
A Model for the Evaluation of Monostable Molecule Signal Energy in Molecular Field-Coupled Nanocomputing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Theoretical Methods
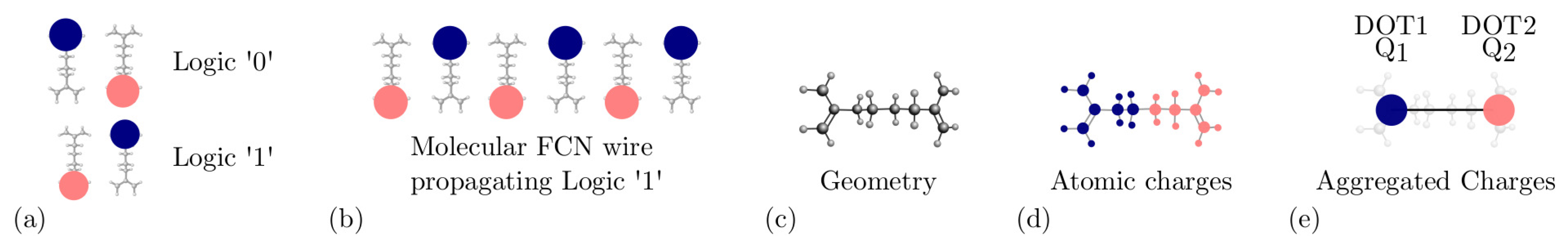
2.1. MoSQuiTo Methodology
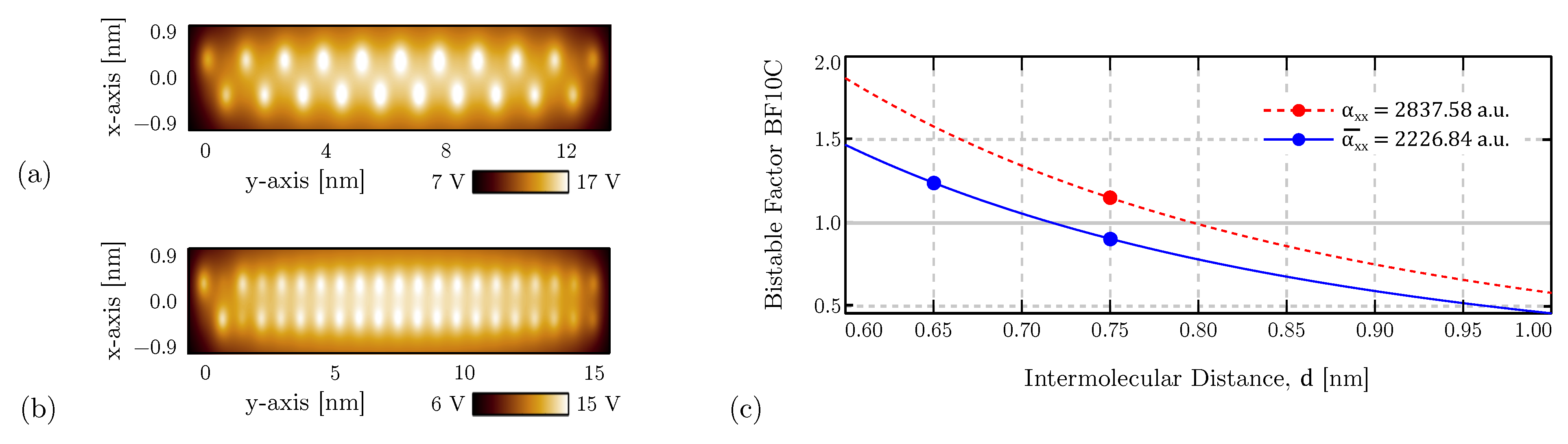
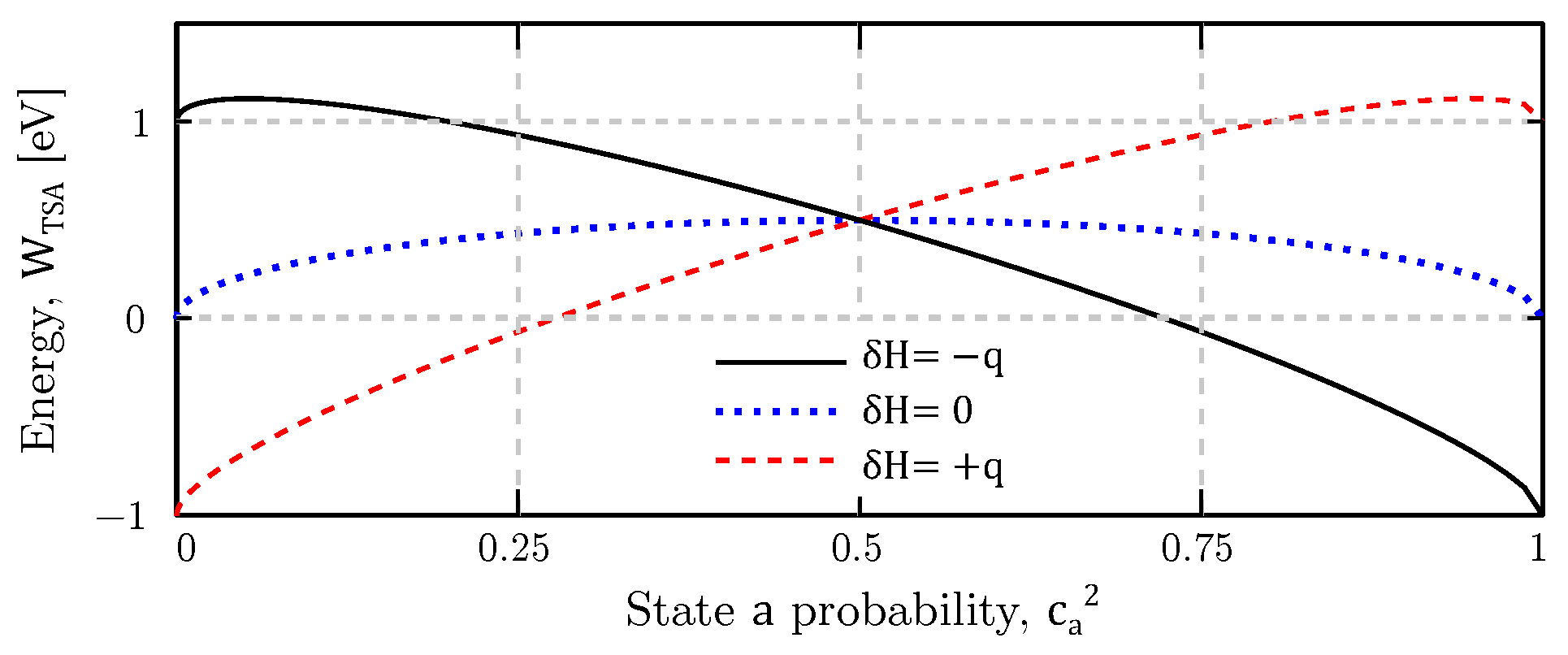
2.2. Bistable Factor
3. Energy Modelling
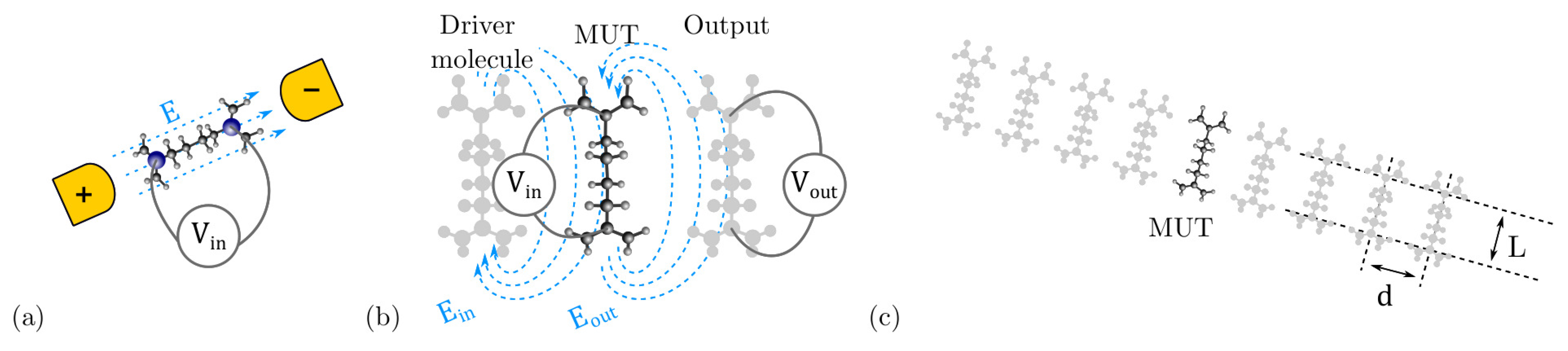
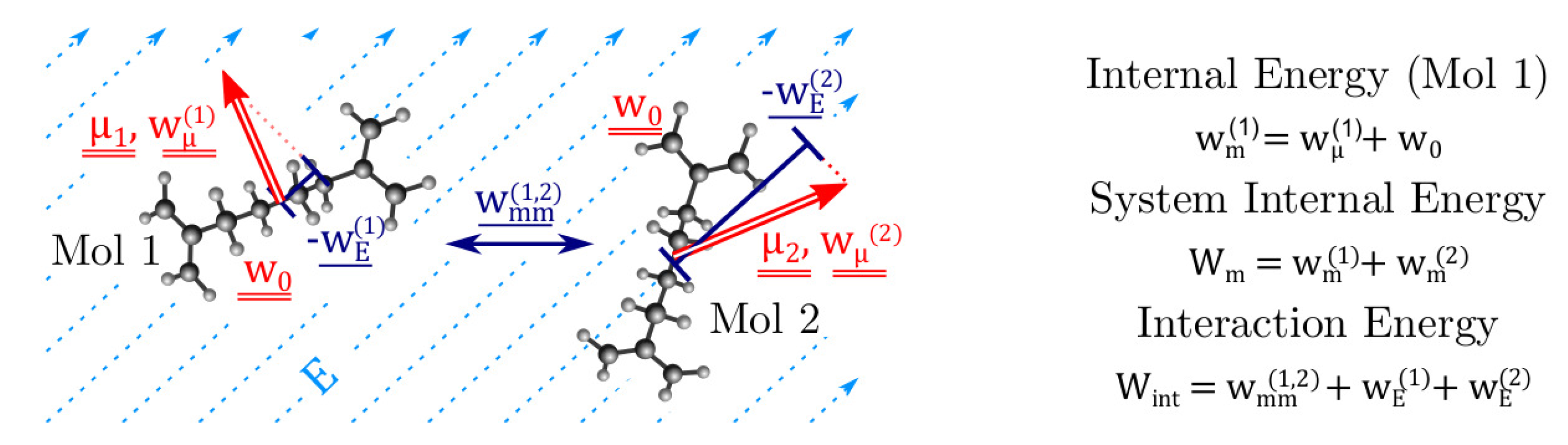
3.1. Model Definition
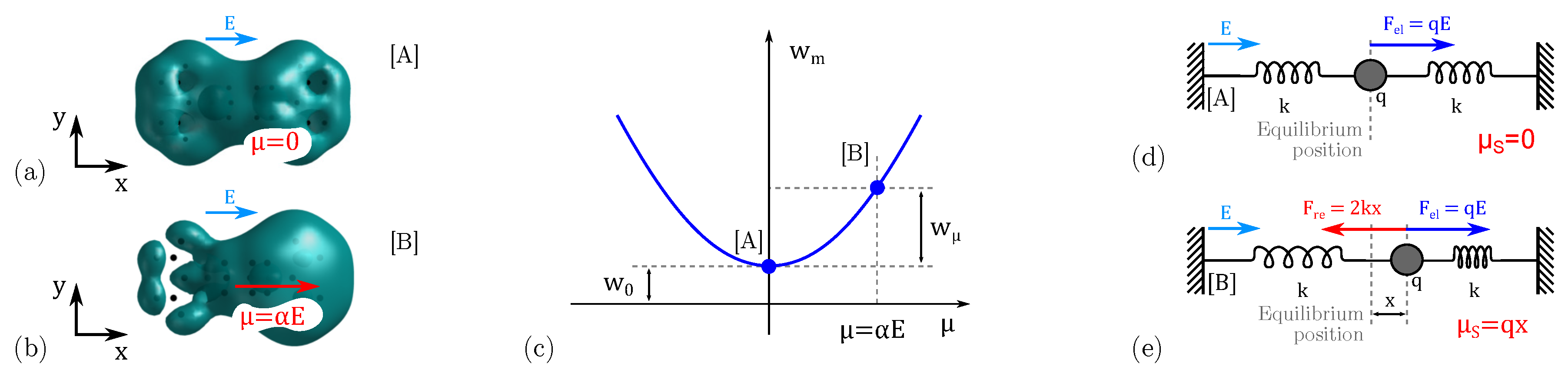
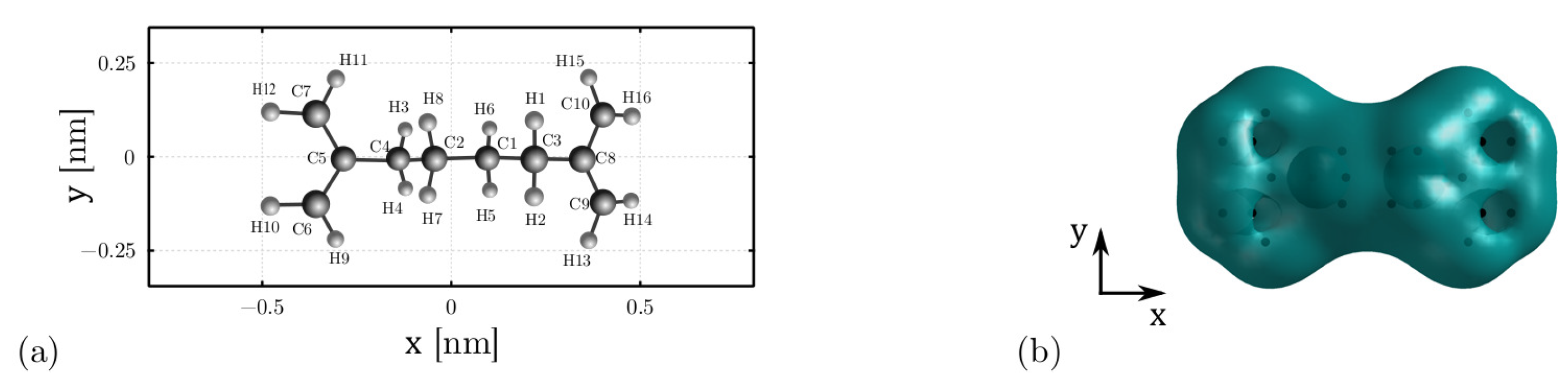
3.2. Internal Energy: The Conformation Energy
3.3. Internal Energy: The Polarization Energy
3.4. The Interaction Energy: Intermolecular Energy
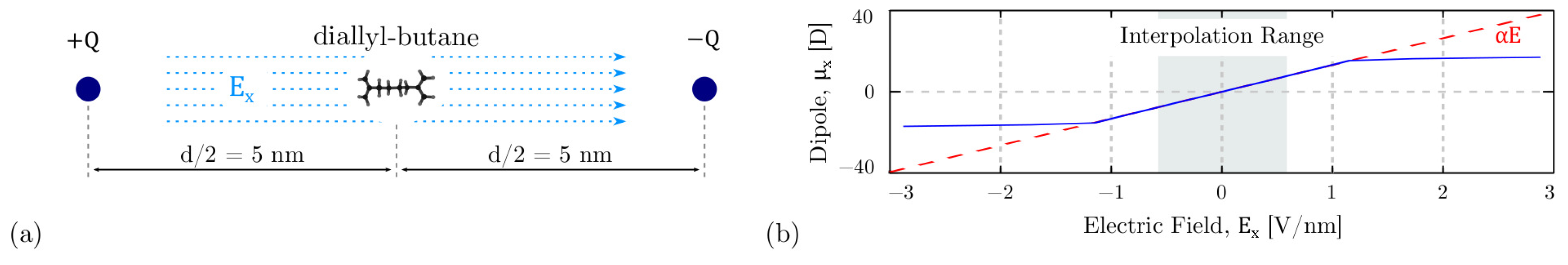
3.5. The Interaction Energy: Electric Field Energy
3.6. Final Expression
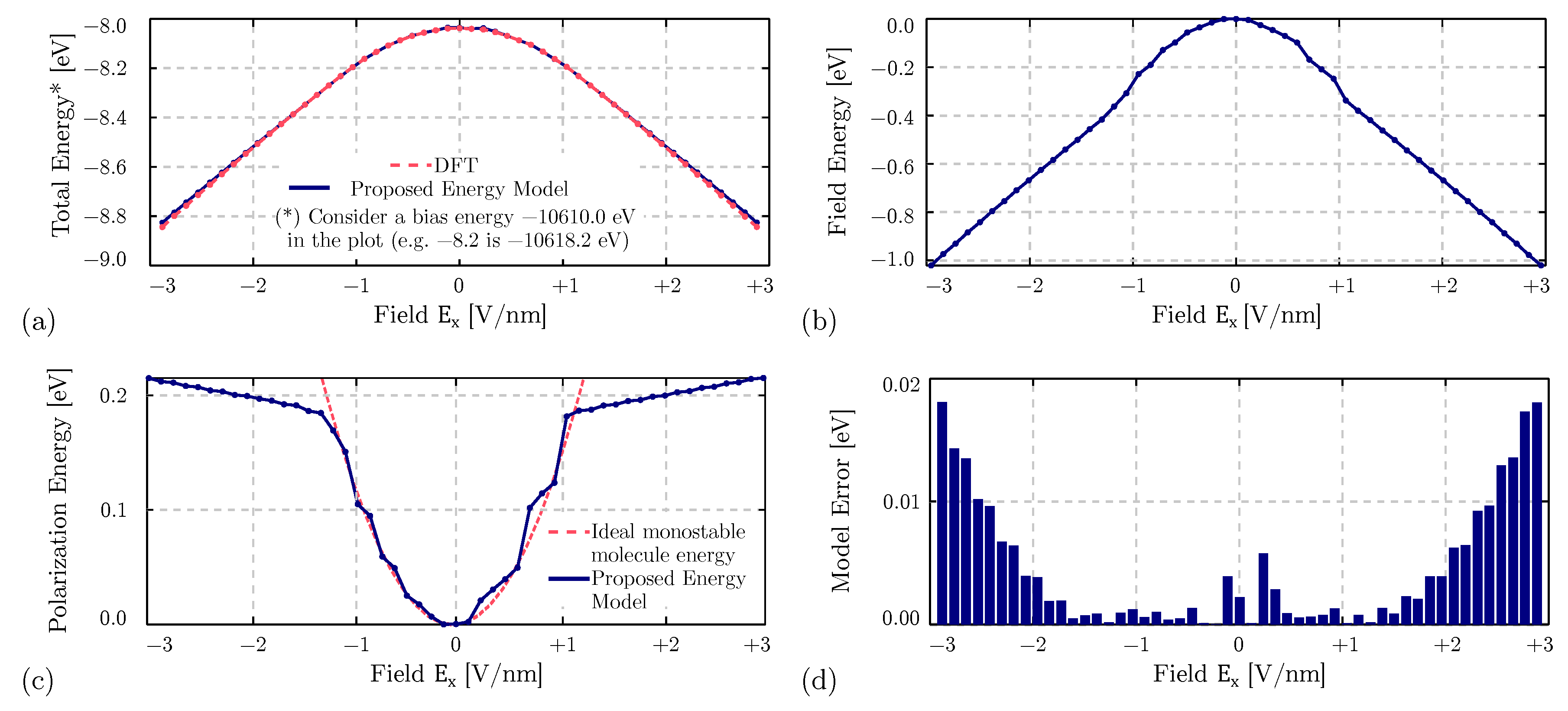
4. Results
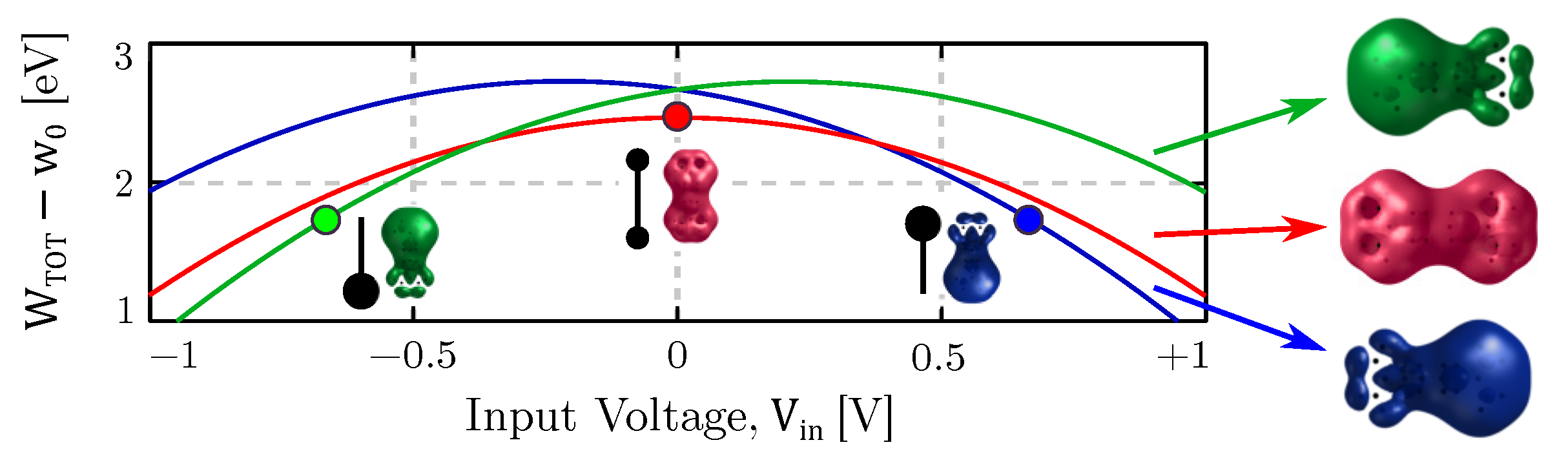
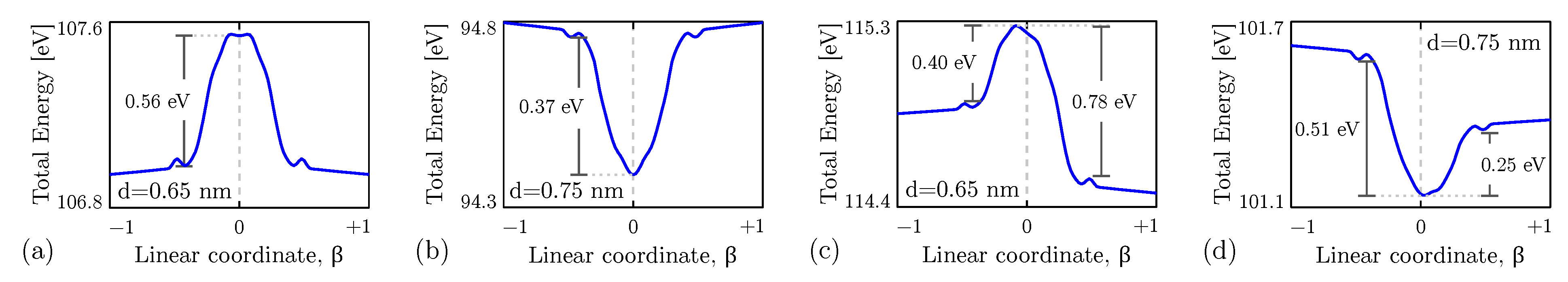
4.1. Equilibrium Analysis
4.2. Field-Induced Polarization of the Diallylbutane
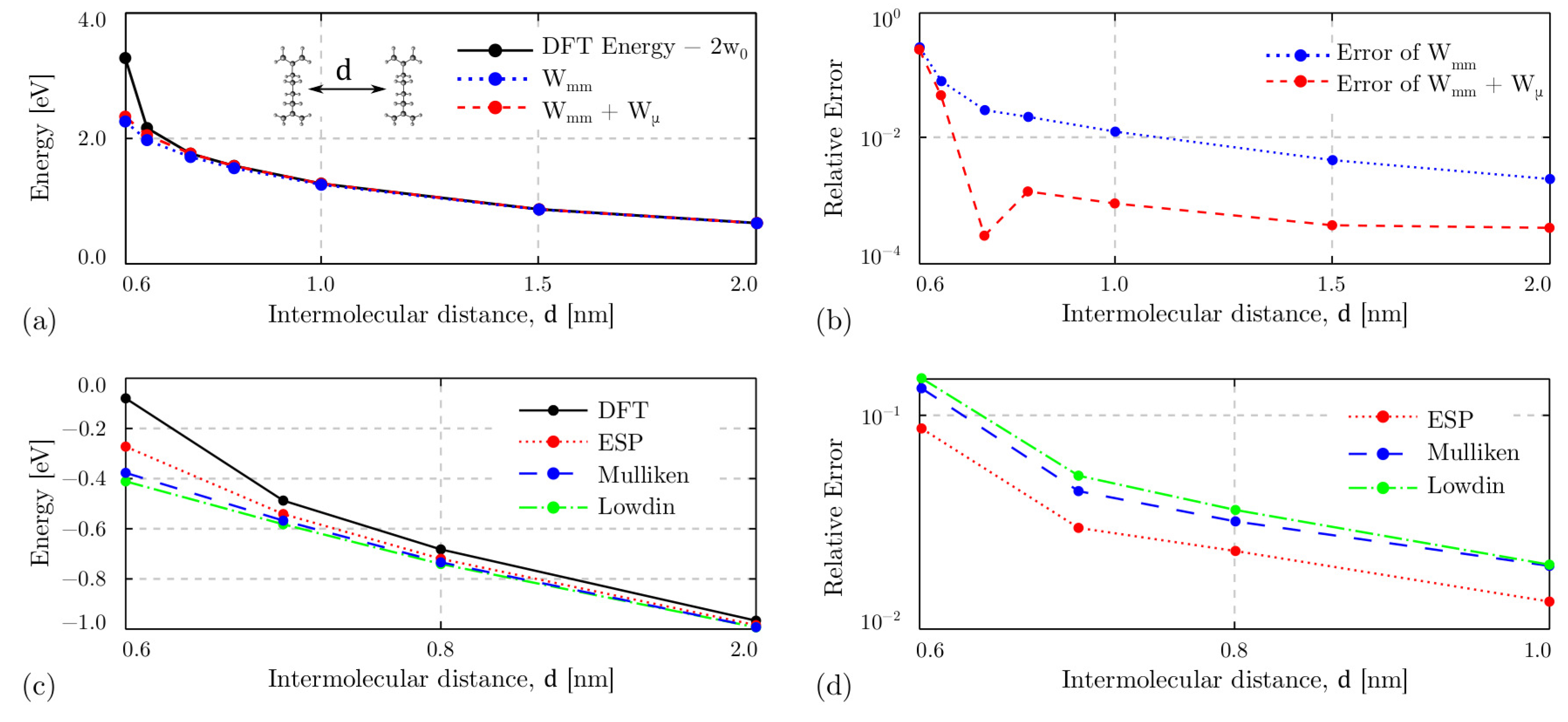
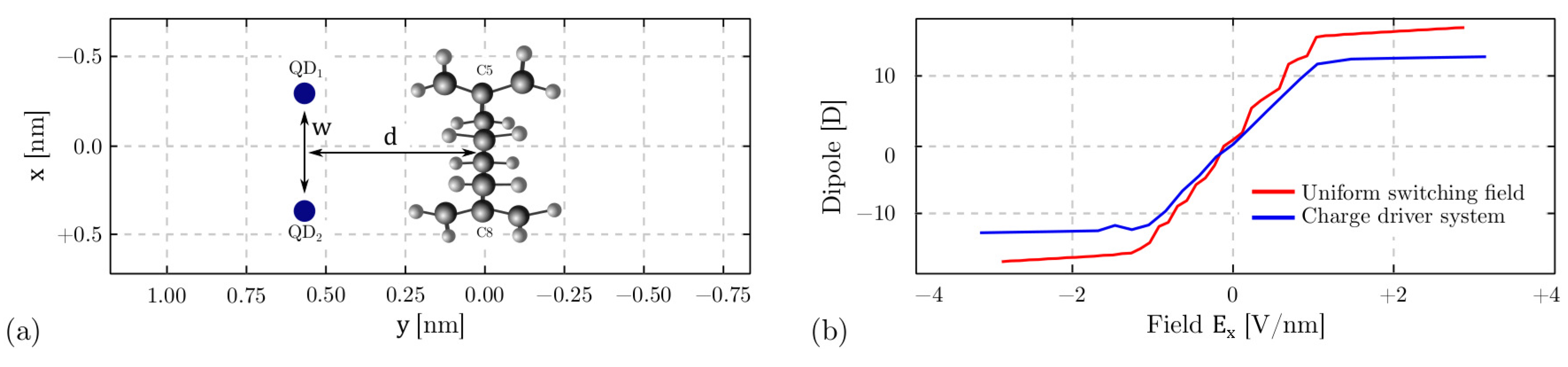
4.3. Intermolecular Interaction
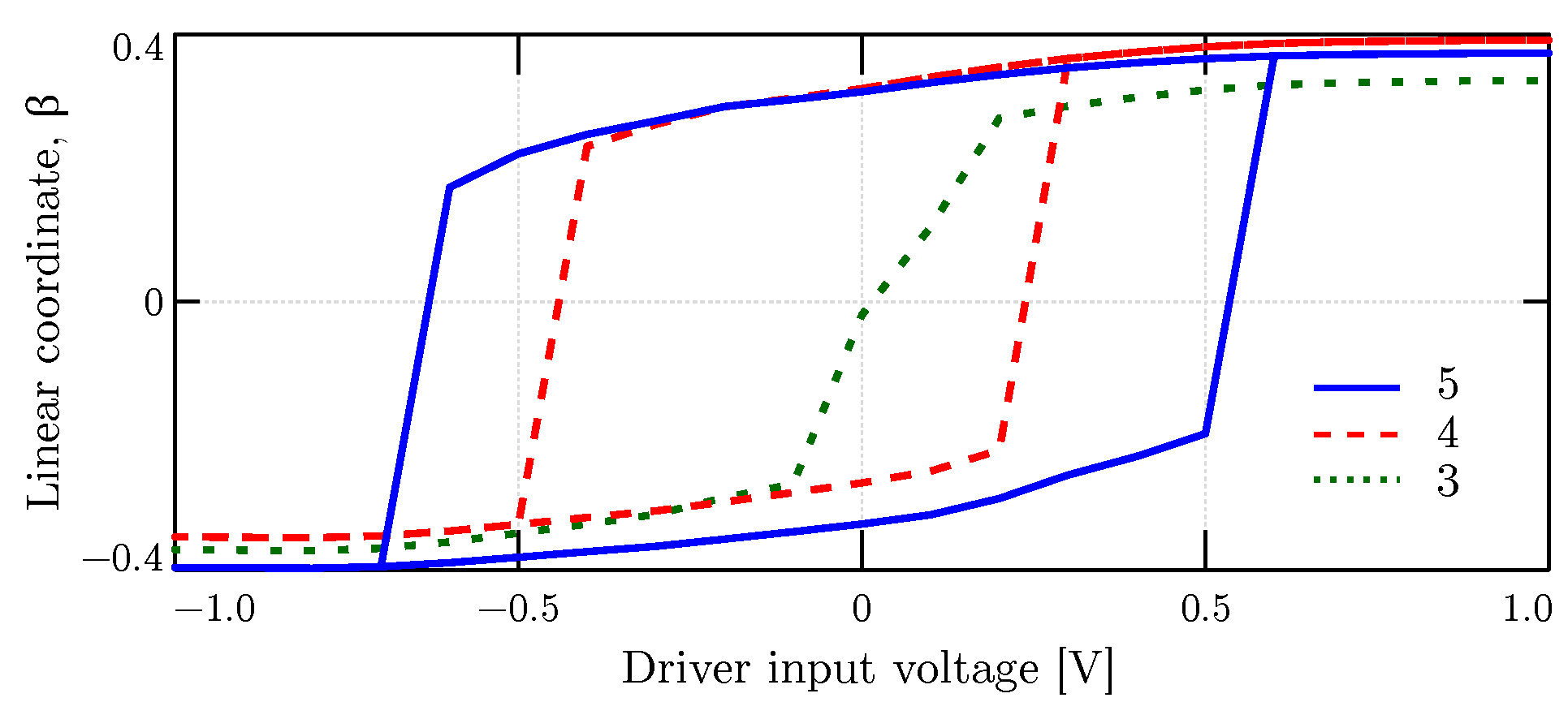
4.3.1. The Driver Response
4.4. Bistability Study
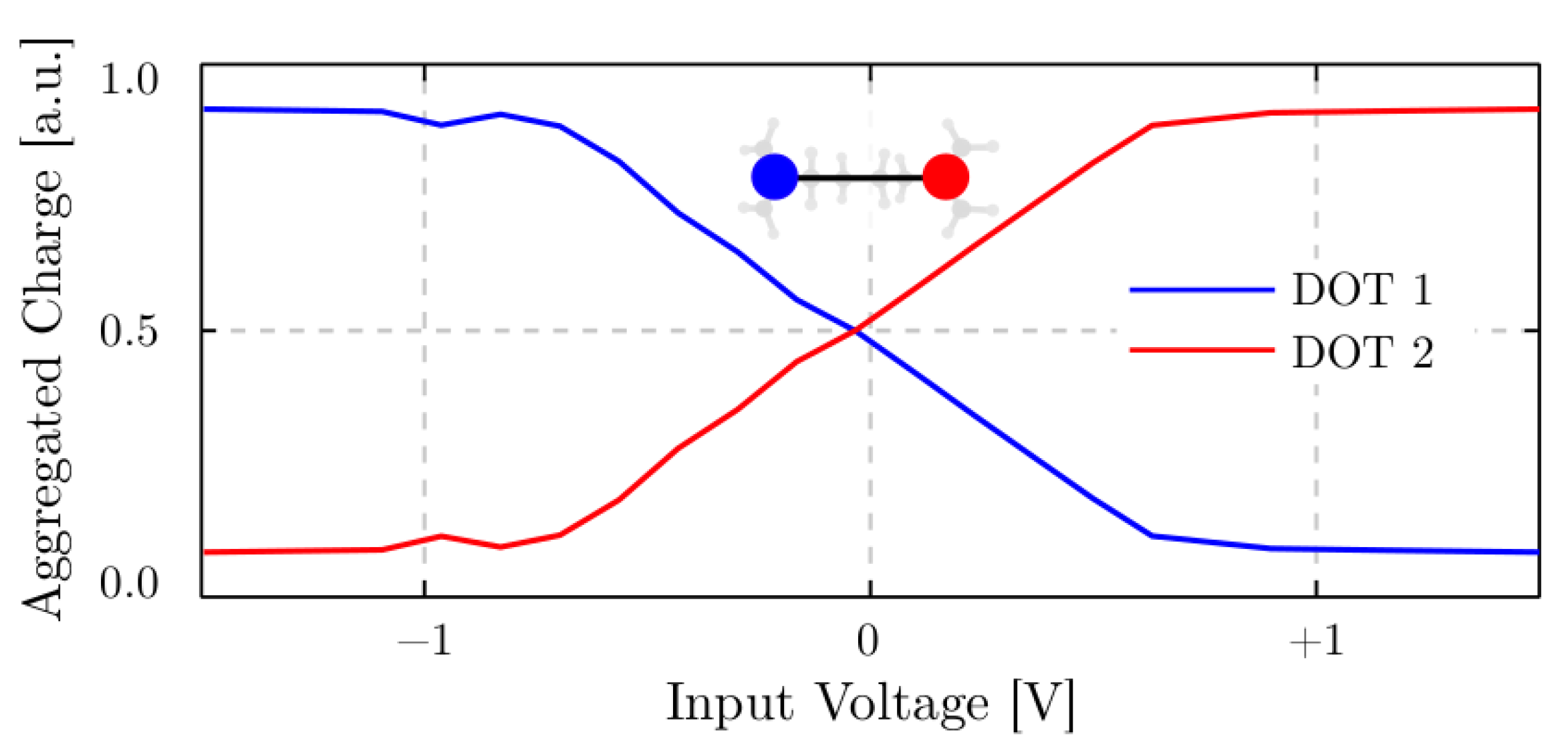
4.5. Memory Effect
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| CAD | Computer-Aided Design |
| CMOS | Complementary Metal-Oxide Semiconductor |
| DFT | Density Functiona Theory |
| ESP | Electrostatic Potential |
