Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing


Simple Summary
Abstract
1. Introduction
2. Types of Annotation
2.1. Structural Annotation
2.1.1. Repeats
2.1.2. Predictions of Gene and Different Features
2.1.3. Databases for Structural Annotation
2.2. Functional Annotation
2.2.1. Automatic Functional Annotation
2.2.2. Databases for Functional Annotation
Gene Ontology (GO)
3. Comparative Annotation Methods
3.1. Ab Initio Annotation
3.2. Homology-Based Annotation
3.3. Variant Annotation
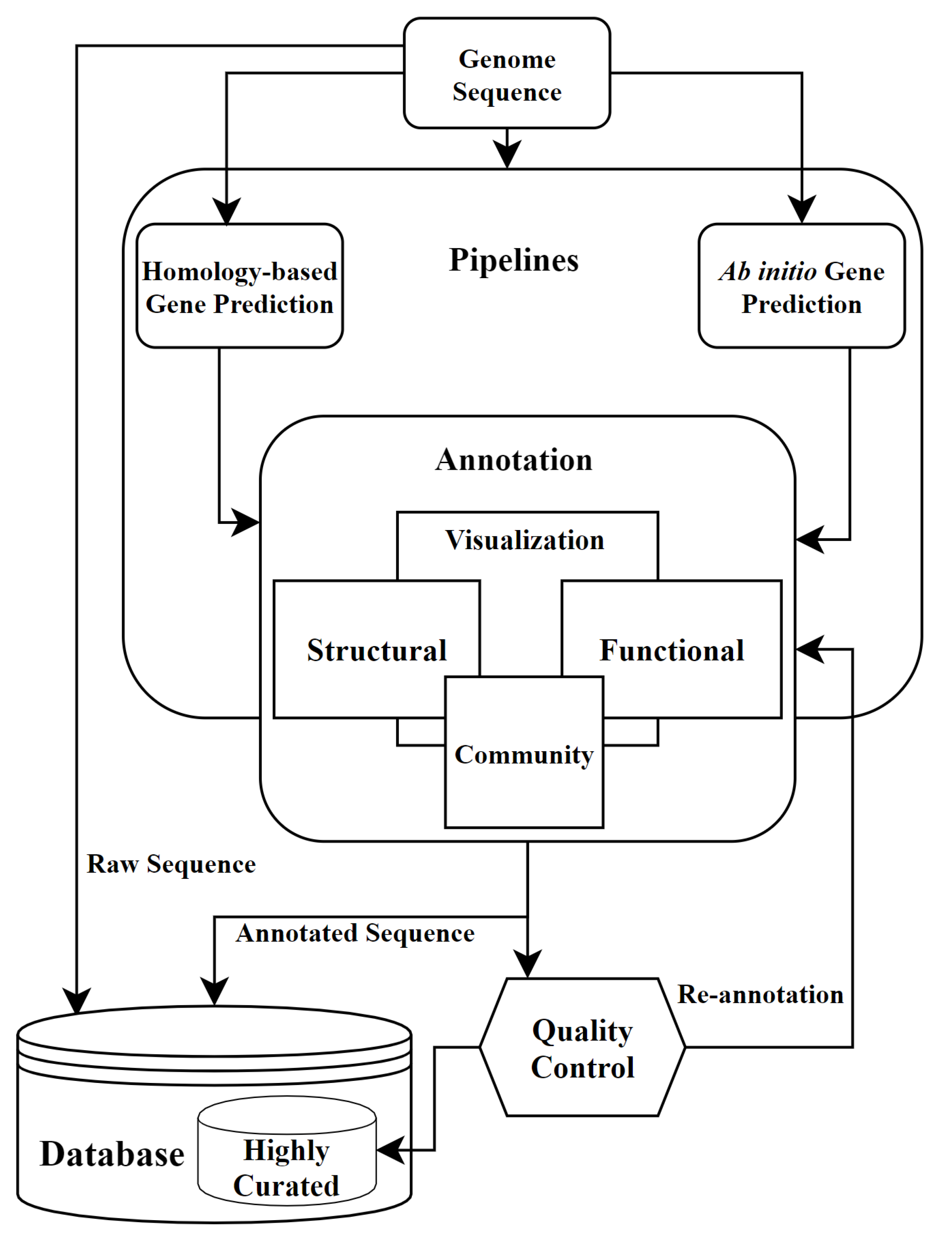
4. Annotation Pipelines
4.1. Structural Pipelines
4.2. Functional Pipelines
4.3. Combined Pipelines
4.4. Variant Pipelines
5. Annotation Visualization
5.1. File Formats
5.2. Genome Browsers
5.3. Functional Analysis Visualization Tools
5.4. Other Visualization Tools
6. Community Annotation and Quality Control in Annotation
6.1. Community Annotation
6.2. Quality Control for Annotation
7. Re-Annotation and Future of Annotation
7.1. Re-Annotation
7.2. The Future of Annotation
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef]
- Steward, C.A.; Parker, A.P.J.; Minassian, B.A.; Sisodiya, S.M.; Frankish, A.; Harrow, J. Genome annotation for clinical genomic diagnostics: Strengths and weaknesses. Genome Med. 2017, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef] [PubMed]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C.; et al. Mind the gap: Upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 2012, 7, e47768. [Google Scholar] [CrossRef]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.; Fiddes, I.T.; Diekhans, M.; Paten, B. Whole-genome alignment and comparative annotation. Annu. Rev. Anim. Biosci. 2019, 7, 41–64. [Google Scholar] [CrossRef]
- Brent, M.R. Genome annotation past, present, and future: How to define an ORF at each locus. Genome Res. 2005, 15, 1777–1786. [Google Scholar] [CrossRef]
- Li, F.; Zhao, X.; Li, M.; He, K.; Huang, C.; Zhou, Y.; Li, Z.; Walters, J.R. Insect genomes: Progress and challenges. Insect Mol. Biol. 2019, 28, 739–758. [Google Scholar] [CrossRef]
- Mishra, S.; Rastogi, Y.P.; Jabin, S.; Kaur, P.; Amir, M.; Khatoon, S. A bacterial phyla dataset for protein function prediction. Data Brief 2020, 28, 105002. [Google Scholar] [CrossRef]
- Spieth, J.; Lawson, D. Overview of gene structure. Genome Biol. Evol. 2005. [Google Scholar] [CrossRef]
- Zhang, B.; Han, D.; Korostelev, Y.; Yan, Z.; Shao, N.; Khrameeva, E.; Velichkovsky, B.M.; Chen, Y.P.P.; Gelfand, M.S.; Khaitovich, P. Changes in snoRNA and snRNA abundance in the human, chimpanzee, macaque, and mouse brain. Genome Biol. Evol. 2016, 8, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Sekhwal, M.K.; Li, P.; Ragupathy, R.; Cloutier, S.; Wang, X.; You, F.M. Pseudogenes and their genome-wide prediction in plants. Int. J. Mol. Sci. 2016, 17, 1991. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Abraham, A.L.; Touchon, M.; Rocha, E.P.C. Genesis, effects and fates of repeats in prokaryotic genomes. FEMS Microbiol. Rev. 2009, 33, 539–571. [Google Scholar] [CrossRef] [PubMed]
- de Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef]
- Barra, V.; Fachinetti, D. The dark side of centromeres: Types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Smit, A.F.; Hubley, R.; Green, P. RepeatMasker, 1996. 4.1.1 Released. Available online: http://www.repeatmasker.org/ (accessed on 3 September 2020).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Yu, Y.; Santat, L.A.; Choi, S. Bioinformatics packages for sequence analysis. In Applied Mycology and Biotechnology; Elsevier: Amsterdam, The Netherlands, 2006; Volume 6, pp. 143–160. [Google Scholar]
