

Mitochondrial Dysfunction in Systemic Lupus Erythematosus: Insights and Therapeutic Potential
 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Anatomy, Role and Activity of Mitochondria
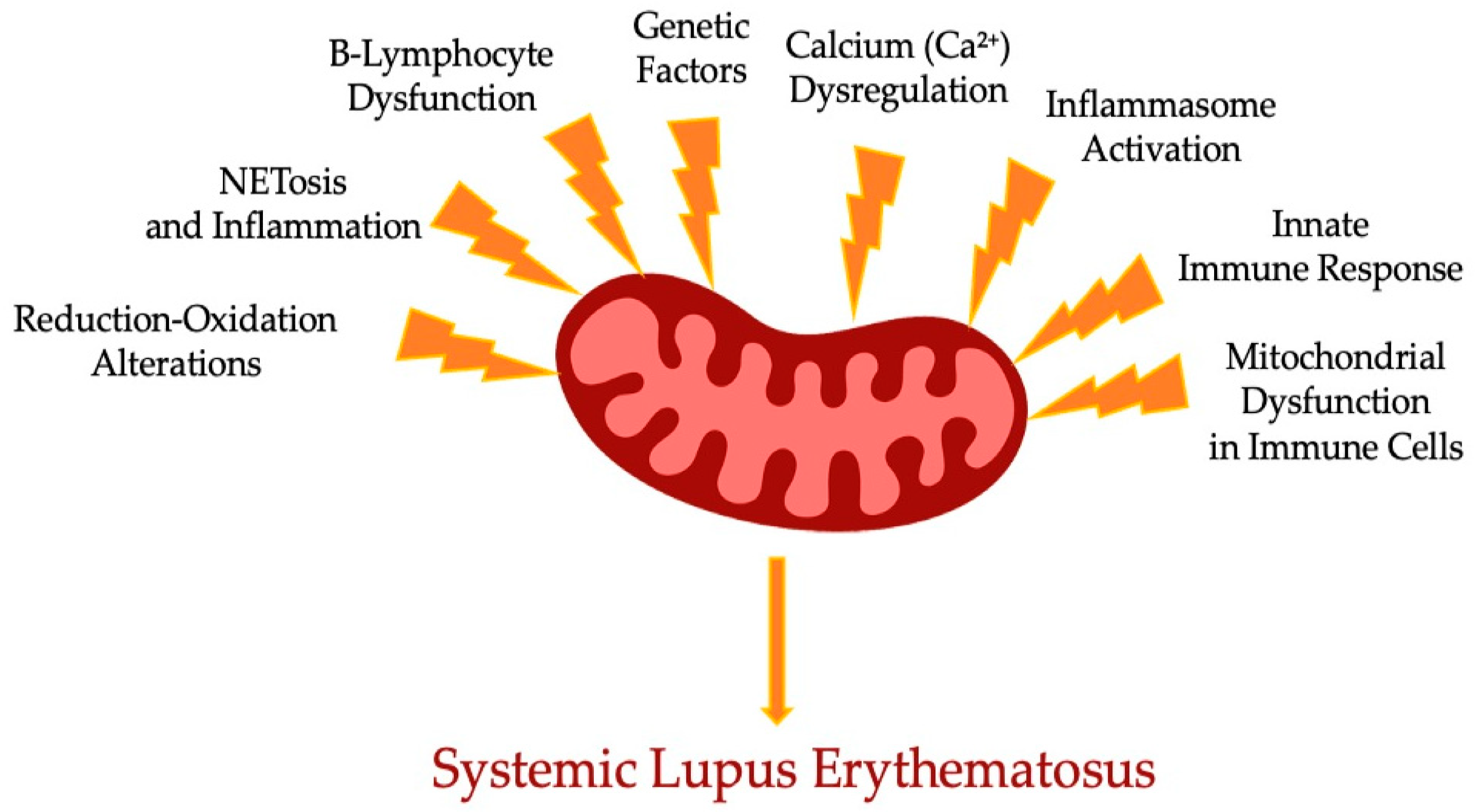
3. Mitochondrial Function Impairment in Systemic Lupus Erythematosus
3.1. Reduction–Oxidation Alterations: A Vicious Cycle
3.2. Genetic Mechanism of Mitochondrial Function Impairment
3.3. Mitochondrial Function Impairment in ICs
4. Mitochondrial Function Impairment in Acquired Immunity
5. Mitochondrial Function Impairment in Innate ICs
6. Prospects of Mt-Targeted Treatment in Systemic Lupus Erythematosus Subjects
6.1. Sirolimus (Rapamycin)
6.2. N-Acetylcysteine (NAC)
6.3. Coenzyme Q10 (CoQ10)
6.4. Metformin
6.5. Hydroxychloroquine (HCQ)
6.6. 3PEHPC
6.7. Pioglitazone
6.8. Piceid
6.9. Nestin
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Justiz Vaillant, A.A.; Goyal, A.; Varacallo, M. Systemic Lupus erythematosus. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK535405/ (accessed on 4 August 2023).
- García-Carrasco, M.; Mendoza Pinto, C.; Solís Poblano, J.C. Systemic lupus erythematosus. In Autoimmunity: From Bench to Bedside; Anaya, J.M., Shoenfeld, Y., Rojas-Villarraga, A., Levy, R.A., Cervera, R., Eds.; El Rosario University Press: Bogota, Colombia, 2013; Chapter 25. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459474/ (accessed on 4 August 2023).