| FCN | Field-Coupled Nanocomputing |
| MoSQuiTo | Molecular Simulator Quantum-dot cellular automata Torino |
| MUT | Molecule Under Test |
| SCERPA | Self-Consistent Electrostatic Potential Algorithm |
| TSA | Two-State Approximation |
| QCA | Quantum-dot Cellular Automata |
| VACT | Vin-Aggregated Charge Transcharacteristics |
Appendix A

References
- Lent, C.S.; Tougaw, P.D.; Porod, W.; Bernstein, G.H. Quantum cellular automata. Nanotechnology 1993, 4, 49. [Google Scholar] [CrossRef]
- Turvani, G.; Tohti, A.; Bollo, M.; Riente, F.; Vacca, M.; Graziano, M.; Zamboni, M. Physical design and testing of Nano Magnetic architectures. In Proceedings of the 2014 9th IEEE International Conference on Design Technology of Integrated Systems in Nanoscale Era (DTIS), Santorini, Greece, 6–8 May 2014; pp. 1–6. [Google Scholar]
- Breitkreutz, S.; Kiermaier, J.; Eichwald, I.; Ju, X.; Csaba, G.; Schmitt-Landsiedel, D.; Becherer, M. Majority Gate for Nanomagnetic Logic With Perpendicular Magnetic Anisotropy. IEEE Trans. Magn. 2012, 48, 4336–4339. [Google Scholar] [CrossRef]
- Riente, F.; Ziemys, G.; Turvani, G.; Schmitt-Landsiedel, D.; Gamm, S.B.; Graziano, M. Towards Logic-In-Memory circuits using 3D-integrated Nanomagnetic logic. In Proceedings of the 2016 IEEE International Conference on Rebooting Computing (ICRC), San Diego, CA, USA, 17–19 October 2016; pp. 1–8. [Google Scholar] [CrossRef]
- Csaba, G.; Imre, A.; Bernstein, G.; Porod, W.; Metlushko, V. Nanocomputing by field-coupled nanomagnets. IEEE Trans. Nanotechnol. 2002, 1, 209–213. [Google Scholar] [CrossRef]