- Modrek, B.; Lee, C. A genomic view of alternative splicing. Nat. Genet. 2002, 30, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Brent, M.R.; Guigo, R. Recent advances in gene structure prediction. Curr. Opin. Struct. Biol. 2004, 14, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.S.; Krogh, A. EasyGene—A prokaryotic gene finder that ranks ORFs by statistical significance. BMC Bioinform. 2003, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.; Kosarev, P.; Seledsov, I.; Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006, 7, S10. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Majoros, W.H.; Pertea, M.; Delcher, A.L.; Salzberg, S.L. Efficient decoding algorithms for generalized hidden Markov model gene finders. BMC Bioinform. 2005, 6, 1–13. [Google Scholar]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Krogh, A. Two methods for improving performance of an HMM and their application for gene finding. Cent. Biol. Seq. Analysis. Phone 1997, 45, 4525. [Google Scholar]
- Schweikert, G.; Zien, A.; Zeller, G.; Behr, J.; Dieterich, C.; Ong, C.S.; Philips, P.; De Bona, F.; Hartmann, L.; Bohlen, A.; et al. mGene: Accurate SVM-based gene finding with an application to nematode genomes. Genome Res. 2009, 19, 2133–2143. [Google Scholar] [CrossRef]
- Hebsgaard, S.M.; Korning, P.G.; Tolstrup, N.; Engelbrecht, J.; Rouzé, P.; Brunak, S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996, 24, 3439–3452. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction; Springer: New York, NY, USA, 2019; pp. 1–14. [Google Scholar]
- Keilwagen, J.; Hartung, F.; Grau, J. GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. In Gene Prediction; Springer: New York, NY, USA, 2019; pp. 161–177. [Google Scholar]
- Gremme, G.; Brendel, V.; Sparks, M.E.; Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inf. Softw. Technol. 2005, 47, 965–978. [Google Scholar] [CrossRef]
- Van Baren, M.J.; Brent, M.R. Iterative gene prediction and pseudogene removal improves genome annotation. Genome Res. 2006, 16, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Carriero, N.; Zheng, D.; Karro, J.; Harrison, P.M.; Gerstein, M. PseudoPipe: An automated pseudogene identification pipeline. Bioinformatics 2006, 22, 1437–1439. [Google Scholar] [CrossRef]
- Korf, I.; Flicek, P.; Duan, D.; Brent, M.R. Integrating genomic homology into gene structure prediction. Bioinformatics 2001, 17, S140–S148. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef]
- Allen, J.E.; Majoros, W.H.; Pertea, M.; Salzberg, S.L. JIGSAW, GeneZilla, and GlimmerHMM: Puzzling out the features of human genes in the ENCODE regions. Genome Biol. 2006, 7, S9. [Google Scholar] [CrossRef]
- Sagot, M.F.; Schiex, T.; Rouze, P.; Mathe, C. Current methods of gene prediction, their strengths and weaknesses. Nucleic Acids Res. 2002, 30, 4103–4117. [Google Scholar]
- Wang, Y.; Chen, L.; Song, N.; Lei, X. GASS: Genome structural annotation for eukaryotes based on species similarity. BMC Genom. 2015, 16, 150. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2019, 47, D94–D99. [Google Scholar] [CrossRef] [PubMed]
- Brooksbank, C.; Bergman, M.T.; Apweiler, R.; Birney, E.; Thornton, J. The european bioinformatics institute’s data resources 2014. Nucleic Acids Res. 2014, 42, D18–D25. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Mashima, J.; Kosuge, T.; Kaminuma, E.; Ogasawara, O.; Okubo, K.; Nakamura, Y.; Takagi, T. DNA data bank of Japan: 30th anniversary. Nucleic Acids Res. 2018, 46, D30–D35. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Sigrist, C.J.A.; De Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2012, 41, D344–D347. [Google Scholar] [CrossRef]
- Haft, D.H.; Selengut, J.D.; Richter, R.A.; Harkins, D.; Basu, M.K.; Beck, E. TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 2012, 41, D387–D395. [Google Scholar] [CrossRef]
- Lewis, T.E.; Sillitoe, I.; Dawson, N.; Lam, S.D.; Clarke, T.; Lee, D.; Orengo, C.; Lees, J. Gene3D: Extensive prediction of globular domains in proteins. Nucleic Acids Res. 2018, 46, D435–D439. [Google Scholar] [CrossRef]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef]