- Zharkova, O.; Celhar, T.; Cravens, P.D.; Satterthwaite, A.B.; Fairhurst, A.M.; Davis, L.S. Pathways leading to an immunological disease: Systemic lupus erythematosus. Rheumatology 2017, 56 (Suppl. 1), i55–i66. [Google Scholar] [CrossRef] [PubMed]
- Accapezzato, D.; Caccavale, R.; Paroli, M.P.; Gioia, C.; Nguyen, B.L.; Spadea, L.; Paroli, M. Advances in the Pathogenesis and Treatment of Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2023, 24, 6578. [Google Scholar] [CrossRef] [PubMed]
- Sciascia, S.; Radin, M.; Roccatello, D.; Sanna, G.; Bertolaccini, M.L. Recent advances in the management of systemic lupus erythematosus. F1000Research 2018, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Katarzyna, P.B.; Wiktor, S.; Ewa, D.; Piotr, L. Current treatment of systemic lupus erythematosus: A clinician’s perspective. Rheumatol. Int. 2023, 43, 1395–1407. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK26894/ (accessed on 4 August 2023).
- Yu, R.; Lendahl, U.; Nistér, M.; Zhao, J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front. Endocrinol. 2020, 11, 374. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress-A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.P.; Dubois, S.M.; Agarwal, S.; Zon, L.I. Mitochondrial function in development and disease. Dis. Models Mech. 2021, 14, dmm048912. [Google Scholar] [CrossRef]
- Hayden, M.R. The Mighty Mitochondria Are Unifying Organelles and Metabolic Hubs in Multiple Organs of Obesity, Insulin Resistance, Metabolic Syndrome, and Type 2 Diabetes: An Observational Ultrastructure Study. Int. J. Mol. Sci. 2022, 23, 4820. [Google Scholar] [CrossRef]
- Vitali, D.G.; Käser, S.; Kolb, A.; Dimmer, K.S.; Schneider, A.; Rapaport, D. Independent evolution of functionally exchangeable mitochondrial outer membrane import complexes. eLife 2018, 7, e34488. [Google Scholar] [CrossRef]
- Wu, Q.; Tsai, H.I.; Zhu, H.; Wang, D. The Entanglement between Mitochondrial DNA and Tumor Metastasis. Cancers 2022, 14, 1862. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int. J. Mol. Sci. 2021, 22, 11338. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. Mitochondrial DNA degradation: A quality control measure for mitochondrial genome maintenance and stress response. Enzymes 2019, 45, 311–341. [Google Scholar] [CrossRef]
- Druzhyna, N.M.; Wilson, G.L.; LeDoux, S.P. Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 2008, 129, 383–390. [Google Scholar] [CrossRef]
- Bi, C.; Wang, L.; Fan, Y.; Yuan, B.; Ramos-Mandujano, G.; Zhang, Y.; Alsolami, S.; Zhou, X.; Wang, J.; Shao, Y.; et al. Single-cell individual full-length mtDNA sequencing by iMiGseq uncovers unexpected heteroplasmy shifts in mtDNA editing. Nucleic Acids Res. 2023, 51, e48. [Google Scholar] [CrossRef]
- Parakatselaki, M.E.; Ladoukakis, E.D. mtDNA Heteroplasmy: Origin, Detection, Significance, and Evolutionary Consequences. Life 2021, 11, 633. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Vona, R.; Pallotta, L.; Cappelletti, M.; Severi, C.; Matarrese, P. The Impact of Oxidative Stress in Human Pathology: Focus on Gastrointestinal Disorders. Antioxidants 2021, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Chandrasekaran, K.; Inoue, T.; Muragundla, A.; Russell, J.W. PGC-1α regulation of mitochondrial degeneration in experimental diabetic neuropathy. Neurobiol. Dis. 2014, 64, 118–130. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef]
- Kubli, D.A.; Gustafsson, Å.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: Roles of calcium and reactive oxygen species. Future Cardiol. 2012, 8, 863–884. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Wu, H. Ubiquitination-Proteasome System (UPS) and Autophagy Two Main Protein Degradation Machineries in Response to Cell Stress. Cells 2022, 11, 851. [Google Scholar] [CrossRef]
- Belousov, D.M.; Mikhaylenko, E.V.; Somasundaram, S.G.; Kirkland, C.E.; Aliev, G. The Dawn of Mitophagy: What Do We Know by Now? Curr. Neuropharmacol. 2021, 19, 170–192. [Google Scholar] [CrossRef]
- Iorio, R.; Celenza, G.; Petricca, S. Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis. Cells 2021, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, Q.; Gao, W.; Sehgal, S.A.; Wu, H. The multifaceted regulation of mitophagy by endogenous metabolites. Autophagy 2022, 18, 1216–1239. [Google Scholar] [CrossRef]
- Venditti, P.; Di Meo, S. The Role of Reactive Oxygen Species in the Life Cycle of the Mitochondrion. Int. J. Mol. Sci. 2020, 21, 2173. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Fenga, C.; Gangemi, S.; Teodoro, M.; Rapisarda, V.; Golokhvast, K.; Docea, A.O.; Tsatsakis, A.M.; Costa, C. 8-Hydroxydeoxyguanosine as a biomarker of oxidative DNA damage in workers exposed to low-dose benzene. Toxicol. Rep. 2017, 4, 291–295. [Google Scholar] [CrossRef]