- Turvani, G.; Riente, F.; Cairo, F.; Vacca, M.; Garlando, U.; Zamboni, M.; Graziano, M. Efficient and reliable fault analysis methodology for nanomagnetic circuits. Int. J. Circuit Theory Appl. 2017, 45, 660–680. [Google Scholar] [CrossRef]
- Cofano, M.; Santoro, G.; Vacca, M.; Pala, D.; Causapruno, G.; Cairo, F.; Riente, F.; Turvani, G.; Roch, M.R.; Graziano, M.; et al. Logic-in-Memory: A Nano Magnet Logic Implementation. In Proceedings of the 2015 IEEE Computer Society Annual Symposium on VLSI, Montpellier, France, 8–10 July 2015; pp. 286–291. [Google Scholar] [CrossRef]
- Ng, S.S.H.; Retallick, J.; Chiu, H.N.; Lupoiu, R.; Livadaru, L.; Huff, T.; Rashidi, M.; Vine, W.; Dienel, T.; Wolkow, R.A.; et al. SiQAD: A Design and Simulation Tool for Atomic Silicon Quantum Dot Circuits. IEEE Trans. Nanotechnol. 2020, 19, 137–146. [Google Scholar] [CrossRef]
- Orlov, A.O.; Amlani, I.; Bernstein, G.H.; Lent, C.S.; Snider, G.L. Realization of a Functional Cell for Quantum-Dot Cellular Automata. Science 1997, 277, 928–930. [Google Scholar] [CrossRef]
- Tóth, G.; Lent, C.S. Quasiadiabatic switching for metal-island quantum-dot cellular automata. J. Appl. Phys. 1999, 85, 2977–2984. [Google Scholar] [CrossRef]
- Wang, R.; Pulimeno, A.; Roch, M.; Turvani, G.; Piccinini, G.; Graziano, M. Effect of a Clock System on Bis-ferrocene Molecular QCA. IEEE Trans. Nanotechnol. 2016, 15, 574–582. [Google Scholar] [CrossRef]