- Fang, S.; Zhang, L.; Guo, J.; Niu, Y.; Wu, Y.; Li, H.; Zhao, L.; Li, X.; Teng, X.; Sun, X.; et al. NONCODEV5: A comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018, 46, D308–D314. [Google Scholar] [CrossRef] [PubMed]
- Karro, J.E.; Yan, Y.; Zheng, D.; Zhang, Z.; Carriero, N.; Cayting, P.; Harrrison, P.; Gerstein, M. Pseudogene. org: A comprehensive database and comparison platform for pseudogene annotation. Nucleic Acids Res. 2007, 35, D55–D60. [Google Scholar] [CrossRef] [PubMed]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.A.; Wheeler, T.J. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016, 44, D81–D89. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Mudge, J.M.; Harrow, J. The state of play in higher eukaryote gene annotation. Nat. Rev. Genet. 2016, 17, 758. [Google Scholar] [CrossRef] [PubMed]
- Cutting, G.R. Annotating DNA variants is the next major goal for human genetics. Am. J. Hum. Genet. 2014, 94, 5–10. [Google Scholar] [CrossRef]
- Butkiewicz, M.; Bush, W.S. In silico functional annotation of genomic variation. Curr. Protoc. Hum. Genet. 2016, 88, 6–15. [Google Scholar] [CrossRef]
- Pavlopoulos, G.A.; Oulas, A.; Iacucci, E.; Sifrim, A.; Moreau, Y.; Schneider, R.; Aerts, J.; Iliopoulos, I. Unraveling genomic variation from next generation sequencing data. BioData Min. 2013, 6, 13. [Google Scholar] [CrossRef]
- Koonin, E.V. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005, 39, 309–338. [Google Scholar] [CrossRef]
- Sasson, O.; Kaplan, N.; Linial, M. Functional annotation prediction: All for one and one for all. Protein Sci. 2006, 15, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; Cherry, J.M.; Ashburner, M.; Ball, C.A.; Blake, J.A.; Butler, H.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar]
- Gene Ontology Consortium. Gene ontology consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Thomas, P.D.; Hill, D.P.; Mi, H.; Osumi-Sutherland, D.; Van Auken, K.; Carbon, S.; Balhoff, J.P.; Albou, L.P.; Good, B.; Gaudet, P.; et al. Gene Ontology Causal Activity Modeling (GO-CAM) moves beyond GO annotations to structured descriptions of biological functions and systems. Nat. Genet. 2019, 51, 1429–1433. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008. [Google Scholar] [CrossRef]
- Chen, T.W.; Gan, R.C.R.; Wu, T.H.; Huang, P.J.; Lee, C.Y.; Chen, Y.Y.M.; Chen, C.C.; Tang, P. FastAnnotator-an efficient transcript annotation web tool. BMC Genom. 2012, 13, S9. [Google Scholar] [CrossRef]
- Araujo, F.A.; Barh, D.; Silva, A.; Guimaraes, L.; Ramos, R.T.J. GO FEAT: A rapid web-based functional annotation tool for genomic and transcriptomic data. Sci. Rep. 2018, 8, 1794. [Google Scholar] [CrossRef]
- Martin, D.M.A.; Berriman, M.; Barton, G.J. GOtcha: A new method for prediction of protein function assessed by the annotation of seven genomes. BMC Bioinform. 2004, 5, 178. [Google Scholar]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef]
- Jung, J.; Yi, G.; Sukno, S.A.; Thon, M.R. PoGO: Prediction of Gene Ontology terms for fungal proteins. BMC Bioinform. 2010, 11, 215. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Morgat, A.; Lombardot, T.; Axelsen, K.B.; Aimo, L.; Niknejad, A.; Hyka-Nouspikel, N.; Coudert, E.; Pozzato, M.; Pagni, M.; Moretti, S.; et al. Updates in Rhea-an expert curated resource of biochemical reactions. Nucleic Acids Res. 2017, 45, D415–D418. [Google Scholar] [CrossRef]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.C.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef]
- Lappalainen, I.; Lopez, J.; Skipper, L.; Hefferon, T.; Spalding, J.D.; Garner, J.; Chen, C.; Maguire, M.; Corbett, M.; Zhou, G.; et al. DbVar and DGVa: Public archives for genomic structural variation. Nucleic Acids Res. 2012, 41, D936–D941. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shaw, K.; Cooper, D.N. The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr. Protoc. Bioinform. 2012, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fredman, D.; Siegfried, M.; Yuan, Y.P.; Bork, P.; Lehväslaiho, H.; Brookes, A.J. HGVbase: A human sequence variation database emphasizing data quality and a broad spectrum of data sources. Nucleic Acids Res. 2002, 30, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Fairley, S.; Lowy-Gallego, E.; Perry, E.; Flicek, P. The international genome sample resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020, 48, D941–D947. [Google Scholar] [CrossRef]
