1. Introduction
Inflammation is a response to injury caused by harmful physical or chemical stimuli or microbiological toxins, and occurs in numerous pathologies, such as asthma, arthritis, multiple sclerosis, atherosclerosis, and inflammatory bowel diseases [
1,
2]. There are three types of mammalian nitric oxide synthase (NOS)—neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). Of these, the iNOS is involved in inflammatory reactions [
3]. Nitric oxide (NO) generated by the iNOS isoform is necessary for the innate immune and inflammatory responses of hosts to various pathogens and microorganisms [
4,
5]. However, excessive production of NO is regarded as a pro-inflammatory mediator that induces inflammation in abnormal physiological situations [
6]. Therefore, therapeutic agents that inhibit iNOS may effectively ameliorate these inflammatory conditions. Lipopolysaccharide (LPS)-induced gene products include pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF)-α and interleukin-6 (IL-6), and adhesion enzymes, such as iNOS and cyclooxygenase-2 (COX)-2 [
7]. COX enzymes are known to produce prostaglandins (PGs) that are involved in many physiological events, such as the progression of inflammation, modulation of the inflammatory response, and transmission of pain [
8]. COX-1 is typically expressed in most tissues, which is expected given that COX-1 maintains housekeeping physiological functions. In contrast, COX-2 is only expressed in some tissues and is transiently induced by growth factors, pro-inflammatory cytokines, tumor promoters, and bacterial toxins [
9]. Nuclear factor-kappa B (NF-κB) is a DNA transcription factor that plays a vital role in the expression of various genes involved in the inflammatory response [
10,
11,
12]. Inactive NF-κB is bound to its inhibitory subunit IκB, and the complexes are sequestered in the cytoplasm. Upon stimulation, IκB proteins are phosphorylated, ubiquitinated, and degraded, allowing NF-κB to translocate to the nucleus, where it can bind to specific DNA sequences located in the promoter regions of target genes, thereby activating gene transcription [
13,
14]. Recently, many studies demonstrated the role of phytochemicals in anti-inflammatory activity via NF-κB pathway down-regulation [
15]. Mitogen-activated protein kinases (MAPKs) have essential functions in the regulation of cell differentiation, cell growth, and cellular responses to cytokines. The MAPK cascades are critical signaling pathways in the immune system and include p38 MAPK, extracellular signal-regulated kinase (ERK), and c-jun N-terminal kinase (JNK) [
16]. One of the main functions of MAPK is the activation of transcription factors, some of which increase the expression of pro-inflammatory cytokines [
17]. Cytokine-mediated host defense mechanisms induce significant cell and organ injury and play crucial roles in the pathophysiology of sepsis [
18].
Sanguisorbae Radix (SR), the dried root of
Sanguisorba officinalis L., has hemostatic, analgesic, and astringent properties and has been used in traditional Chinese medicine for the treatment of burns, scalds, inflammation, and internal hemorrhage [
19,
20,
21]. SR is also used to control bloody pus, treat boils, repair damaged tissue, relieve alcohol poisoning, quench one’s thirst, and relieve eye pain [
22]. Until now, saponin components such as triterpenes and their glycosides (e.g., ziyuglycoside I) and disaccharide (5-O-alpha-D-(3-C-hydroxymethyl)lyxofuranosyl-beta-D-(2-C-hydroxymethyl) arabino furanose) have been reported as the major active compounds of SR that confer these in vitro and in vivo pharmacological effects [
22,
23,
24,
25]. Although numerous activities of this plant have been reported and published to date, research on the safety of SR has been limited. Zebrafish are economical with regards to maintenance because it requires simpler breeding facilities, and its feed is less expensive than that of other model species. It produces 200–300 embryos with single external fertilization, which makes it suitable for toxicity assessment [
26]. Thus, we used the zebrafish (
Danio rerio) embryo model to investigate the potential anti-inflammatory mechanism of SR against LPS-induced inflammatory response in macrophages, to evaluate the root as a potential safe cosmetic material.
2. Materials and Methods
2.1. Materials and Reagents
RAW264.7 cells were obtained from the Korean Cell Line Bank (KCLB, Seoul, Korea). Dulbecco’s Modified Eagle <edium (DMEM), fetal bovine serum (FBS), and penicillin/streptomycin were purchased from Gibco BRL Co. (Grand Island, NY, USA). Lipopolysaccharide (LPS), 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT), acrylamide, and N,N’-bis-methylene-acrylamide were purchased from Sigma-Aldrich Corp. (St, Louis, MO, USA). The iNOS, COX-2, IKK, p-IKK, IκB, p-IκB, ERK, p-ERK, p-38, p-p-38, JNK, p-JNK, and β-actin monoclonal antibodies and the secondary antibody were purchased from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA, USA). Prostaglandin (PGE2) and TNF-α for ELISA Kits were purchased from R&D Systems Inc. (Minneapolis, MN, USA). Halt™ Protease Inhibitor Cocktail Kits and BSA kits were purchased from Thermo Fisher Scientific (Rockford, IL, USA). GoScript™ Reverse Transcriptase and GoTaq® Flexi DNA Polymerase were purchased from Promega Corporation (Madison, WI, USA).