- Ardesi, Y.; Turvani, G.; Graziano, M.; Piccinini, G. SCERPA Simulation of Clocked Molecular Field-Coupling Nanocomputing. IEEE Trans. Very Large Scale Integr. (VLSI) Syst. 2021, 29, 558–567. [Google Scholar] [CrossRef]
- Mo, F.; Spano, C.E.; Ardesi, Y.; Piccinini, G.; Graziano, M. Beyond-CMOS Artificial Neuron: A Simulation- Based Exploration of the Molecular-FET. IEEE Trans. Nanotechnol. 2021, 20, 903–911. [Google Scholar] [CrossRef]
- Blair, E.P.; Liu, M.; Lent, C.S. Signal Energy in Quantum-Dot Cellular Automata Bit Packets. J. Comput. Theor. Nanosci. 2011, 8, 972–982. [Google Scholar] [CrossRef]
- Timler, J.; Lent, C.S. Power gain and dissipation in quantum-dot cellular automata. J. Appl. Phys. 2002, 91, 823–831. [Google Scholar] [CrossRef]
- Sill Torres, F.; Wille, R.; Niemann, P.; Drechsler, R. An Energy-Aware Model for the Logic Synthesis of Quantum-Dot Cellular Automata. IEEE Trans. Comput.-Aided Des. Integr. Circuits Syst. 2018, 37, 3031–3041. [Google Scholar] [CrossRef]
- Blair, E.P.; Blair, E.P.; Lent, C.S. Power dissipation in clocking wires for clocked molecular quantum-dot cellular automata. J. Comput. Electron. 2010, 9, 49–55. [Google Scholar] [CrossRef]
- Vacca, M.; Frache, S.; Graziano, M.; Riente, F.; Turvani, G.; Roch, M.R.; Zamboni, M. ToPoliNano: NanoMagnet Logic Circuits Design and Simulation. In Field-Coupled Nanocomputing: Paradigms, Progress, and Perspectives; Springer: Berlin/Heidelberg, Germany, 2014; pp. 274–306. [Google Scholar] [CrossRef]