- Clarke, L.; Zheng-Bradley, X.; Smith, R.; Kulesha, E.; Xiao, C.; Toneva, I.; Vaughan, B.; Preuss, D.; Leinonen, R.; Shumway, M.; et al. The 1000 Genomes Project: Data management and community access. Nat. Methods 2012, 9, 459–462. [Google Scholar] [CrossRef]
- Sharma, V.; Hiller, M. Increased alignment sensitivity improves the usage of genome alignments for comparative gene annotation. Nucleic Acids Res. 2017, 45, 8369–8377. [Google Scholar] [CrossRef]
- Tian, R.; Basu, M.K.; Capriotti, E. Computational methods and resources for the interpretation of genomic variants in cancer. BMC Genom. 2015, 16, S7. [Google Scholar] [CrossRef]
- Coghlan, A.; Fiedler, T.J.; McKay, S.J.; Flicek, P.; Harris, T.W.; Blasiar, D.; Stein, L.D.; nGASP Consortium. nGASP–the nematode genome annotation assessment project. BMC Bioinform. 2008, 9, 549. [Google Scholar] [CrossRef]
- Salamov, A.A.; Solovyev, V.V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000, 10, 516–522. [Google Scholar] [CrossRef]
- Solovyev, V. Statistical approaches in eukaryotic gene prediction. In Handbook of Statistical Genetics; Wiley: New York, NY, USA, 2004. [Google Scholar]
- Yeh, R.F.; Lim, L.P.; Burge, C.B. Computational inference of homologous gene structures in the human genome. Genome Res. 2001, 11, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.P.; Pazdernik, N.J.; McGehee, M.R. Chapter 29—Molecular Evolution. In Molecular Biology, 3rd ed.; Academic Press: London, UK, 2019; pp. 925–969. [Google Scholar] [CrossRef]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, B. DIALIGN: Multiple DNA and protein sequence alignment at BiBiServ. Nucleic Acids Res. 2004, 32, W33–W36. [Google Scholar] [CrossRef]
- Taher, L.; Rinner, O.; Garg, S.; Sczyrba, A.; Brudno, M.; Batzoglou, S.; Morgenstern, B. AGenDA: Homology-based gene prediction. Bioinformatics 2003, 19, 1575–1577. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Agarwal, P.; Abril, J.F.; Wiehe, T.; Fickett, J.W.; Guigó, R. Comparative gene prediction in human and mouse. Genome Res. 2003, 13, 108–117. [Google Scholar] [CrossRef]
- Guigó, R.; Flicek, P.; Abril, J.F.; Reymond, A.; Lagarde, J.; Denoeud, F.; Antonarakis, S.; Ashburner, M.; Bajic, V.B.; Birney, E.; et al. EGASP: The human ENCODE genome annotation assessment project. Genome Biol. 2006, 7, S2. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Paila, U.; Chapman, B.A.; Kirchner, R.; Quinlan, A.R. GEMINI: Integrative exploration of genetic variation and genome annotations. PLoS Comput. Biol. 2013, 9, e1003153. [Google Scholar] [CrossRef]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar] [CrossRef]
- Dayem Ullah, A.Z.; Lemoine, N.R.; Chelala, C. A practical guide for the functional annotation of genetic variations using SNPnexus. Brief. Bioinform. 2013, 14, 437–447. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Coldren, C.; Karunamurthy, A.; Kip, N.S.; Klee, E.W.; Lincoln, S.E.; Leon, A.; Pullambhatla, M.; Temple-Smolkin, R.L.; Voelkerding, K.V.; et al. Standards and guidelines for validating next-generation sequencing bioinformatics pipelines: A joint recommendation of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagn. 2018, 20, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef] [PubMed]
- hibaud-Nissen, F.; Souvorov, A.; Murphy, T.; DiCuccio, M.; Kitts, P. Eukaryotic genome annotation pipeline. In The NCBI Handbook, 2nd ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2013. Available online: https://www.ncbi.nlm.nih.gov/sites/books/NBK169439/ (accessed on 14 November 2013).