- Torres-Gonzalez, M.; Gawlowski, T.; Kocalis, H.; Scott, B.T.; Dillmann, W.H. Mitochondrial 8-oxoguanine glycosylase decreases mitochondrial fragmentation and improves mitochondrial function in H9C2 cells under oxidative stress conditions. Am. J. Physiol. Cell Physiol. 2014, 306, C221–C229. [Google Scholar] [CrossRef]
- Elghzaly, A.A.; Sun, C.; Looger, L.L.; Hirose, M.; Salama, M.; Khalil, N.M.; Behiry, M.E.; Hegazy, M.T.; Hussein, M.A.; Salem, M.N.; et al. Genome-wide association study for systemic lupus erythematosus in an egyptian population. Front. Genet. 2022, 13, 948505. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.S.; Brown, E.E.; Kimberly, R.P.; Langefeld, C.D. Genetic factors predisposing to systemic lupus erythematosus and lupus nephritis. Semin. Nephrol. 2010, 30, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Becker, Y.; Marcoux, G.; Allaeys, I.; Julien, A.S.; Loignon, R.C.; Benk-Fortin, H.; Rollet-Labelle, E.; Rauch, J.; Fortin, P.R.; Boilard, E. Autoantibodies in Systemic Lupus Erythematosus Target Mitochondrial RNA. Front. Immunol. 2019, 10, 1026. [Google Scholar] [CrossRef] [PubMed]
- Becker, Y.; Loignon, R.C.; Julien, A.S.; Marcoux, G.; Allaeys, I.; Lévesque, T.; Rollet-Labelle, E.; Benk-Fortin, H.; Cloutier, N.; Melki, I.; et al. Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations. Sci. Rep. 2019, 9, 4530. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Yan, Z.; Yang, A. Mitochondria in innate immunity signaling and its therapeutic implications in autoimmune diseases. Front. Immunol. 2023, 14, 1160035. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Zuo, Z.; Jing, K.; Wu, H.; Wang, S.; Ye, L.; Li, Z.; Yang, C.; Pan, Q.; Liu, W.J.; Liu, H.F. Mechanisms and Functions of Mitophagy and Potential Roles in Renal Disease. Front. Physiol. 2020, 11, 935. [Google Scholar] [CrossRef]
- Zhao, L.; Hu, X.; Xiao, F.; Zhang, X.; Zhao, L.; Wang, M. Mitochondrial impairment and repair in the pathogenesis of systemic lupus erythematosus. Front. Immunol. 2022, 13, 929520. [Google Scholar] [CrossRef]
- Zhao, W.; Zhuang, P.; Chen, Y.; Wu, Y.; Zhong, M.; Lun, Y. “Double-edged sword” effect of reactive oxygen species (ROS) in tumor development and carcinogenesis. Physiol. Res. 2023, 72, 301–307. [Google Scholar] [CrossRef]
- Kyttaris, V.C.; Zhang, Z.; Kampagianni, O.; Tsokos, G.C. Calcium signaling in systemic lupus erythematosus T cells: A treatment target. Arthritis Rheum. 2011, 63, 2058–2066. [Google Scholar] [CrossRef]
- Fisher, W.G.; Yang, P.C.; Medikonduri, R.K.; Jafri, M.S. NFAT and NFkappaB activation in T lymphocytes: A model of differential activation of gene expression. Ann. Biomed. Eng. 2006, 34, 1712–1728. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Yoo, S.A.; Kim, M.; Kim, W.U. The Role of Calcium-Calcineurin-NFAT Signaling Pathway in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 195. [Google Scholar] [CrossRef]
- Lai, Z.W.; Hanczko, R.; Bonilla, E.; Caza, T.N.; Clair, B.; Bartos, A.; Miklossy, G.; Jimah, J.; Doherty, E.; Tily, H.; et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012, 64, 2937–2946. [Google Scholar] [CrossRef]
- Abbasifard, M.; Khorramdelazad, H.; Rostamian, A.; Rezaian, M.; Askari, P.S.; Sharifi, G.T.K.; Parizi, M.K.; Sharifi, M.T.K.; Najafizadeh, S.R. Effects of N-acetylcysteine on systemic lupus erythematosus disease activity and its associated complications: A randomized double-blind clinical trial study. Trials 2023, 24, 129. [Google Scholar] [CrossRef]
- Torres Acosta, M.A.; Mambetsariev, N.; Reyes Flores, C.P.; Helmin, K.A.; Liu, Q.; Joudi, A.M.; Morales-Nebreda, L.; Gurkan, J.; Cheng, K.; Abdala-Valencia, H.; et al. AMP-activated protein kinase is necessary for Treg cell functional adaptation to microenvironmental stress. bioRxiv 2023. [Google Scholar] [CrossRef]
- Hurtado, C.; Rojas-Gualdrón, D.F.; Urrego, R.; Cashman, K.; Vásquez-Trespalacios, E.M.; Díaz-Coronado, J.C.; Rojas, M.; Jenks, S.; Vásquez, G.; Sanz, I. Altered B cell phenotype and CD27+ memory B cells are associated with clinical features and environmental exposure in Colombian systemic lupus erythematosus patients. Front. Med. 2022, 9, 950452. [Google Scholar] [CrossRef]