- Riente, F.; Garlando, U.; Turvani, G.; Vacca, M.; Ruo Roch, M.; Graziano, M. MagCAD: Tool for the Design of 3-D Magnetic Circuits. IEEE J. Explor. Solid-State Comput. Devices Circuits 2017, 3, 65–73. [Google Scholar] [CrossRef]
- Wille, R.; Walter, M.; Sill Torres, F.; Große, D.; Drechsler, R. Ignore Clocking Constraints: An Alternative Physical Design Methodology for Field-Coupled Nanotechnologies. In Proceedings of the 2019 IEEE Computer Society Annual Symposium on VLSI (ISVLSI), Miami, FL, USA, 15–17 July 2019; pp. 651–656. [Google Scholar] [CrossRef]
- Garlando, U.; Walter, M.; Wille, R.; Riente, F.; Torres, F.S.; Drechsler, R. ToPoliNano and fiction: Design Tools for Field-coupled Nanocomputing. In Proceedings of the 2020 23rd Euromicro Conference on Digital System Design (DSD), Kranj, Slovenia, 26–28 August 2020; pp. 408–415. [Google Scholar] [CrossRef]
- Walter, M.; Wille, R.; Torres, F.S.; Große, D.; Drechsler, R. Verification for Field-coupled Nanocomputing Circuits. In Proceedings of the 2020 57th ACM/IEEE Design Automation Conference (DAC), San Francisco, CA, USA, 20–24 July 2020; pp. 1–6. [Google Scholar] [CrossRef]
- Luz, L.O.; Nacif, J.A.M.; Ferreira, R.S.; Neto, O.P.V. NMLib: A Nanomagnetic Logic Standard Cell Library. In Proceedings of the 2021 IEEE International Symposium on Circuits and Systems (ISCAS), Daegu, Korea, 22–28 May 2021; pp. 1–5. [Google Scholar] [CrossRef]