- Kapustin, Y.; Souvorov, A.; Tatusova, T.; Lipman, D. Splign: Algorithms for computing spliced alignments with identification of paralogs. Biol. Direct 2008, 3, 20. [Google Scholar] [CrossRef]
- Fiddes, I.T.; Armstrong, J.; Diekhans, M.; Nachtweide, S.; Kronenberg, Z.N.; Underwood, J.G.; Gordon, D.; Earl, D.; Keane, T.; Eichler, E.E.; et al. Comparative Annotation Toolkit (CAT)—simultaneous clade and personal genome annotation. Genome Res. 2018, 28, 1029–1038. [Google Scholar] [CrossRef]
- Paten, B.; Earl, D.; Nguyen, N.; Diekhans, M.; Zerbino, D.; Haussler, D. Cactus: Algorithms for genome multiple sequence alignment. Genome Res. 2011, 21, 1512–1528. [Google Scholar] [CrossRef]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Lilue, J.; Doran, A.G.; Fiddes, I.T.; Abrudan, M.; Armstrong, J.; Bennett, R.; Chow, W.; Collins, J.; Collins, S.; Czechanski, A.; et al. Sixteen diverse laboratory mouse reference genomes define strain-specific haplotypes and novel functional loci. Nat. Genet. 2018, 50, 1574–1583. [Google Scholar] [CrossRef]
- Kronenberg, Z.N.; Fiddes, I.T.; Gordon, D.; Murali, S.; Cantsilieris, S.; Meyerson, O.S.; Underwood, J.G.; Nelson, B.J.; Chaisson, M.J.P.; Dougherty, M.L.; et al. High-resolution comparative analysis of great ape genomes. Science 2018, 360, 6343. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Lomsadze, A.; Burns, P.D.; Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 2014, 42, e119. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Whole-genome annotation with BRAKER. In Gene Prediction; Springer: New York, NY, USA, 2019; pp. 65–95. [Google Scholar]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 1–15. [Google Scholar] [CrossRef]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Tanizawa, Y.; Fujisawa, T.; Nakamura, Y. DFAST: A flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 2018, 34, 1037–1039. [Google Scholar] [CrossRef]
- Suzuki, S.; Kakuta, M.; Ishida, T.; Akiyama, Y. GHOSTX: An improved sequence homology search algorithm using a query suffix array and a database suffix array. PLoS ONE 2014, 9, e103833. [Google Scholar] [CrossRef] [PubMed]
- Kiełbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Boratyn, G.M.; Schäffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct 2012, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Humann, J.L.; Lee, T.; Ficklin, S.; Main, D. Structural and functional annotation of eukaryotic genomes with GenSAS. In Gene Prediction; Springer: New York, NY, USA, 2019; pp. 29–51. [Google Scholar] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Chang, X.; Wang, K. wANNOVAR: Annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef]
- Sheng, Q.; Yu, H.; Oyebamiji, O.; Wang, J.; Chen, D.; Ness, S.; Zhao, Y.Y.; Guo, Y. AnnoGen: Annotating genome-wide pragmatic features. Bioinformatics 2020, 36, 2899–2901. [Google Scholar] [CrossRef]
- Cavalcante, R.G.; Sartor, M.A. Annotatr: Genomic regions in context. Bioinformatics 2017, 33, 2381–2383. [Google Scholar] [CrossRef]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef]
- Norling, M.; Jareborg, N.; Dainat, J. EMBLmyGFF3: A converter facilitating genome annotation submission to European Nucleotide Archive. BMC Res. Notes 2018, 11, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kong, L.; Gao, G.; Luo, J. A brief introduction to web-based genome browsers. Brief. Bioinform. 2013, 14, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Haeussler, M.; Zweig, A.S.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Hinrichs, A.S.; Gonzalez, J.N.; et al. The UCSC genome browser database: 2019 update. Nucleic Acids Res. 2019, 47, D853–D858. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef]
- Harris, T.W.; Arnaboldi, V.; Cain, S.; Chan, J.; Chen, W.J.; Cho, J.; Davis, P.; Gao, S.; Grove, C.A.; Kishore, R.; et al. WormBase: A modern model organism information resource. Nucleic Acids Res. 2020, 48, D762–D767. [Google Scholar] [CrossRef]
- Thurmond, J.; Goodman, J.L.; Strelets, V.B.; Attrill, H.; Gramates, L.S.; Marygold, S.J.; Matthews, B.B.; Millburn, G.; Antonazzo, G.; Trovisco, V.; et al. FlyBase 2.0: The next generation. Nucleic Acids Res. 2019, 47, D759–D765. [Google Scholar] [CrossRef]
- Portwood, J.L.; Woodhouse, M.R.; Cannon, E.K.; Gardiner, J.M.; Harper, L.C.; Schaeffer, M.L.; Walsh, J.R.; Sen, T.Z.; Cho, K.T.; Schott, D.A.; et al. MaizeGDB 2018: The maize multi-genome genetics and genomics database. Nucleic Acids Res. 2019, 47, D1146–D1154. [Google Scholar] [CrossRef]
- Stein, L.D.; Mungall, C.; Shu, S.; Caudy, M.; Mangone, M.; Day, A.; Nickerson, E.; Stajich, J.E.; Harris, T.W.; Arva, A.; et al. The generic genome browser: A building block for a model organism system database. Genome Res. 2002, 12, 1599–1610. [Google Scholar] [CrossRef]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L.; et al. JBrowse: A dynamic web platform for genome visualization and analysis. Genome Biol. 2016, 17, 1–12. [Google Scholar] [CrossRef]