2.2. Sample Preparation
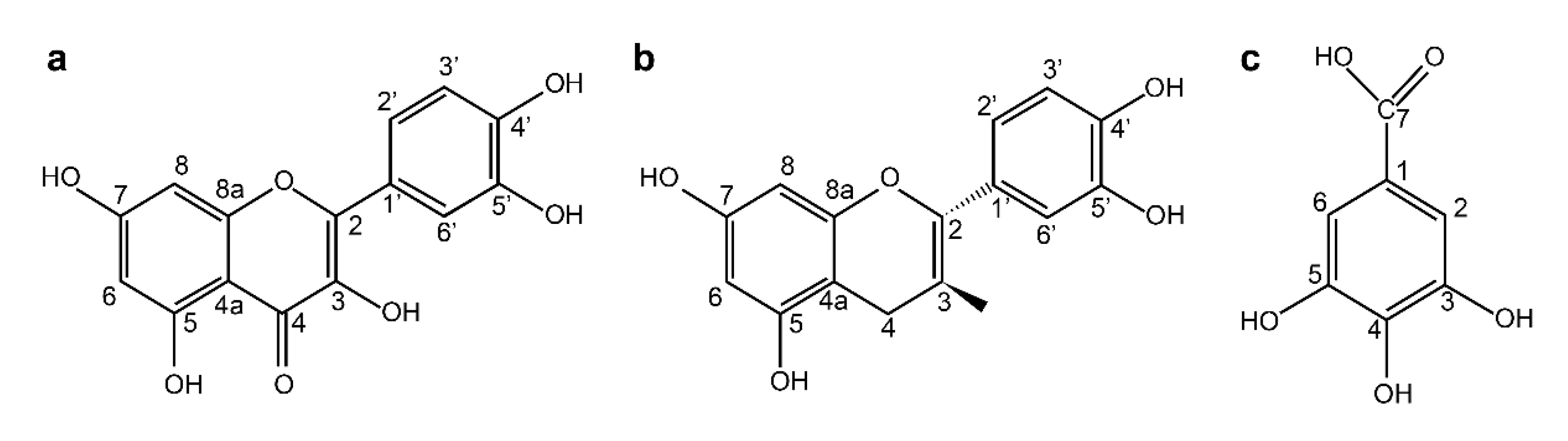
SR was obtained from Andong City, Kyung-Buk, Korea. The powdered roots of SR were extracted with 70% acetone at 20 °C for 3 days; the solution was filtered using a filter paper (Whatman No. 2, Tokyo, Japan) and the obtained acetone extract was concentrated under reduced pressure. The acetone extract was then dissolved in chloroform in a separatory funnel, and further fractionated by successive solvent extraction with ethyl acetate and n-butanol saturated with H2O. The n-butanol fraction was fractionated by Diaion HP-20 column chromatography, with stepwise elution of H2O–MeOH (100:1–1:100) to obtain separated active fractions. To determine the structure of the compounds obtained, 1H- and 13C-NMR were measured, and the structure of individual substances was determined Quercetin (1). Yellow amorphous powder 1H-NMR (500 MHz, MeOH-d6): δ 7.79 (1H, d, J = 2.3 Hz, H-2′), 7.69 (1H, dd, J = 2.3, 8.5 Hz, H-6′), 6.99 (1H, d, J = 8.5 Hz, H-5′) 6.55 (1H, d, J = 2.0 Hz, H-6) 6.25 (1H, d, J = 2.0 Hz, H-8). 13C-NMR (125 MHz, MeOH-d6): δ 175.9 (C-4), 163.9 (C-7), 160.8 (C-5′), 156.2 (C-9), 147.7 (C-4′), 146.8 (C-2), 145.1 (C-3′), 135.8 (C-3), 121.9 (C-1′), 120.0 (C-6′), 115.6 (C-5′), 115.1 (C-2′), 103.1 (C-10), 98.2 (C-6), 93.4 (C-8). (+)-Catechin (2). Brown amorphous powder 1H-NMR (500 MHz, MeOH-d6): δ 6.84 (1H, dd, J = 8.0, 2.0 Hz, H-6′), 6.76 (1H, dd, J = 2.0 Hz, H-2′), 6.75 (1H, d, J = 8.0 Hz, H-5′), 5.93 (1H, d, J = 2.0 Hz, H-6), 5.86 (1H, d, J = 2.0 Hz, H-8), 4.57 (1H, d, J = 7.5 Hz, H-2), 3.98 (1H, m, H-3), 2.85 (1H, dd, J = 16.0, 5.5 Hz, H-4ax), 2.50 (1H, dd, J = 16.0, 5.5 Hz, H-4eq). 13C-NMR (125 MHz, MeOH-d6): δ 157.9 (C-7), 157.6 (C-5), 157.0 (C-9), 146.3 (C-3′), 146.3 (C-4′), 132.3 (C-1′), 120.1 (C-6′) 116.1 (C-5′), 115.3 (C-2′), 100.9 (C-10), 96.3 (C-6), 95.9 (C-8), 83.0 (C-2), 68.9 (C-3), 28.6 (C-4). Gallic acid (3). White amorphous powder 1H-NMR (500 MHz, MeOH-d6): δ 7.15 (2H, s, H-2, 6). 13C-NMR (125 MHz, MeOH-d6): δ 169.1 (C-7), 145.7 (C-3, 5), 138.7 (C-4), 121.4 (C-1), 109.9(C-2, 6). In this study, three compounds—quercetin (QC), (+)- catechin (CC), and gallic acid (GA)—obtained from SR were used. These compounds were stored in a refrigerator at 4 °C until use.