- Crispin, J.C.; Hedrich, C.M.; Suárez-Fueyo, A.; Comte, D.; Tsokos, G.C. SLE-Associated Defects Promote Altered T Cell Function. Crit. Rev. Immunol. 2017, 37, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Li, D.; Zhang, D.; Xu, Y.; Pan, Y.; Lu, L.; Li, J.; Xia, X.; Dou, H.; Hou, Y. IRF-8/miR-451a regulates M-MDSC differentiation via the AMPK/mTOR signal pathway during lupus development. Cell Death Discov. 2021, 7, 179. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef]
- Decker, P. Neutrophils and interferon-α-producing cells: Who produces interferon in lupus? Arthritis Res. Ther. 2011, 13, 118. [Google Scholar] [CrossRef]
- Infante, B.; Mercuri, S.; Dello Strologo, A.; Franzin, R.; Catalano, V.; Troise, D.; Cataldo, E.; Pontrelli, P.; Alfieri, C.; Binda, V.; et al. Unraveling the Link between Interferon-α and Systemic Lupus Erythematosus: From the Molecular Mechanisms to Target Therapies. Int. J. Mol. Sci. 2022, 23, 15998. [Google Scholar] [CrossRef]
- Miao, N.; Wang, Z.; Wang, Q.; Xie, H.; Yang, N.; Wang, Y.; Wang, J.; Kang, H.; Bai, W.; Wang, Y.; et al. Oxidized mitochondrial DNA induces gasdermin D oligomerization in systemic lupus erythematosus. Nat. Commun. 2023, 14, 872. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, T.; Nurbaeva, K. The Role of Neutrophil Extracellular Traps (NETs) in the Pathogenesis of Systemic Lupus Erythematosus and Antiphospholipid Syndrome. Int. J. Mol. Sci. 2023, 24, 13581. [Google Scholar] [CrossRef] [PubMed]
- Gkirtzimanaki, K.; Kabrani, E.; Nikoleri, D.; Polyzos, A.; Blanas, A.; Sidiropoulos, P.; Makrigiannakis, A.; Bertsias, G.; Boumpas, D.T.; Verginis, P. IFNα Impairs Autophagic Degradation of mtDNA Promoting Autoreactivity of SLE Monocytes in a STING-Dependent Fashion. Cell Rep. 2018, 25, 921–933.e5. [Google Scholar] [CrossRef]
- Di Domizio, J.; Cao, W. Fueling autoimmunity: Type I interferon in autoimmune diseases. Expert Rev. Clin. Immunol. 2013, 9, 201–210. [Google Scholar] [CrossRef]
- Sharma, M.; de Alba, E. Structure, Activation and Regulation of NLRP3 and AIM2 Inflammasomes. Int. J. Mol. Sci. 2021, 22, 872. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.J.; DeVylder, J.E.; Fedina, L. Police Violence among Adults Diagnosed with Mental Disorders. Health Soc. Work 2020, 45, 81–89. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Westerterp, M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J. Lipid Res. 2019, 60, 721–727. [Google Scholar] [CrossRef]
- Liu, Y.; Tao, X.; Tao, J. Strategies of Targeting Inflammasome in the Treatment of Systemic Lupus Erythematosus. Front. Immunol. 2022, 13, 894847. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Kaplan, M.J. The inflammasome and lupus: Another innate immune mechanism contributing to disease pathogenesis? Curr. Opin. Rheumatol. 2014, 26, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Amissah-Arthur, M.B.; Gordon, C. Contemporary treatment of systemic lupus erythematosus: An update for clinicians. Ther. Adv. Chronic Dis. 2010, 1, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Bhaoighill, M.N.; Dunlop, E.A. Mechanistic target of rapamycin inhibitors: Successes and challenges as cancer therapeutics. Cancer Drug Resist. 2019, 2, 1069–1085. [Google Scholar] [CrossRef] [PubMed]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Sig. Transduct. Target Ther. 2023, 8, 375. [Google Scholar] [CrossRef]
- Ganesh, S.K.; Subathra Devi, C. Molecular and therapeutic insights of rapamycin: A multi-faceted drug from Streptomyces hygroscopicus. Mol. Biol. Rep. 2023, 50, 3815–3833. [Google Scholar] [CrossRef]
- Peng, L.; Wu, C.; Hong, R.; Sun, Y.; Qian, J.; Zhao, J.; Wang, Q.; Tian, X.; Wang, Y.; Li, M.; et al. Clinical efficacy and safety of sirolimus in systemic lupus erythematosus: A real-world study and meta-analysis. Ther. Adv. Musculoskelet. Dis. 2020, 12, 1759720X20953336. [Google Scholar] [CrossRef]
- Piranavan, P.; Perl, A. Improvement of renal and non-renal SLE outcome measures on sirolimus therapy—A 21-year follow-up study of 73 patients. Clin. Immunol. 2021, 229, 108781. [Google Scholar] [CrossRef]
- Ding, M.; Jin, L.; Zhao, J.; Yang, L.; Cui, S.; Wang, X.; He, J.; Chang, F.; Shi, M.; Ma, J.; et al. Add-on sirolimus for the treatment of mild or moderate systemic lupus erythematosus via T lymphocyte subsets balance. Lupus Sci. Med. 2024, 11, e001072. [Google Scholar] [CrossRef]
- Reis-Neto, E.T.; Seguro, L.P.; Sato, E.I.; Borba, E.F.; Klumb, E.M.; Costallat, L.T.; Medeiros, M.M.; Bonfá, E.; Araújo, N.C.; Appenzeller, S.; et al. II Brazilian Society of Rheumatology consensus for lupus nephritis diagnosis and treatment. Adv. Rheumatol. 2024, 64, 48. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.C.; Kim, H.W.; Chang, T.I.; Kang, E.W.; Lim, B.J.; Park, J.T.; Yoo, T.H.; Jeong, H.J.; Kang, S.W.; Han, S.H. Reduction in proteinuria after immunosuppressive therapy and long-term kidney outcomes in patients with immunoglobulin A nephropathy. Korean J. Intern. Med. 2021, 36, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Yung, S.; Chan, T.M. Mechanisms of Kidney Injury in Lupus Nephritis—The Role of Anti-dsDNA Antibodies. Front. Immunol. 2015, 6, 475. [Google Scholar] [CrossRef]