- Frache, S.; Chiabrando, D.; Graziano, M.; Riente, F.; Turvani, G.; Zamboni, M. ToPoliNano: Nanoarchitectures design made real. In Proceedings of the 2012 IEEE/ACM International Symposium on Nanoscale Architectures (NANOARCH), Amsterdam, The Netherlands, 4–6 July 2012; pp. 160–167. [Google Scholar] [CrossRef]
- Lu, Y.; Lent, C.S. A metric for characterizing the bistability of molecular quantum-dot cellular automata. Nanotechnology 2008, 19, 155703. [Google Scholar] [CrossRef]
- Ardesi, Y.; Gaeta, A.; Beretta, G.; Piccinini, G.; Graziano, M. Ab initio Molecular Dynamics Simulations of Field-Coupled Nanocomputing Molecules. J. Integr. Circuits Syst. 2021, 16, 1–8. [Google Scholar] [CrossRef]
- Rahimi, E.; Reimers, J.R. Molecular quantum cellular automata cell design trade-offs: Latching vs. power dissipation. Phys. Chem. Chem. Phys. 2018, 20, 17881–17888. [Google Scholar] [CrossRef]
- Pulimeno, A.; Graziano, M.; Antidormi, A.; Wang, R.; Zahir, A.; Piccinini, G. Understanding a Bisferrocene Molecular QCA Wire. In Field-Coupled Nanocomputing: Paradigms, Progress, and Perspectives; Springer: Berlin/Heidelberg, Germany, 2014; pp. 307–338. [Google Scholar] [CrossRef]
- Ardesi, Y.; Pulimeno, A.; Graziano, M.; Riente, F.; Piccinini, G. Effectiveness of Molecules for Quantum Cellular Automata as Computing Devices. J. Low Power Electron. Appl. 2018, 8, 24. [Google Scholar] [CrossRef]
- Ardesi, Y.; Wang, R.; Graziano, M.; Piccinini, G. SCERPA: A Self-Consistent Algorithm for the Evaluation of the Information Propagation in Molecular Field-Coupled Nanocomputing. IEEE Trans. Comput.-Aided Des. Integr. Circuits Syst. 2019, 39, 2749–2760. [Google Scholar] [CrossRef]