2.3. MTT Assay
Cells were uniformly dispensed in a 96-well plate (0.18 mL) at a density of 5 × 103 cells/well. QC, CC, and GA were prepared at concentrations of 5, 10, 25, 50, 75, and 100 μg/mL, and 0.02 mL of the solution was added. Then, the cells were cultured in a 5% CO2 incubator at 37 °C for 24 h. Next, 0.02 mL MTT solution prepared at a concentration of 5 mg/mL was added, and the mixture was cultured for 4 h. Following incubation, the culture solution was removed, and 0.15 mL DMSO was added to each well. After incubation at room temperature for 15 min, absorbance was measured at 540 nm with a Microplate Reader (Winooski, VT, USA). Cell viability was calculated as a percentage of control absorbance.
2.4. Measurement of NO, PGE2, and TNF-α
Cells were plated at a density of 2 × 105 cells/well in a 24-well plate. Cells were induced with LPS (1 μg/mL) and treated with either the control, QC, CC, or GA at concentrations of 10, 25, and 50 μg/mL and cultured. The supernatant was collected, and the inhibitory effect of samples on NO, PGE2, and TNF-α cytokine production was determined using the Griess reagent enzyme and ELISA kit (Minneapolis, MN, USA), according to the manufacturer’s instructions.
2.5. Western Blotting
RAW264.7 cells were uniformly dispensed in a 6-well plate at a density of 1 × 105 cells/well and cultured for 24 h. Then, cells were induced with LPS (1 μg/mL) and treated with the control or QC, CC, or GA at concentrations of 10, 25, and 50 μg/mL and cultured. Protein lysates were obtained by incubating the culture with RIPA lysis buffer (Pierce, IL, USA) for 1 h, followed by centrifugation at 12,000 rpm for 30 min at 4 °C. Then, proteins were quantified by Bradford assay (Hercules, CA, USA) according to the manufacturer’s instruction. Equal proteins were electrophoresed using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a PVDF membrane (Amersham Pharmacia Biotech, Buckinghamshire, UK) at 60 V for 3 h. After blocking with tris buffer solution containing 5% skim milk for 1 h, the membrane was incubated with dilution of polyclonal antibodies for iNOS, COX-2, IKK, p-IKK, IκB, p-IκB, ERK, p-ERK, p-38, p-p-38, JNK, p-JNK, and β-actin. After extensive washes, the membrane was incubated with the secondary antibody (1:1000 dilution) for 1 h at room temperature. Next, the cells were reacted with Immobilon Western Chemiluminescent HRP substrate (Millipore, MA, USA) and protein levels were analyzed using EZ-Capture MG (ATTO Corporation, Tokyo, Japan).
2.6. RNA Isolation and RT-PCR
Total RNA was isolated using the TRIsolution™ Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA), according to the manufacturer’s instructions. One milliliter of Trizol reagent was added to the cells. After the cells were dissolved and kept at room temperature for 5 min, 0.2 mL of chloroform was added and mixed well. The cells were then centrifuged at 12,000× g for 10 min. The isolated RNA was dissolved in distilled water containing diethyl pyrocarbonate and the purity of RNA was measured using the ELISA reader. Then, the concentration of RNA was calculated and used for the experiment. GoScript™ Reverse Transcriptase was used for cDNA synthesis, and PCR was performed using GoTaq® Flexi DNA Polymerase and specific primers. The amplified cDNA was separated by electrophoresis on a 1.5% agarose gel and visualized by the electrophoresis EZ-Capture MG image analyzer.
2.7. Zebrafish Breeding Conditions and Embryo Acquisition
Zebrafish were obtained from the Division of Biomedical & Cosmetics, Mokwon University, Korea, and were used during the breeding period. All experimental protocols were approved and conducted according to the guidelines and regulations of the Animal Ethics Committee of Mokwon University. Wild-type zebrafish were provided by the Zebrafish Center for Disease Modeling, Korea. The rearing condition was kept constant at 28 ± 1 °C with a 16 h/8 h light/dark cycle (Rotifer continuous culture system, GDBC, Korea). They were fed brine shrimp (Ocean Star International, Utah, USA). Female and male fish with at least five spawning experiences were fed adequate feed the day before spawning; 24 h later, irradiation of light was used to produce embryos. The produced embryos were incubated at 28 ± 1 °C for 2 days in pre-manufactured egg water (sea salt solution, Aquarium Systems, France) to avoid exceeding a maximum of 50 eggs in a 100-mm petri dish, and the egg water was replaced periodically.
Figure 1 shows the Zebrafish model.