- Dunn, N.A.; Unni, D.R.; Diesh, C.; Munoz-Torres, M.; Harris, N.L.; Yao, E.; Rasche, H.; Holmes, I.H.; Elsik, C.G.; Lewis, S.E. Apollo: Democratizing genome annotation. PLoS Comput. Biol. 2019, 15, e1006790. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Wang, J.; Zhao, S.; Gu, X.; Luo, J.; Gao, G. ABrowse-a customizable next-generation genome browser framework. BMC Bioinform. 2012, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Medina, I.; Salavert, F.; Sanchez, R.; de Maria, A.; Alonso, R.; Escobar, P.; Bleda, M.; Dopazo, J. Genome Maps, a new generation genome browser. Nucleic Acids Res. 2013, 41, W41–W46. [Google Scholar] [CrossRef]
- Pak, T.R.; Roth, F.P. ChromoZoom: A flexible, fluid, web-based genome browser. Bioinformatics 2013, 29, 384–386. [Google Scholar] [CrossRef]
- Szot, P.S.; Yang, A.; Wang, X.; Parsania, C.; Röhm, U.; Wong, K.H.; Ho, J.W.K. PBrowse: A web-based platform for real-time collaborative exploration of genomic data. Nucleic Acids Res. 2017, 45, e67. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, 1–11. [Google Scholar] [CrossRef]
- Reimand, J.; Arak, T.; Adler, P.; Kolberg, L.; Reisberg, S.; Peterson, H.; Vilo, J. g: Profiler—A web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016, 44, W83–W89. [Google Scholar] [CrossRef] [PubMed]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Scala, G.; Serra, A.; Marwah, V.S.; Saarimäki, L.A.; Greco, D. FunMappOne: A tool to hierarchically organize and visually navigate functional gene annotations in multiple experiments. BMC Bioinform. 2019, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Huynh, T.; Xu, S. Gene Annotation Easy Viewer (GAEV): Integrating KEGG’s Gene Function Annotations and Associated Molecular Pathways. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kim, J.I.; Jeong, Y.S.; Yi, G. AGORA: Organellar genome annotation from the amino acid and nucleotide references. Bioinformatics 2018, 34, 2661–2663. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Pabinger, S.; Dander, A.; Fischer, M.; Snajder, R.; Sperk, M.; Efremova, M.; Krabichler, B.; Speicher, M.R.; Zschocke, J.; Trajanoski, Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief. Bioinform. 2014, 15, 256–278. [Google Scholar] [CrossRef]
- Drori, E.; Levy, D.; Smirin-Yosef, P.; Rahimi, O.; Salmon-Divon, M. CircosVCF: Circos visualization of whole-genome sequence variations stored in VCF files. Bioinformatics 2017, 33, 1392–1393. [Google Scholar] [CrossRef]
- Simonetti, F.L.; Teppa, E.; Chernomoretz, A.; Nielsen, M.; Marino Buslje, C. MISTIC: Mutual information server to infer coevolution. Nucleic Acids Res. 2013, 41, W8–W14. [Google Scholar] [CrossRef]
- An, J.; Lai, J.; Sajjanhar, A.; Batra, J.; Wang, C.; Nelson, C.C. J-Circos: An interactive Circos plotter. Bioinformatics 2015, 31, 1463–1465. [Google Scholar] [CrossRef]
- Yu, Y.; Ouyang, Y.; Yao, W. shinyCircos: An R/Shiny application for interactive creation of Circos plot. Bioinformatics 2018, 34, 1229–1231. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Huss III, J.W.; Orozco, C.; Goodale, J.; Wu, C.; Batalov, S.; Vickers, T.J.; Valafar, F.; Su, A.I. A gene wiki for community annotation of gene function. PLoS Biol. 2008, 6, e175. [Google Scholar] [CrossRef] [PubMed]
- Stein, L. Genome annotation: From sequence to biology. Nat. Rev. Genet. 2001, 2, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. Ideas fly at gene-finding jamboree. Science 2000, 287, 2182–2184. [Google Scholar] [CrossRef]
- Kawai, J.; Shinagawa, A.; Shibata, K.; Yoshino, M.; Itoh, M.; Ishii, Y.; Arakawa, T.; Hara, A.; Fukunishi, Y.; Konno, H.; et al. Functional annotation of a full-length mouse cDNA collection. Nature 2001, 409, 685–689. [Google Scholar]
- Loveland, J.E.; Gilbert, J.G.R.; Griffiths, E.; Harrow, J.L. Community gene annotation in practice. Database 2012, 2012. [Google Scholar] [CrossRef]
- Mazumder, R.; Natale, D.A.; Julio, J.A.E.; Yeh, L.S.; Wu, C.H. Community annotation in biology. Biol. Direct 2010, 5, 1–7. [Google Scholar] [CrossRef]
- Madoui, M.A.; Dossat, C.; d’Agata, L.; van Oeveren, J.; van der Vossen, E.; Aury, J.M. MaGuS: A tool for quality assessment and scaffolding of genome assemblies with Whole Genome Profiling™ Data. BMC Bioinform. 2016, 17, 115. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, I.; Tsoka, S.; Andrade, M.A.; Enright, A.J.; Carroll, M.; Poullet, P.; Promponas, V.; Liakopoulos, T.; Palaios, G.; Pasquier, C.; et al. Evaluation of annotation strategies using an entire genome sequence. Bioinformatics 2003, 19, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Kasukawa, T.; Furuno, M.; Nikaido, I.; Bono, H.; Hume, D.A.; Bult, C.; Hill, D.P.; Baldarelli, R.; Gough, J.; Kanapin, A.; et al. Development and evaluation of an automated annotation pipeline and cDNA annotation system. Genome Res. 2003, 13, 1542–1551. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gilbert, D.; Kim, S. Annotation confidence score for genome annotation: A genome comparison approach. Bioinformatics 2010, 26, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, H.; Goryanin, I. A semi-automated genome annotation comparison and integration scheme. BMC Bioinform. 2013, 14, 1–12. [Google Scholar] [CrossRef]