- Ardesi, Y.; Gnoli, L.; Graziano, M.; Piccinini, G. Bistable Propagation of Monostable Molecules in Molecular Field-Coupled Nanocomputing. In Proceedings of the 15th Conference on PhD Research in Microelectronics and Electronics (PRIME), Lausanne, Switzerland, 15–18 July 2019. [Google Scholar]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Pulimeno, A.; Graziano, M.; Abrardi, C.; Demarchi, D.; Piccinini, G. Molecular QCA: A write-in system based on electric fields. In Proceedings of the 4th IEEE International NanoElectronics Conference, Tao-Yuan, Taiwan, 21–24 June 2011; pp. 1–2. [Google Scholar] [CrossRef]
- Ardesi, Y.; Beretta, G.; Vacca, M.; Piccinini, G.; Graziano, M. Impact of Molecular Electrostatics on Field-Coupled Nanocomputing and Quantum-Dot Cellular Automata Circuits. Electronics 2022, 11, 276. [Google Scholar] [CrossRef]
- Sutcliffe, B.T.; Woolley, R.G. On the quantum theory of molecules. J. Chem. Phys. 2012, 137, 22A544. [Google Scholar] [CrossRef] [PubMed]
- Graziano, M.; Wang, R.; Roch, M.R.; Ardesi, Y.; Riente, F.; Piccinini, G. Characterisation of a bis-ferrocene molecular QCA wire on a non-ideal gold surface. Micro Nano Lett. 2019, 14, 22–27. [Google Scholar] [CrossRef]
- Lent, C.S.; Isaksen, B. Clocked molecular quantum-dot cellular automata. IEEE Trans. Electron Devices 2003, 50, 1890–1896. [Google Scholar] [CrossRef]