2.8. Zebra Fish Embryo Toxicity Test
Mature male and female zebrafish were separated in the tank for egg collection. After 24 h, they were put together, and eggs were collected. The culture solution was used as a control. To each well of the 24-well plate, 3 zebrafish eggs were added and treated with different concentrations of QC: 10, 25, and 50 μL/mL. The plate was then placed in the incubator at 28 °C, and the toxicity level of the samples treated with QC was analyzed for coagulation rate and hatching rate at 24, 48, and 72 h after the QC treatment and compared with control.
2.9. Statistical Analysis
Statistical analysis was performed using SPSS 12.0, and differences were considered significant when p < 0.05, 0.01 after a t-test using analysis of variance (ANOVA).
3. Results and Discussion
Inflammation is a defensive pathway primarily caused by the action of phagocytic cells, such as macrophages and dendritic cells, which work to protect the body from exogenous pathogens [
27]. The innate immune response is part of an appropriate inflammatory reaction that contributes to the development of adaptive immunity. However, excessive and inappropriate inflammatory reactions cause cell necrosis and various chronic diseases [
28]. Skin-related diseases are often accompanied by inflammation, which causes aggravation, such as acne and atopy. Studies are being actively conducted to identify natural materials that can alleviate the inflammatory reaction [
29]. Herein, we identified three compounds from SR extracts (
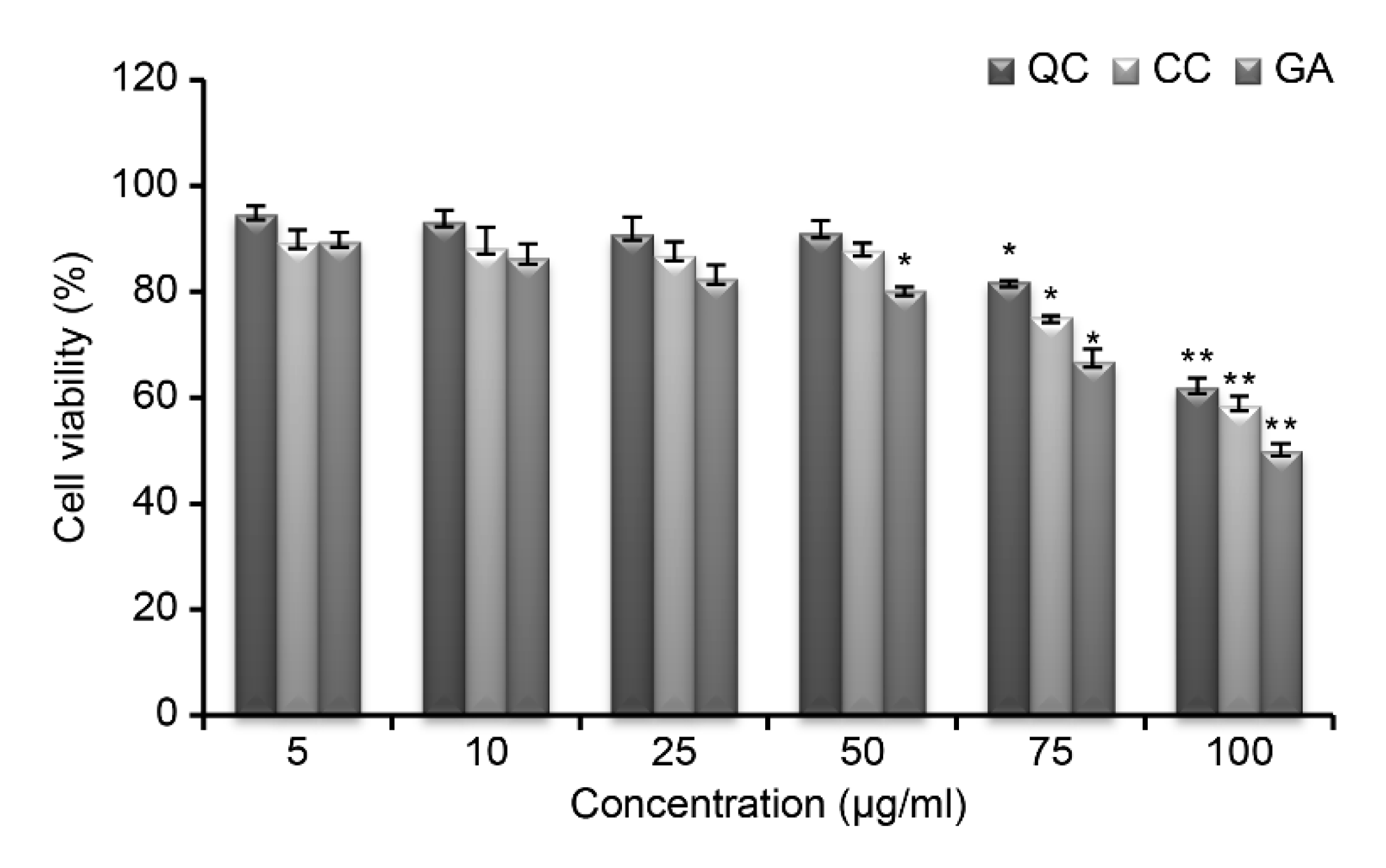
Figure 2) and then evaluated the toxicity of SR compounds (QC, CC, and GA) at different concentrations against RAW264.7 cells (
Figure 3); the cells showed a viability of >85% at all concentrations below 50 μg/mL. Thus, these compounds were considered non-toxic, and further experiments were conducted using a concentration of 50 μg/mL.