- Chan, J.; Walters, G.D.; Puri, P.; Jiang, S.H. Safety and efficacy of biological agents in the treatment of Systemic Lupus Erythematosus (SLE). BMC Rheumatol. 2023, 7, 37. [Google Scholar] [CrossRef]
- Zhang, F.; Lau, S.S.; Monks, T.J. The cytoprotective effect of N-acetyl-L-cysteine against ROS-induced cytotoxicity is independent of its ability to enhance glutathione synthesis. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 120, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y. N-acetylcysteine, reactive oxygen species and beyond. Cancer Biol. Ther. 2010, 9, 109–110. [Google Scholar] [CrossRef]
- Tenório, M.C.D.S.; Graciliano, N.G.; Moura, F.A.; Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef]
- Sahasrabudhe, S.A.; Terluk, M.R.; Kartha, R.V. N-acetylcysteine Pharmacology and Applications in Rare Diseases-Repurposing an Old Antioxidant. Antioxidants 2023, 12, 1316. [Google Scholar] [CrossRef]
- Doherty, E.; Oaks, Z.; Perl, A. Increased mitochondrial electron transport chain activity at complex I is regulated by N-acetylcysteine in lymphocytes of patients with systemic lupus erythematosus. Antioxid. Redox Signal. 2014, 21, 56–65. [Google Scholar] [CrossRef]
- Nasr, S.; Perl, A. Principles behind SLE treatment with N-acetylcysteine. Immunometabolism 2022, 4, e00010. [Google Scholar] [CrossRef]
- Schwalfenberg, G.K. N-Acetylcysteine: A Review of Clinical Usefulness (an Old Drug with New Tricks). J. Nutr. Metab. 2021, 2021, 9949453. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Liu, J.; Sun, L.; Meng, Z. The role of N-acetylcysteine in osteogenic microenvironment for bone tissue engineering. Front. Cell Dev. Biol. 2024, 12, 1435125. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme Q10 Analogues: Benefits and Challenges for Therapeutics. Antioxidants 2021, 10, 236. [Google Scholar] [CrossRef]
- Pallotti, F.; Bergamini, C.; Lamperti, C.; Fato, R. The Roles of Coenzyme Q in Disease: Direct and Indirect Involvement in Cellular Functions. Int. J. Mol. Sci. 2021, 23, 128. [Google Scholar] [CrossRef]
- Pham, T.; MacRae, C.L.; Broome, S.C.; D’Souza, R.F.; Narang, R.; Wang, H.W.; Mori, T.A.; Hickey, A.J.R.; Mitchell, C.J.; Merry, T.L. MitoQ and CoQ10 supplementation mildly suppresses skeletal muscle mitochondrial hydrogen peroxide levels without impacting mitochondrial function in middle-aged men. Eur. J. Appl. Physiol. 2020, 120, 1657–1669. [Google Scholar] [CrossRef]
- Lee, R.; Lee, W.Y.; Park, H.J. Anticancer Effects of Mitoquinone via Cell Cycle Arrest and Apoptosis in Canine Mammary Gland Tumor Cells. Int. J. Mol. Sci. 2024, 25, 4923. [Google Scholar] [CrossRef] [PubMed]
- López-Pedrera, C.; Villalba, J.M.; Patiño-Trives, A.M.; Luque-Tévar, M.; Barbarroja, N.; Aguirre, M.Á.; Escudero-Contreras, A.; Pérez-Sánchez, C. Therapeutic Potential and Immunomodulatory Role of Coenzyme Q10 and Its Analogues in Systemic Autoimmune Diseases. Antioxidants 2021, 10, 600. [Google Scholar] [CrossRef]
- Blanco, L.P.; Pedersen, H.L.; Wang, X.; Lightfoot, Y.L.; Seto, N.; Carmona-Rivera, C.; Yu, Z.X.; Hoffmann, V.; Yuen, P.S.T.; Kaplan, M.J. Improved Mitochondrial Metabolism and Reduced Inflammation Following Attenuation of Murine Lupus With Coenzyme Q10 Analog Idebenone. Arthritis Rheumatol. 2020, 72, 454–464. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Arenas-de Larriva, A.P.; Limia-Perez, L.; Romero-Cabrera, J.L.; Yubero-Serrano, E.M.; López-Miranda, J. Coenzyme Q10 Supplementation for the Reduction of Oxidative Stress: Clinical Implications in the Treatment of Chronic Diseases. Int. J. Mol. Sci. 2020, 21, 7870. [Google Scholar] [CrossRef]
- Mantle, D.; Hargreaves, I.P. Coenzyme Q10 and Autoimmune Disorders: An Overview. Int. J. Mol. Sci. 2024, 25, 4576. [Google Scholar] [CrossRef]
- Arnaud, L.; Parodis, I.; Devilliers, H.; Chasset, F. Clinical trial outcomes for SLE: What we have and what we need. Lupus Sci. Med. 2024, 11, e001114. [Google Scholar] [CrossRef] [PubMed]