- Kalkatawi, M.; Alam, I.; Bajic, V.B. BEACON: Automated tool for bacterial GEnome annotation ComparisON. BMC Genom. 2015, 16, 616. [Google Scholar] [CrossRef]
- Eilbeck, K.; Moore, B.; Holt, C.; Yandell, M. Quantitative measures for the management and comparison of annotated genomes. BMC Bioinform. 2009, 10, 67. [Google Scholar] [CrossRef]
- Cochrane, G.; Karsch-Mizrachi, I.; Takagi, T.; Sequence Database Collaboration, I.N. The international nucleotide sequence database collaboration. Nucleic Acids Res. 2016, 44, D48–D50. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Schnoes, A.M.; Brown, S.D.; Dodevski, I.; Babbitt, P.C. Annotation error in public databases: Misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol. 2009, 5, e1000605. [Google Scholar] [CrossRef]
- Jones, C.E.; Brown, A.L.; Baumann, U. Estimating the annotation error rate of curated GO database sequence annotations. BMC Bioinform. 2007, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Klimke, W.; O’Donovan, C.; White, O.; Brister, J.R.; Clark, K.; Fedorov, B.; Mizrachi, I.; Pruitt, K.D.; Tatusova, T. Solving the problem: Genome annotation standards before the data deluge. Stand. Genom. Sci. 2011, 5, 168–193. [Google Scholar] [CrossRef] [PubMed]
- Nobre, T.; Campos, M.D.; Lucic-Mercy, E.; Arnholdt-Schmitt, B. Misannotation awareness: A tale of two gene-groups. Front. Plant Sci. 2016, 7, 868. [Google Scholar] [CrossRef] [PubMed]
- Ouzounis, C.A.; Karp, P.D. The past, present and future of genome-wide re-annotation. Genome Biol. 2002, 3. [Google Scholar] [CrossRef]
- Siezen, R.J.; Van Hijum, S.A.F.T. Genome (re-) annotation and open-source annotation pipelines. Microb. Biotechnol. 2010, 3, 362. [Google Scholar] [CrossRef]
- Yang, H.; Jaime, M.; Polihronakis, M.; Kanegawa, K.; Markow, T.; Kaneshiro, K.; Oliver, B. Re-annotation of eight Drosophila genomes. Life Sci. Alliance 2018, 1. [Google Scholar] [CrossRef]
- Cormier, A.; Avia, K.; Sterck, L.; Derrien, T.; Wucher, V.; Andres, G.; Monsoor, M.; Godfroy, O.; Lipinska, A.; Perrineau, M.M.; et al. Re-annotation, improved large-scale assembly and establishment of a catalogue of noncoding loci for the genome of the model brown alga Ectocarpus. New Phytol. 2017, 214, 219–232. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2017, 89, 789–804. [Google Scholar] [CrossRef]
- Tamaki, S.; Arakawa, K.; Kono, N.; Tomita, M. Restauro-G: A rapid genome re-annotation system for comparative genomics. Genom. Proteom. Bioinform. 2007, 5, 53–58. [Google Scholar] [CrossRef]
- Salzberg, S.L. Genome re-annotation: A wiki solution? Genome Biol. 2007, 8, 1–5. [Google Scholar] [CrossRef]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M.; et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, J.; Uszczynska-Ratajczak, B.; Carbonell, S.; Pérez-Lluch, S.; Abad, A.; Davis, C.; Gingeras, T.R.; Frankish, A.; Harrow, J.; Guigo, R.; et al. High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing. Nat. Genet. 2017, 49, 1731–1740. [Google Scholar] [CrossRef]
- Robert, C.; Kapetanovic, R.; Beraldi, D.; Watson, M.; Archibald, A.L.; Hume, D.A. Identification and annotation of conserved promoters and macrophage-expressed genes in the pig genome. BMC Genom. 2015, 16, 970. [Google Scholar] [CrossRef]
- Li, W.; Yang, W.; Wang, X.J. Pseudogenes: Pseudo or real functional elements? J. Genet. Genom. 2013, 40, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Workman, R.E.; Tang, A.D.; Tang, P.S.; Jain, M.; Tyson, J.R.; Razaghi, R.; Zuzarte, P.C.; Gilpatrick, T.; Payne, A.; Quick, J.; et al. Nanopore native RNA sequencing of a human poly (A) transcriptome. Nat. Methods 2019, 16, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.L. Next-generation genome annotation: We still struggle to get it right, 2019. Genome Biol. 2019, 20. [Google Scholar] [CrossRef]
- Danchin, A.; Ouzounis, C.; Tokuyasu, T.; Zucker, J.D. No wisdom in the crowd: Genome annotation in the era of big data–current status and future prospects. Microb. Biotechnol. 2018, 11, 588–605. [Google Scholar] [CrossRef]
- Reed, J.L.; Famili, I.; Thiele, I.; Palsson, B.O. Towards multidimensional genome annotation. Nat. Rev. Genet. 2006, 7, 130–141. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Buske, O.J.; Wang, J.; Weng, Z.; Bilmes, J.A.; Noble, W.S. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods 2012, 9, 473. [Google Scholar] [CrossRef]
- Yip, K.Y.; Cheng, C.; Gerstein, M. Machine learning and genome annotation: A match meant to be? Genome Biol. 2013, 14, 1–10. [Google Scholar] [CrossRef]
- Nakano, F.K.; Lietaert, M.; Vens, C. Machine learning for discovering missing or wrong protein function annotations. BMC Bioinform. 2019, 20, 485. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Method | Program | Description | URL | Ref |
|---|---|---|---|---|
| Ab initio | EasyGene | HMM-based automatic gene predictor for prokaryotes that ranks open reading frames (ORFs) by statistical significance | https://services.healthtech.dtu.dk/service.php?EasyGene-1.2 | [25] |
| FGENESH | HMM-based gene structure prediction | http://www.softberry.com/berry.phtml?topic=fgenesh&group=programs&subgroup=gfind | [26] | |