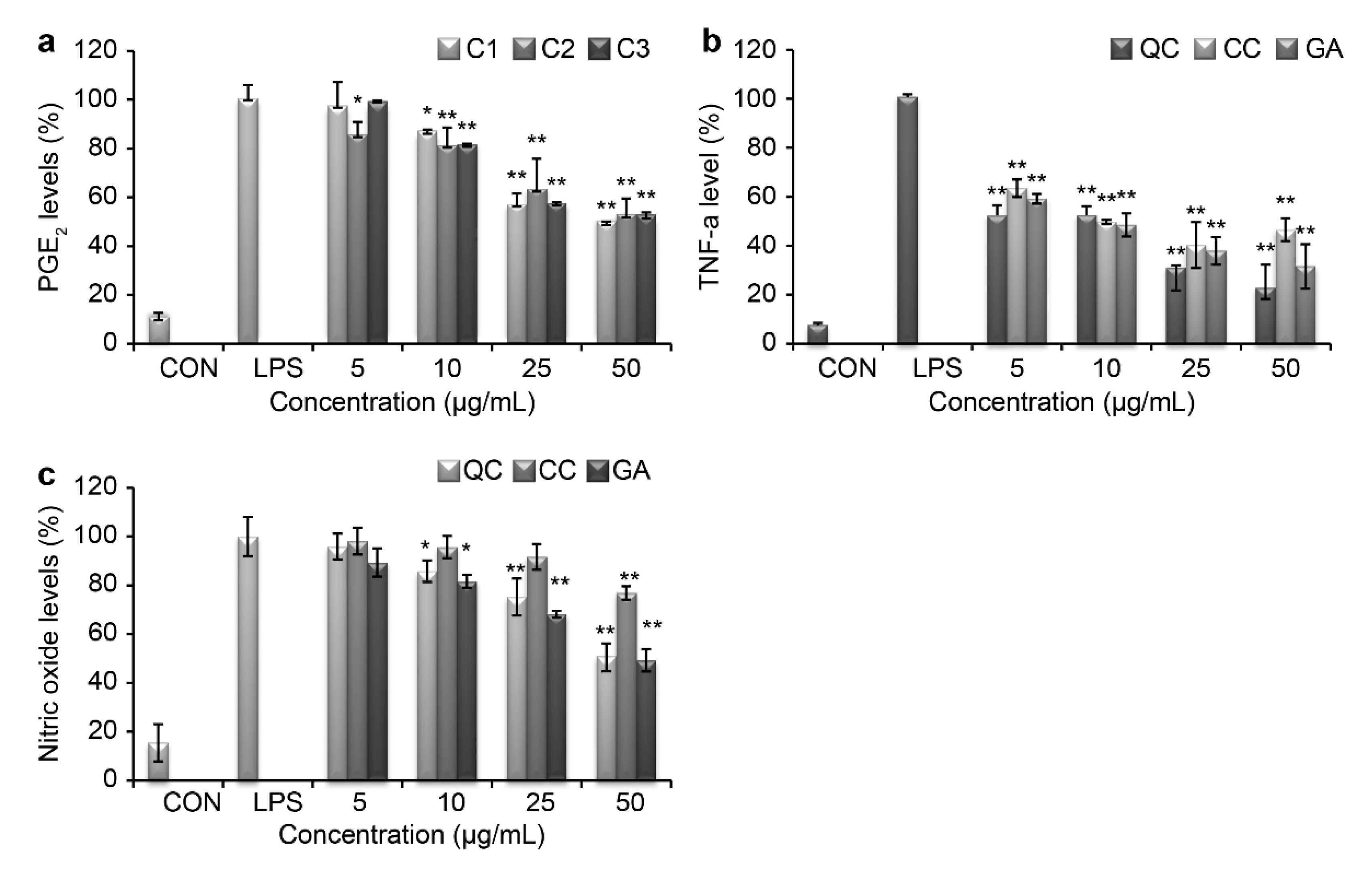
Several pro-inflammatory cytokines, such as TNF-α, PGE
2, and IL-8, promote systemic inflammation. We measured the production levels of TNF-α and PGE
2 using an ELISA Kit and examined the production of NO using the Griess reagent. As shown in
Figure 4a,b, SR markedly blocked the production of these inflammatory mediators in LPS-treated RAW264.7 cells. At 50 µg/mL of QC, PGE
2 levels decreased by 38.8% and TNF-α levels decreased by 21.9%, which was the highest cytokine inhibition activity observed. TNF-α and PGE
2 production were also suppressed by CC and GA treatments in a dose-dependent manner. Additionally, QC was effective at suppressing the release of NO from LPS-induced peritoneal macrophages (
Figure 4c).