- Touma, Z.; Gladman, D.D. Current and future therapies for SLE: Obstacles and recommendations for the development of novel treatments. Lupus Sci. Med. 2017, 4, e000239. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.K.X.; Heng, T.Y.J.; Mak, A. The Potential Use of Metformin, Dipyridamole, N-Acetylcysteine and Statins as Adjunctive Therapy for Systemic Lupus Erythematosus. Cells 2019, 8, 323. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Moret, Y.A.; Lo, K.B.; Tan, I.J. Metformin in Systemic Lupus Erythematosus: Investigating Cardiovascular Impact and Nephroprotective Effects in Lupus Nephritis. ACR Open Rheumatol. 2024, 6, 497–503. [Google Scholar] [CrossRef]
- Lee, S.Y.; Moon, S.J.; Kim, E.K.; Seo, H.B.; Yang, E.J.; Son, H.J.; Kim, J.K.; Min, J.K.; Park, S.H.; Cho, M.L. Metformin Suppresses Systemic Autoimmunity in Roquinsan/san Mice through Inhibiting B Cell Differentiation into Plasma Cells via Regulation of AMPK/mTOR/STAT3. J. Immunol. 2017, 198, 2661–2670. [Google Scholar] [CrossRef]
- Plowman, T.J.; Christensen, H.; Aiges, M.; Fernandez, E.; Shah, M.H.; Ramana, K.V. Anti-Inflammatory Potential of the Anti-Diabetic Drug Metformin in the Prevention of Inflammatory Complications and Infectious Diseases Including COVID-19: A Narrative Review. Int. J. Mol. Sci. 2024, 25, 5190. [Google Scholar] [CrossRef]
- Nojima, I.; Wada, J. Metformin and Its Immune-Mediated Effects in Various Diseases. Int. J. Mol. Sci. 2023, 24, 755. [Google Scholar] [CrossRef]
- Sun, F.; Geng, S.; Wang, H.; Wang, H.; Liu, Z.; Wang, X.; Li, T.; Wan, W.; Lu, L.; Teng, X.; et al. Effects of metformin on disease flares in patients with systemic lupus erythematosus: Post hoc analyses from two randomised trials. Lupus Sci. Med. 2020, 7, e000429. [Google Scholar] [CrossRef] [PubMed]
- Dima, A.; Jurcut, C.; Chasset, F.; Felten, R.; Arnaud, L. Hydroxychloroquine in systemic lupus erythematosus: Overview of current knowledge. Ther. Adv. Musculoskelet. Dis. 2022, 14, 1759720X211073001. [Google Scholar] [CrossRef]
- Basta, F.; Fasola, F.; Triantafyllias, K.; Schwarting, A. Systemic Lupus Erythematosus (SLE) Therapy: The Old and the New. Rheumatol. Ther. 2020, 7, 433–446. [Google Scholar] [CrossRef]
- Echavarria, R.; Cardona-Muñoz, E.G.; Ortiz-Lazareno, P.; Andrade-Sierra, J.; Gómez-Hermosillo, L.F.; Casillas-Moreno, J.; Campos-Bayardo, T.I.; Román-Rojas, D.; García-Sánchez, A.; Miranda-Díaz, A.G. The Role of the Oxidative State and Innate Immunity Mediated by TLR7 and TLR9 in Lupus Nephritis. Int. J. Mol. Sci. 2023, 24, 15234. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Liu, C.; Su, C.; Liu, L.; Chen, P.; Li, X.; Zhang, X.; Yuan, B.; Wang, H.; Du, X. Role of reactive oxygen species and mitochondrial damage in rheumatoid arthritis and targeted drugs. Front. Immunol. 2023, 14, 1107670. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Xue, L.; Xiao, J.; Li, P.; Xue, W.; Li, C.; Guo, H.; Chen, Y. The effect of the cyclic GMP-AMP synthase-stimulator of interferon genes signaling pathway on organ inflammatory injury and fibrosis. Front. Pharmacol. 2022, 13, 1033982. [Google Scholar] [CrossRef]
- Ben-Zvi, I.; Kivity, S.; Langevitz, P.; Shoenfeld, Y. Hydroxychloroquine: From malaria to autoimmunity. Clin. Rev. Allergy Immunol. 2012, 42, 145–153. [Google Scholar] [CrossRef]
- Niemann, B.; Puleo, A.; Stout, C.; Markel, J.; Boone, B.A. Biologic Functions of Hydroxychloroquine in Disease: From COVID-19 to Cancer. Pharmaceutics 2022, 14, 2551. [Google Scholar] [CrossRef]
- Brant, E.J.; Rietman, E.A.; Klement, G.L.; Cavaglia, M.; Tuszynski, J.A. Personalized therapy design for systemic lupus erythematosus based on the analysis of protein-protein interaction networks. PLoS ONE 2020, 15, e0226883. [Google Scholar] [CrossRef]
- Corbeel, L.; Freson, K. Rab proteins and Rab-associated proteins: Major actors in the mechanism of protein-trafficking disorders. Eur. J. Pediatr. 2008, 167, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, D.; Capili, A.; Choi, M.E. Mitochondrial dysfunction in kidney injury, inflammation, and disease: Potential therapeutic approaches. Kidney Res. Clin. Pract. 2020, 39, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Cravedi, P.; Remuzzi, G. Pathophysiology of proteinuria and its value as an outcome measure in chronic kidney disease. Br. J. Clin. Pharmacol. 2013, 76, 516–523. [Google Scholar] [CrossRef]
- Choi, M.Y.; Costenbader, K.H. Understanding the Concept of Pre-Clinical Autoimmunity: Prediction and Prevention of Systemic Lupus Erythematosus: Identifying Risk Factors and Developing Strategies Against Disease Development. Front. Immunol. 2022, 13, 890522. [Google Scholar] [CrossRef]
