Gygax, M.; Schmid, S.; van der Sluis, I.; l’Allemand, D. A daily comprehensive muscle training programme increases lean mass and spontaneous activity in children with Prader-Willi syndrome after 6 months. J. Pediatr. Endocrinol. Metab. 2006, 19, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Angulo, M.A.; Castro-Magana, M.; Lamerson, M.; Arguello, R.; Accacha, S.; Khan, A. Final adult height in children with Prader-Willi syndrome with and without human growth hormone treatment. Am. J. Med. Genet. A 2007, 143A, 1456–1461. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Lee, J.; Cox, D.M.; Manzardo, A.M.; Gold, J.A.; Miller, J.L.; Roof, E.; Dykens, E.; Kimonis, V.; Driscoll, D.J. Growth Charts for Prader-Willi Syndrome during Growth Hormone Treatment. Clin. Pediatr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Reus, L.; Pillen, S.; Pelzer, B.J.; van Alfen-van der Velden, J.A.; Hokken-Koelega, A.C.; Zwarts, M.; Otten, B.J.; Nijhuis-van der Sanden, M.W. Growth hormone therapy, muscle thickness, and motor development in Prader-Willi syndrome: An RCT. Pediatrics 2014, 134, e1619–e1627. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.E.; Whitman, B.Y.; Carrel, A.L.; Moerchen, V.; Bekx, M.T.; Allen, D.B. Two years of growth hormone therapy in young children with Prader-Willi syndrome: Physical and neurodevelopmental benefits. Am. J. Med. Genet. A 2007, 143A, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Siemensma, E.P.; Tummers-de Lind van Wijngaarden, R.F.; Festen, D.A.; Troeman, Z.C.; van Alfen-van der Velden, A.A.; Otten, B.J.; Rotteveel, J.; Odink, R.J.; Bindels-de Heus, G.C.; van Leeuwen, M.; et al. Beneficial effects of growth hormone treatment on cognition in children with Prader-Willi syndrome: A randomized controlled trial and longitudinal study. J. Clin. Endocrinol. Metab. 2012, 97, 2307–2314. [Google Scholar] [CrossRef] [PubMed]
- Osório, J. Growth and development: Growth hormone therapy improves cognition in children with Prader-Willi syndrome. Nat. Rev. Endocrinol. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Höybye, C.; Thorén, M.; Böhm, B. Cognitive, emotional, physical and social effects of growth hormone treatment in adults with Prader-Willi syndrome. J. Intellect. Disabil. Res. 2005, 49, 245–252. [Google Scholar] [CrossRef] [PubMed]
- GeneReviews® [Internet]. Prader-Willi Syndrome. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1330/ (accessed on 10 March 2016).
- Sode-Carlsen, R.; Farholt, S.; Rabben, K.F.; Bollerslev, J.; Schreiner, T.; Jurik, A.G.; Christiansen, J.S.; Höybye, C. One year of growth hormone treatment in adults with Prader-Willi syndrome improves body composition: Results from a randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 2010, 95, 4943–4950. [Google Scholar] [CrossRef] [PubMed]
- Imura, H. Life course health care and preemptive approach to non-communicable diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Fuchikami, M.; Morinobu, S.; Segawa, M.; Okamoto, Y.; Yamawaki, S.; Ozaki, N.; Inoue, T.; Kusumi, I.; Koyama, T.; Tsuchiyama, K.; et al. DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PLoS ONE 2011, 6, e23881. [Google Scholar] [CrossRef] [PubMed]
- Kundakovic, M.; Gudsnuk, K.; Herbstman, J.B.; Tang, D.; Perera, F.P.; Champagne, F.A. DNA methylation of BDNF as a biomarker of early-life adversity. Proc. Natl. Acad. Sci. USA 2015, 112, 6807–6813. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, H.; Yu, M.; Zhang, X.; Zhang, Y.; Liu, H.; Wilson, J.X.; Huang, G. Folic Acid Alters Methylation Profile of JAK-STAT and Long-Term Depression Signaling Pathways in Alzheimer’s Disease Models. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Mitchell, A.; Schneider, A.; Halene, T.; Akbarian, S. Epigenetic dysregulation in schizophrenia: Molecular and clinical aspects of histone deacetylase inhibitors. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Brodie, S.A.; Brandes, J.C. Could valproic acid be an effective anticancer agent? The evidence so far. Expert Rev. Anticancer Ther. 2014, 14, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Whittle, N.; Singewald, N. HDAC inhibitors as cognitive enhancers in fear, anxiety and trauma therapy: Where do we stand? Biochem. Soc. Trans. 2014, 42, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Schmauss, C. An HDAC-dependent epigenetic mechanism that enhances the efficacy of the antidepressant drug fluoxetine. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubota, T.; Miyake, K.; Hariya, N.; Tran Nguyen Quoc, V.; Mochizuki, K. Prader-Willi Syndrome: The Disease that Opened up Epigenomic-Based Preemptive Medicine. Diseases 2016, 4, 15. https://doi.org/10.3390/diseases4010015
Kubota T, Miyake K, Hariya N, Tran Nguyen Quoc V, Mochizuki K. Prader-Willi Syndrome: The Disease that Opened up Epigenomic-Based Preemptive Medicine. Diseases. 2016; 4(1):15. https://doi.org/10.3390/diseases4010015
Chicago/Turabian StyleKubota, Takeo, Kunio Miyake, Natsuyo Hariya, Vuong Tran Nguyen Quoc, and Kazuki Mochizuki. 2016. "Prader-Willi Syndrome: The Disease that Opened up Epigenomic-Based Preemptive Medicine" Diseases 4, no. 1: 15. https://doi.org/10.3390/diseases4010015
APA StyleKubota, T., Miyake, K., Hariya, N., Tran Nguyen Quoc, V., & Mochizuki, K. (2016). Prader-Willi Syndrome: The Disease that Opened up Epigenomic-Based Preemptive Medicine. Diseases, 4(1), 15. https://doi.org/10.3390/diseases4010015