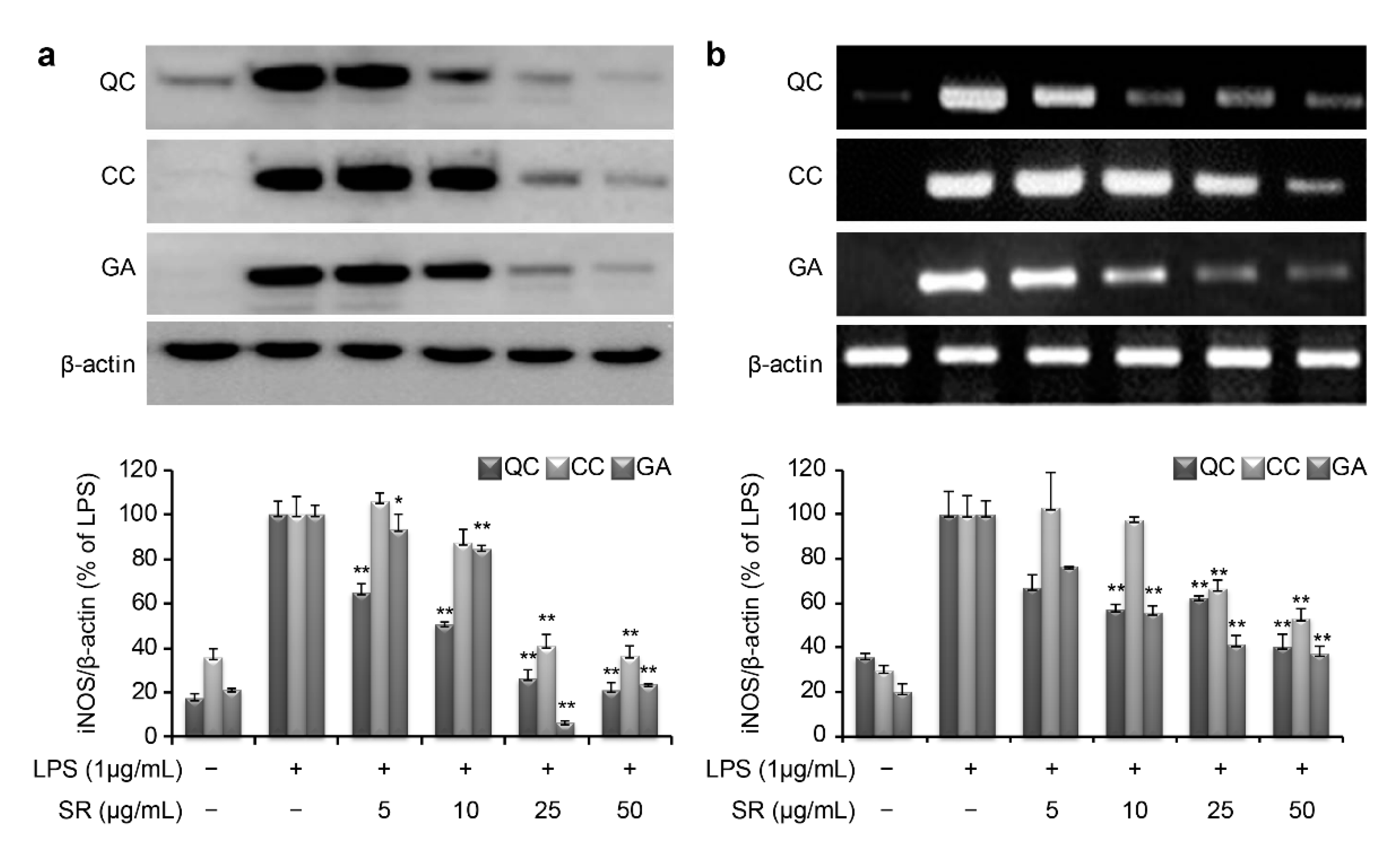
To investigate the molecular mechanism through which these compounds suppress inflammation, we measured the mRNA and protein expression levels of iNOS and COX-2. LPS stimulation of RAW264.7 cells markedly increased iNOS and COX-2 production compared with that in the negative control. As shown in
Figure 5a, the protein expression level of iNOS was found to be 20.7%, 36.2%, and 23.8% upon treatment with 50 μg/mL QC, CC, and GA, respectively. Similarly, the mRNA expression level of iNOS was found to be 40.6%, 53.3%, and 37.3% after treatment with 50 μg/mL of QC, CC, and GA, respectively (
Figure 5b). Thus, treatment with the SR compounds markedly inhibited the LPS-mediated induction of iNOS mRNA and protein expression.