- Moulton, V.R.; Suarez-Fueyo, A.; Meidan, E.; Li, H.; Mizui, M.; Tsokos, G.C. Pathogenesis of Human Systemic Lupus Erythematosus: A Cellular Perspective. Trends Mol. Med. 2017, 23, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Won, K.C. Pioglitazone-induced AMPK-Glutaminase-1 prevents high glucose-induced pancreatic β-cell dysfunction by glutathione antioxidant system. Redox Biol. 2021, 45, 102029. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E.; Daniele, G.; Galindo, C.; Abdul-Ghani, M.A.; DeFronzo, R.A.; Norton, L. Pioglitazone inhibits mitochondrial pyruvate metabolism and glucose production in hepatocytes. FEBS J. 2017, 284, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhao, M.; Jia, S. Macrophage: Key player in the pathogenesis of autoimmune diseases. Front. Immunol. 2023, 14, 1080310. [Google Scholar] [CrossRef]
- Hasni, S.; Temesgen-Oyelakin, Y.; Davis, M.; Chu, J.; Poncio, E.; Naqi, M.; Gupta, S.; Wang, X.; Oliveira, C.; Claybaugh, D.; et al. Peroxisome proliferator activated receptor-γ agonist pioglitazone improves vascular and metabolic dysfunction in systemic lupus erythematosus. Ann. Rheum. Dis. 2022, 81, 1576–1584, Advance online publication. [Google Scholar] [CrossRef]
- Orasanu, G.; Ziouzenkova, O.; Devchand, P.R.; Nehra, V.; Hamdy, O.; Horton, E.S.; Plutzky, J. The peroxisome proliferator-activated receptor-gamma agonist pioglitazone represses inflammation in a peroxisome proliferator-activated receptor-alpha-dependent manner in vitro and in vivo in mice. J. Am. Coll. Cardiol. 2008, 52, 869–881. [Google Scholar] [CrossRef]
- Juárez-Rojas, J.G.; Medina-Urrutia, A.X.; Jorge-Galarza, E.; Caracas-Portilla, N.A.; Posadas-Sánchez, R.; Cardoso-Saldaña, G.C.; Goycochea-Robles, M.V.; Silveira, L.H.; Lino-Pérez, L.; Mas-Oliva, J.; et al. Pioglitazone improves the cardiovascular profile in patients with uncomplicated systemic lupus erythematosus: A double-blind randomized clinical trial. Lupus 2012, 21, 27–35. [Google Scholar] [CrossRef]
- Salemme, R.; Peralta, L.N.; Meka, S.H.; Pushpanathan, N.; Alexander, J.J. The Role of NETosis in Systemic Lupus Erythematosus. J. Cell. Immunol. 2019, 1, 33–42. [Google Scholar] [CrossRef]
- Pannu, N.; Bhatnagar, A. Oxidative stress and immune complexes: Pathogenic mechanisms in pristane induced murine model of lupus. Immunobiology 2020, 225, 151871. [Google Scholar] [CrossRef]
- Liao, P.; He, Y.; Yang, F.; Luo, G.; Zhuang, J.; Zhai, Z.; Zhuang, L.; Lin, Z.; Zheng, J.; Sun, E. Polydatin effectively attenuates disease activity in lupus-prone mouse models by blocking ROS-mediated NET formation. Arthritis Res. Ther. 2018, 20, 254. [Google Scholar] [CrossRef]
- Tian, Y.; Guo, H.; Miao, X.; Xu, J.; Yang, R.; Zhao, L.; Liu, J.; Yang, L.; Gao, F.; Zhang, W.; et al. Nestin protects podocyte from injury in lupus nephritis by mitophagy and oxidative stress. Cell Death Dis. 2020, 11, 319. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Chen, J.; Yang, H.; You, L.; Xu, L.; Wang, X.; Li, R.; Gao, L.; Gu, Y.; Lin, S.; et al. Expression of nestin in the podocytes of normal and diseased human kidneys. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1761–R1767. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Klionsky, D.J.; Zhang, H. Podocytes and autophagy: A potential therapeutic target in lupus nephritis. Autophagy 2019, 15, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Halfon, M.; Tankeu, A.T.; Ribi, C. Mitochondrial Dysfunction in Systemic Lupus Erythematosus with a Focus on Lupus Nephritis. Int. J. Mol. Sci. 2024, 25, 6162. [Google Scholar] [CrossRef]

{kind=link}
| Main Process | Mechanism/Process | Implications | Therapeutic Strategies |
|---|---|---|---|
| Reduction–Oxidation Alterations | Elevated mitochondrial reactive oxygen species (ROS) production | Impairment leads to cellular injury and release of mtDNA; perpetuates oxidative stress (OS) | Anti-oxidant therapies (e.g., NAC), targeting ROS production |
| Genetic Factors | Mitochondrial single nucleotide polymorphisms (SNPs) | Genetic susceptibilities to SLE; accumulation of autoantibodies and immune complexes (ICs) | Gene-targeted therapies; modulating mitochondrial functions |
| Mitochondrial Dysfunction in Immune Cells | Impaired mitophagy and increased mtROS in T-lymphocytes | Alters T-cell-signaling pathways; leads to T-lymphocyte exhaustion and autoantigen expression | mTORC1 inhibitors (e.g., rapamycin), Metformin |
| Innate Immune Response | Increased interferon alpha from activated immune complexes | Drives autoreactivity and inflammation; promotes autoantibody generation | Targeting interferon pathways; restoring mitophagy |
| NETosis and Inflammation | Mitochondrial DNA (mtDNA) release linked to NET formation | Contributes to inflammation via immune complex formation and exacerbates disease pathology | Scavenging agents for mtROS; controlling NET formation |