| GeneMark | A family of self-training gene prediction programs for bacteria, archaea, metagenomes, metatranscriptomes and eukaryotes | http://opal.biology.gatech.edu/GeneMark/ | [27] | |
| GeneZilla | Generalized hidden Markov model (GHMM) eukaryotic gene finder (formerly known as TIGRscan) | http://www.genezilla.org/ | [28] | |
| GenScan | Algorithm for ab initio prediction of complete gene structures in vertebrate, Drosophila, and plant genomic sequences | http://hollywood.mit.edu/GENSCAN.html | [29] | |
| GlimmerHMM | GHMM-based eukaryotic gene finder that incorporates splice sites from GeneSplicer and decision tree from GlimmerM in Unix environment | http://ccb.jhu.edu/software/glimmerhmm/ | [30] | |
| HMMgene | HMM-based gene predictor for vertebrates and C. elegans, full as well as partial genes | https://services.healthtech.dtu.dk/service.php?HMMgene-1.1 | [31] | |
| mGene | Web service for predicting eukaryotic gene structures, including protein-coding genes and untranslated region (UTR) with pre-trained models | https://galaxy.inf.ethz.ch/tool_runner?tool_id=mgenepredict | [32] | |
| NetGene | Predicts splice sites in human, C. elegans and A. thaliana DNA | https://services.healthtech.dtu.dk/service.php?NetGene2-2.42 | [33] | |
| RNAmmer | A two level HMM-based predictor of rRNA genes in full genome sequences | http://www.cbs.dtu.dk/services/RNAmmer/ | [34] | |
| SNAP | Semi-HMM general-purpose gene finding program suitable for both eukaryotic and prokaryotic genomes | https://github.com/KorfLab/SNAP | [35] | |
| tRNAscan-SE | A covariance model-based program that provides genomic coordinates, predicted function, and secondary structure of tRNA genes | http://lowelab.ucsc.edu/tRNAscan-SE/ | [36] | |
| Homology | GeMoMa | A program that uses annotated genes to infer protein-coding genes in a target genome | http://galaxy.informatik.uni-halle.de/ | [37] |
| GenomeThreader | Uses cDNA, EST and protein sequences to predict gene structures via spliced alignments | http://genomethreader.org/ | [38] | |
| PPFINDER | Identifier of processed pseudogenes incorporated in mammalian genome annotation | https://mblab.wustl.edu/software.html | [39] | |
| PseudoPipe | A computational pipeline that searches a mammalian genome and identifies pseudogene sequences | http://www.pseudogene.org/pseudopipe/ | [40] | |
| TWINSCAN | GenScan extension, gene structure prediction system that exploits homology of related genomes | https://mblab.wustl.edu/software.html | [41] | |
| Combined | AUGUSTUS | An ab initio gene prediction program that can also incorporate extrinsic sources, e.g., EST alignment, protein alignments and syntetic genome alignments | http://bioinf.uni-greifswald.de/augustus/ | [42] |
| JIGSAW | Gene model predictor that combines outputs from other gene finders, splice site predictors, and sequence alignments | http://www.cbcb.umd.edu/software/jigsaw/ | [43] |
| Program | Description | URL | Ref |
|---|---|---|---|
| BLAST2GO | A comprehensive bioinformatics tool for functional annotation of sequences and data mining on annotation results | https://www.blast2go.com/ | [70] |
| FastAnnotator | An integration of well-established annotation tools for annotation of transcripts, which assigns GO terms, enzyme commission numbers, and functional domains | - | [71] |
| GO FEAT | Homology-based functional annotation tool for genomic and transcriptomic data | http://computationalbiology.ufpa.br/gofeat/ | [72] |
| GOtcha | A method that predicts gene product function by annotation with GO terms | http://www.compbio.dundee.ac.uk/gotcha/gotcha.php | [73] |
| PANNZER2 | A fully automated service for functional annotation of prokaryotic and eukaryotic proteins of unknown function that provides both GO annotations and free text description predictions | http://ekhidna2.biocenter.helsinki.fi/sanspanz/ | [74] |
| PoGO | A statistical pattern recognition method that assigns GO terms for fungal proteins | - | [75] |
| Gene Prediction | Source of Data | Evolutionary Distance Effect | Strength | |
|---|---|---|---|---|
| Ab initio | Rely on statistical model and gene signal | Models (HMM, GHMM, WAM) that can be trained supervised or unsupervised | Medium | Fast and easy means to identify and novel genes |
| Homology | Rely on sequence alignment | Proteins, EST, cDNA | High | Better accuracy, suitable for functional annotations |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ejigu, G.F.; Jung, J. Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing. Biology 2020, 9, 295. https://doi.org/10.3390/biology9090295
Ejigu GF, Jung J. Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing. Biology. 2020; 9(9):295. https://doi.org/10.3390/biology9090295
Chicago/Turabian StyleEjigu, Girum Fitihamlak, and Jaehee Jung. 2020. "Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing" Biology 9, no. 9: 295. https://doi.org/10.3390/biology9090295
APA StyleEjigu, G. F., & Jung, J. (2020). Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing. Biology, 9(9), 295. https://doi.org/10.3390/biology9090295