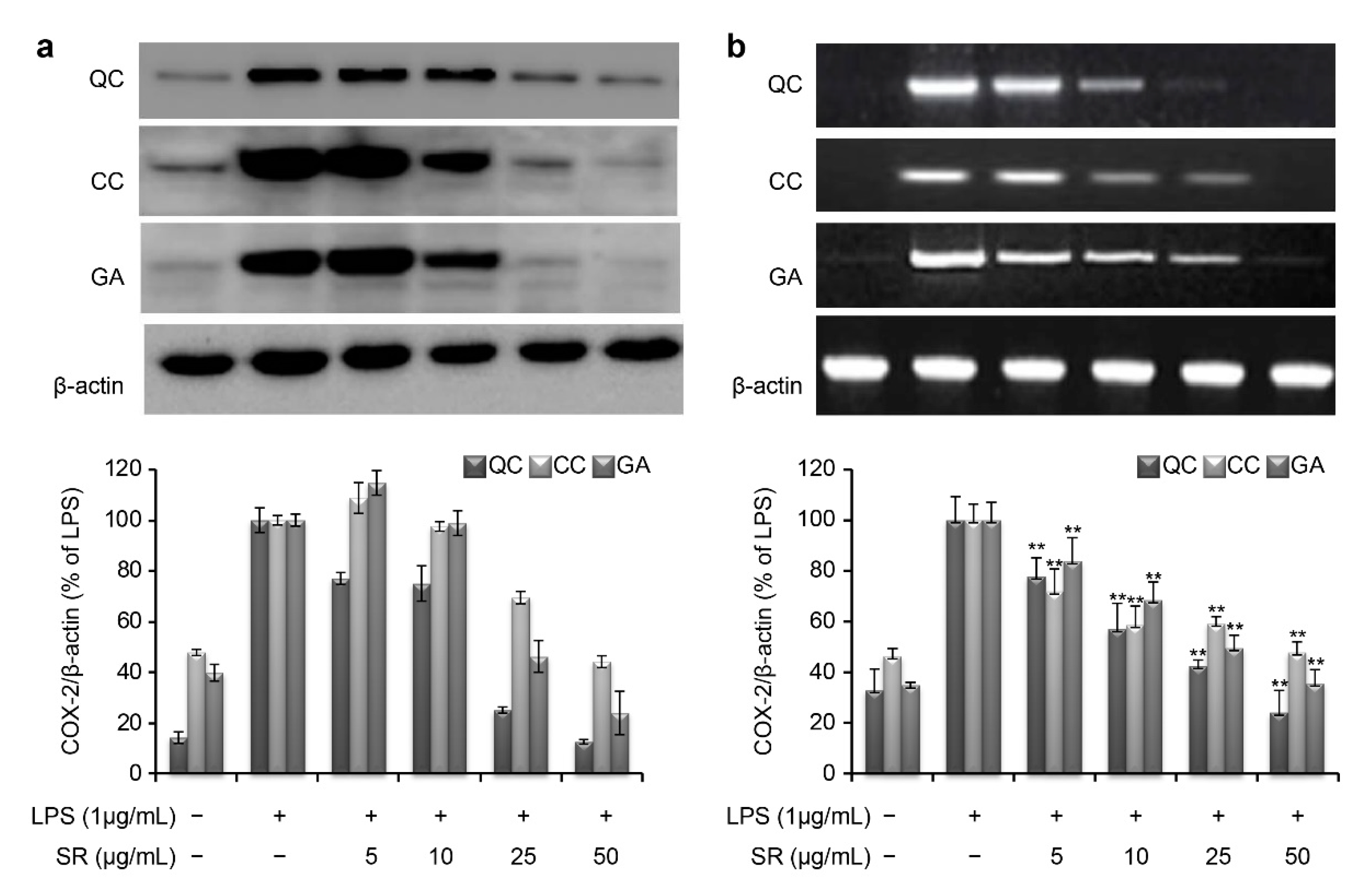
Similar to iNOS, the COX-2 mRNA and protein levels were markedly induced by LPS stimulation (
Figure 6). However, the protein expression level of COX-2 was reduced to 12.4%, 44.1%, and 23.8% upon simultaneous treatment with 50 μg/mL QC, CC, and GA, respectively (
Figure 6a). Consistent with this, the corresponding COX-2 mRNA expression level was reduced to 23.9%, 47.8%, and 35.5% after treatment with 50 μg/mL QC, CC, and GA, respectively (
Figure 6b).
Thus, LPS-stimulated macrophages show the rapid induction of genes responsible for production of pro-inflammatory cytokines, such as TNF-α and PGE2, and inflammatory mediators, such as NO. Our data demonstrate that SR markedly inhibited or suppressed these LPS-induced pro-inflammatory effects in RAW264.7 cells at both the mRNA and protein levels.
A previous study reported that LPS increased activation of NF-κB and regulated the expression of iNOS, COX-2, and other cytokines [
30]; thus, NF-κB plays an important role in inflammation. NF-κB binds to inhibitory κBα (IκBα) molecules in the promoter region of an inflammatory response and only becomes active after IκBα is phosphorylated and subsequently degraded; this modification of IκBα occurs at serine 32 and 36 by the IκB kinase (IKK) complex [
31]. Thus, we examined the effects of SR on phosphorylation of IκBα and IKK in LPS-induced cells. As a result, treatment with QC, CC, or GA attenuated the phosphorylation of IκBα and IKK in a dose-dependent manner (
Figure 7).