| Inflammasome Activation | Cryopyrin inflammasome linked to ROS and mtDNA release | Activates proinflammatory cytokines; exacerbates SLE progression | Targeting inflammasome pathways; enhancing mitophagy |
| B-Lymphocyte Dysfunction | mTORC1 activation linked to excessive autoantibody production | Drives differentiation of B-cells into plasmablasts, furthering autoimmune response | Inhibiting mTORC1; Metformin to regulate B-cell activity |
| Calcium (Ca2+) Dysregulation | Altered calcium efflux in T-lymphocytes | Disrupts normal immune signaling and function, contributing to T-cell dysregulation | Targeting pathways that regulate Ca2+ homeostasis |
| Immune Cell Type | Mitochondrial Dysfunction Mechanism | Impact on Immune Function | Implications for SLE Pathogenesis |
|---|---|---|---|
| T-lymphocytes | Increased ROS production, impaired mitophagy | T-cell exhaustion, skewed immune response | Increased autoreactivity, disease exacerbation |
| B-lymphocytes | mTORC1 activation linked to excessive autoantibody production | Enhanced differentiation into plasmablasts | Amplified humoral response |
| Neutrophils | Impaired NETosis, excessive ROS generation | Dysfunctional clearance of pathogens, persistent inflammation | Sustained inflammatory response, tissue damage |
| Plasmacytoid dendritic cells | Increased interferon-alpha production linked to mitochondrial stress | Heightened type I interferon response and autoimmunity | Promotion of inflammation, anti-dsDNA autoantibody production |
| Therapy | Mechanism of Action | Demonstrated Effects |
|---|---|---|
| Sirolimus (Rapamycin) | m-TOR suppression, reducing T-lymphocyte function | Improved mitochondrial function; decreased protein levels in urine; enhanced renal function; reduced anti-ds-DNA antibody titers |
| N-acetylcysteine (NAC) | GSH precursor; anti-oxidant and ROS removal; m-TORC-1 suppression | Reduced anti-double stranded DNA antibody levels; alleviated SLE nephritis symptoms; decreased lupus activity |
| Coenzyme Q10 (CoQ10) | Anti-oxidant; neutralizes free radicals; facilitates electron transport | Decreased mitochondrial ROS production; reduced NETs activation and interferon alpha generation; improved immune response |
| Metformin | Normalizes redox balance; AMPK–m-TOR–STAT-3 pathway modulation | Decreased ROS generation; reduced mitochondrial DNA leakage; ameliorated SLE conditions; lowered corticosteroid dependence |
| Hydroxychloroquine (HCQ) | Targets mitochondrial anti-oxidant systems; immunomodulator | Decreased proliferation of T-helper cells; inhibited proinflammatory cytokines; potentially improved SLE symptoms |
| 3PEHPC | Suppresses Rab-protein geranylgeranyltransferase | Improved mitochondrial dynamics; decreased anti-nuclear antibody production; alleviated nephritis symptoms |
| Pioglitazone | Binds to mitochondrial electron transport chain complex | Inhibited autoimmune response; improved nephritis symptoms; promoted T-reg activity |
| Piceid | Suppresses ROS-mediated NET formation | Alleviated SLE symptoms in murine models |
| Nestin | Modulates mitochondrial autophagy and oxidative stress | Protects podocytes from injury; potentially alleviates symptoms associated with SLE nephritis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poznyak, A.V.; Orekhov, N.A.; Churov, A.V.; Starodubtseva, I.A.; Beloyartsev, D.F.; Kovyanova, T.I.; Sukhorukov, V.N.; Orekhov, A.N. Mitochondrial Dysfunction in Systemic Lupus Erythematosus: Insights and Therapeutic Potential. Diseases 2024, 12, 226. https://doi.org/10.3390/diseases12090226
Poznyak AV, Orekhov NA, Churov AV, Starodubtseva IA, Beloyartsev DF, Kovyanova TI, Sukhorukov VN, Orekhov AN. Mitochondrial Dysfunction in Systemic Lupus Erythematosus: Insights and Therapeutic Potential. Diseases. 2024; 12(9):226. https://doi.org/10.3390/diseases12090226
Chicago/Turabian StylePoznyak, Anastasia V., Nikolay A. Orekhov, Alexey V. Churov, Irina A. Starodubtseva, Dmitry F. Beloyartsev, Tatiana I. Kovyanova, Vasily N. Sukhorukov, and Alexander N. Orekhov. 2024. "Mitochondrial Dysfunction in Systemic Lupus Erythematosus: Insights and Therapeutic Potential" Diseases 12, no. 9: 226. https://doi.org/10.3390/diseases12090226
APA StylePoznyak, A. V., Orekhov, N. A., Churov, A. V., Starodubtseva, I. A., Beloyartsev, D. F., Kovyanova, T. I., Sukhorukov, V. N., & Orekhov, A. N. (2024). Mitochondrial Dysfunction in Systemic Lupus Erythematosus: Insights and Therapeutic Potential. Diseases, 12(9), 226. https://doi.org/10.3390/diseases12090226