- Atkins, P.W.; Paula, J.D. Physical Chemistry, 8th ed.; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Frydel, D. Mean Field Electrostatics Beyond the Point Charge Description. In Advances in Chemical Physics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2016; pp. 209–260. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, M.; Lent, C. Molecular quantum-dot cellular automata: From molecular structure to circuit dynamics. J. Appl. Phys. 2007, 102, 034311. [Google Scholar] [CrossRef]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ardesi, Y.; Graziano, M.; Piccinini, G. A Model for the Evaluation of Monostable Molecule Signal Energy in Molecular Field-Coupled Nanocomputing. J. Low Power Electron. Appl. 2022, 12, 13. https://doi.org/10.3390/jlpea12010013
Ardesi Y, Graziano M, Piccinini G. A Model for the Evaluation of Monostable Molecule Signal Energy in Molecular Field-Coupled Nanocomputing. Journal of Low Power Electronics and Applications. 2022; 12(1):13. https://doi.org/10.3390/jlpea12010013
Chicago/Turabian StyleArdesi, Yuri, Mariagrazia Graziano, and Gianluca Piccinini. 2022. "A Model for the Evaluation of Monostable Molecule Signal Energy in Molecular Field-Coupled Nanocomputing" Journal of Low Power Electronics and Applications 12, no. 1: 13. https://doi.org/10.3390/jlpea12010013
APA StyleArdesi, Y., Graziano, M., & Piccinini, G. (2022). A Model for the Evaluation of Monostable Molecule Signal Energy in Molecular Field-Coupled Nanocomputing. Journal of Low Power Electronics and Applications, 12(1), 13. https://doi.org/10.3390/jlpea12010013