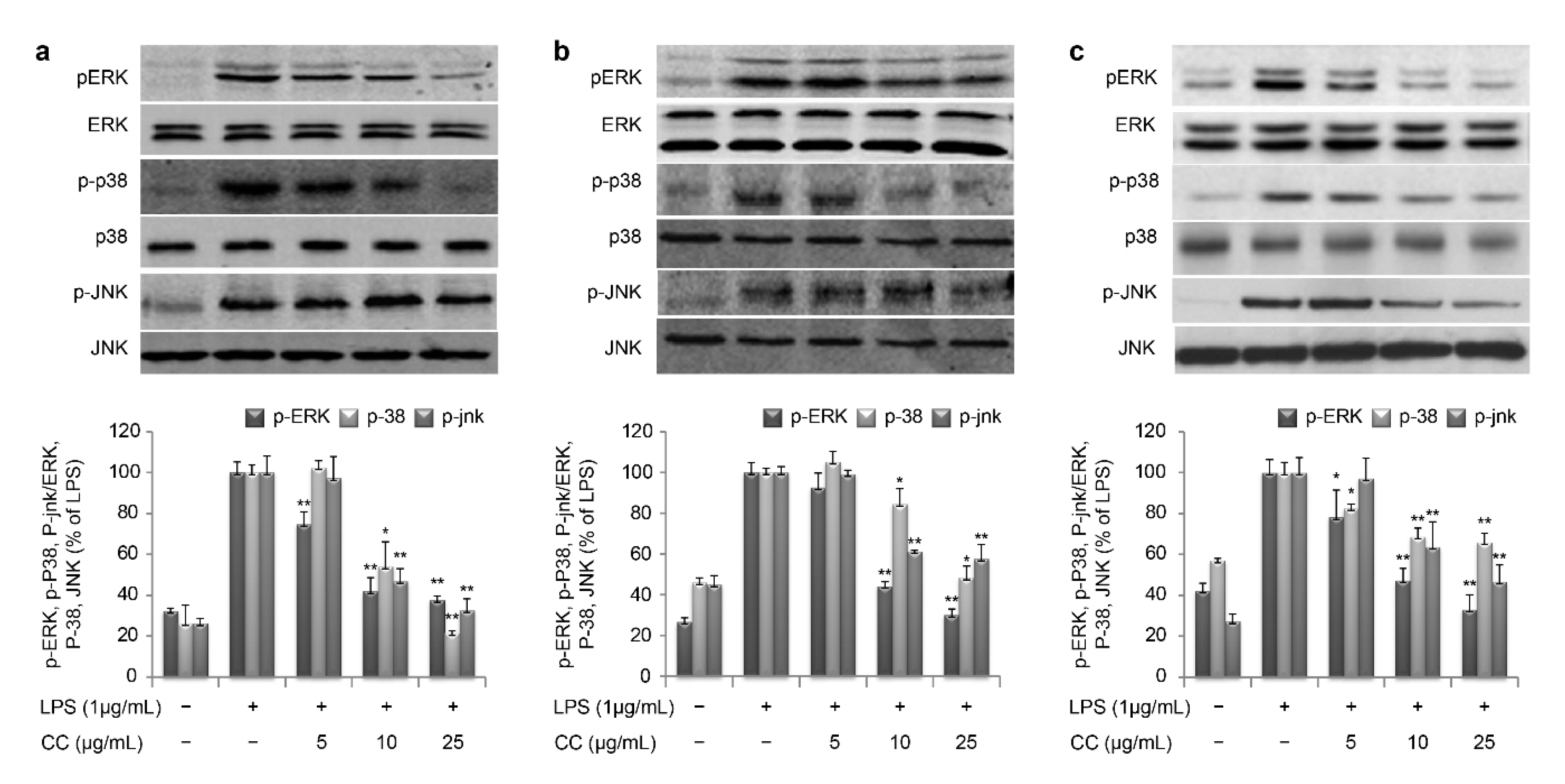
The MAPK family consists of ERK, p38, and JNK [
32]. In our study, SR effectively suppressed the phosphorylation of ERK, p38, and JNK, which is a critical event in the MAPK signal transduction pathway (
Figure 8). These data suggest that SR inhibits the production of inflammation by regulating gene expression involved in the NF-κB and MAPK-mediated inflammatory process.
To examine the toxicity of QC, zebrafish embryos were treated with 0, 10, 25, and 50 ppm of QC 12 h post-fertilization, and the coagulation rate and hatching rate of embryos were monitored for 72 h (
Figure 9a,b). In the control group, the embryos were treated with egg water 24 and 48 h post-fertilization, and the coagulation rate was 0%. In contrast, the coagulation rate of the group treated with 50 ppm QC for 72 h was 33%. The hatching rate of the embryos from the control group after 48 h was 100%, while that of the embryos treated with 10, 25, and 50 ppm of QC was 58%, 41%, and 33%, respectively, indicating a delay in hatching. According to the results, QC qualified as a safe material at a concentration of 50 μg/ml in the safety experiment using the zebrafish model. Nonetheless, the hatching rate was 100% after 72 h in these treatment groups, indicating that the QC extract is safe. Moreover, zebrafish embryos treated with 10 ppm and 25 ppm of QC did not coagulate and underwent normal embryogenesis, similar to that in the control group.
Although analysis of the development of zebrafish, when exposed to different concentrations of QC, showed no morphological abnormality at 10 ppm and 25 ppm, yolk swelling and melanin loss were observed at 50 ppm (
Figure 9c).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








