A Putative Prophylactic Solution for COVID-19: Development of Novel Multiepitope Vaccine Candidate against SARS-COV-2 by Comprehensive Immunoinformatic and Molecular Modelling Approach


 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Coronavirus Protein Sequences and Structural Information
2.2. Prediction of Linear and Conformational B-Cell Epitopes
2.3. Prediction of Potential Cytotoxic T-Lymphocyte (CTL) Epitopes
2.4. Epitope Prediction of Helper T-Cell
2.5. Multiepitope Vaccine Designing
2.6. Antigenicity and Allergenicity Estimation of the MVC
2.7. Physiochemical Parameters Evaluation
2.8. Tertiary Structure Prediction and Refinement of MVC
2.9. Stability Enhancement of MVC by Disulfide Engineering
2.10. Molecular Docking of Vaccine Constructs with TLR4
2.11. Molecular Dynamics Simulation for TLRs/MVC Complex
2.12. Codon Adaptation and In Silico Cloning
2.13. In Silico Immune Simulation
3. Results
3.1. Antigenic B-Cell Epitope Prediction
3.2. Prediction of Cytotoxic T-Lymphocyte (CTL) Epitopes
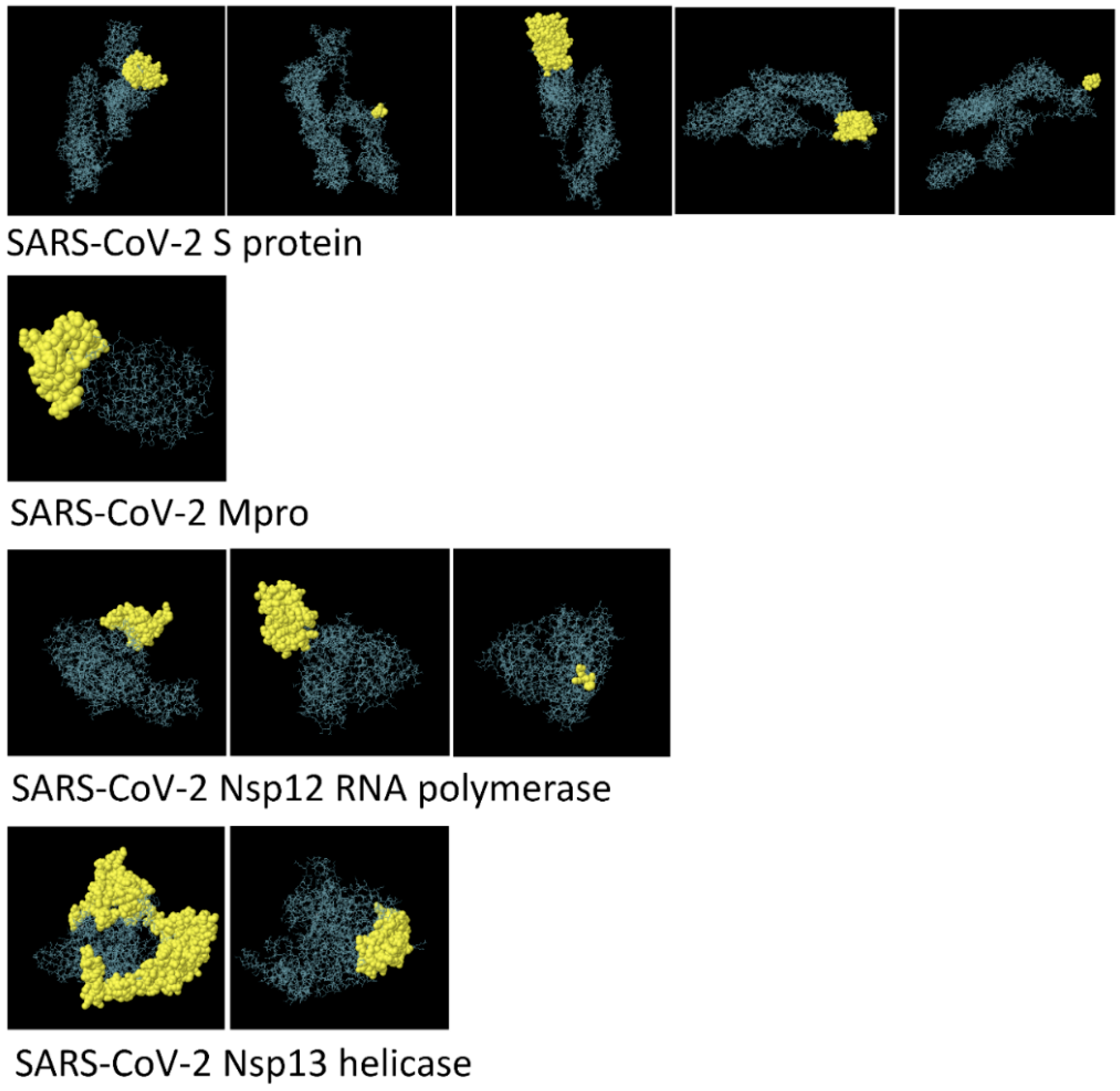
3.3. Structure-Based Epitope Prediction
3.4. Epitope Prediction for (HTL) Helper T Lymphocytes
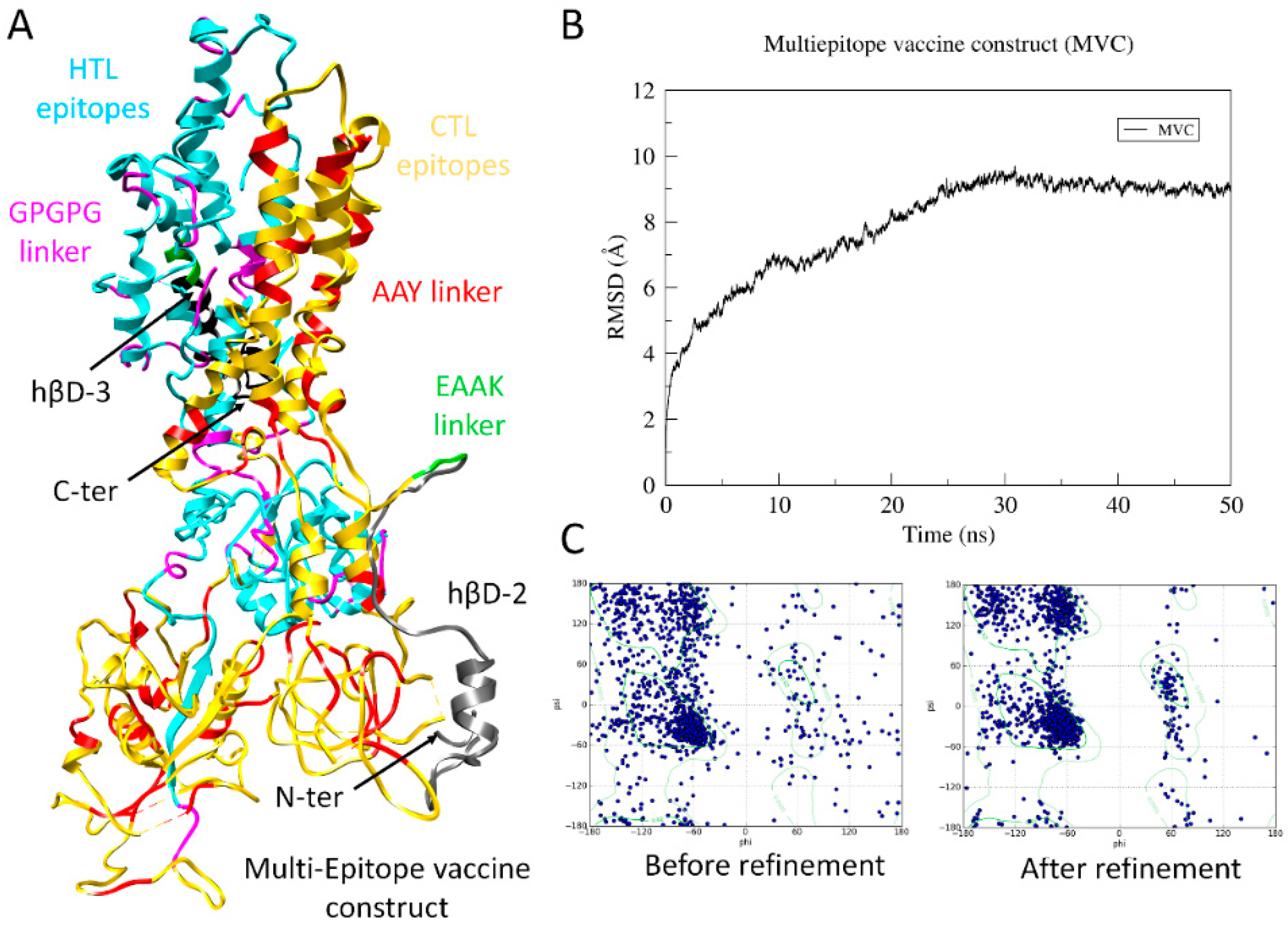
3.5. Design and Construction of Final Multiepitope Vaccine
3.6. Parametric Evaluation of Physiochemical Properties
3.7. Assessment of Allergenicity and Immunogenicity
3.8. Structure Prediction and Validation of MVC
3.9. Disulfide Engineering for Vaccine Stability
3.10. Molecular Docking of Vaccine Constructs with TLR3 and TLR4
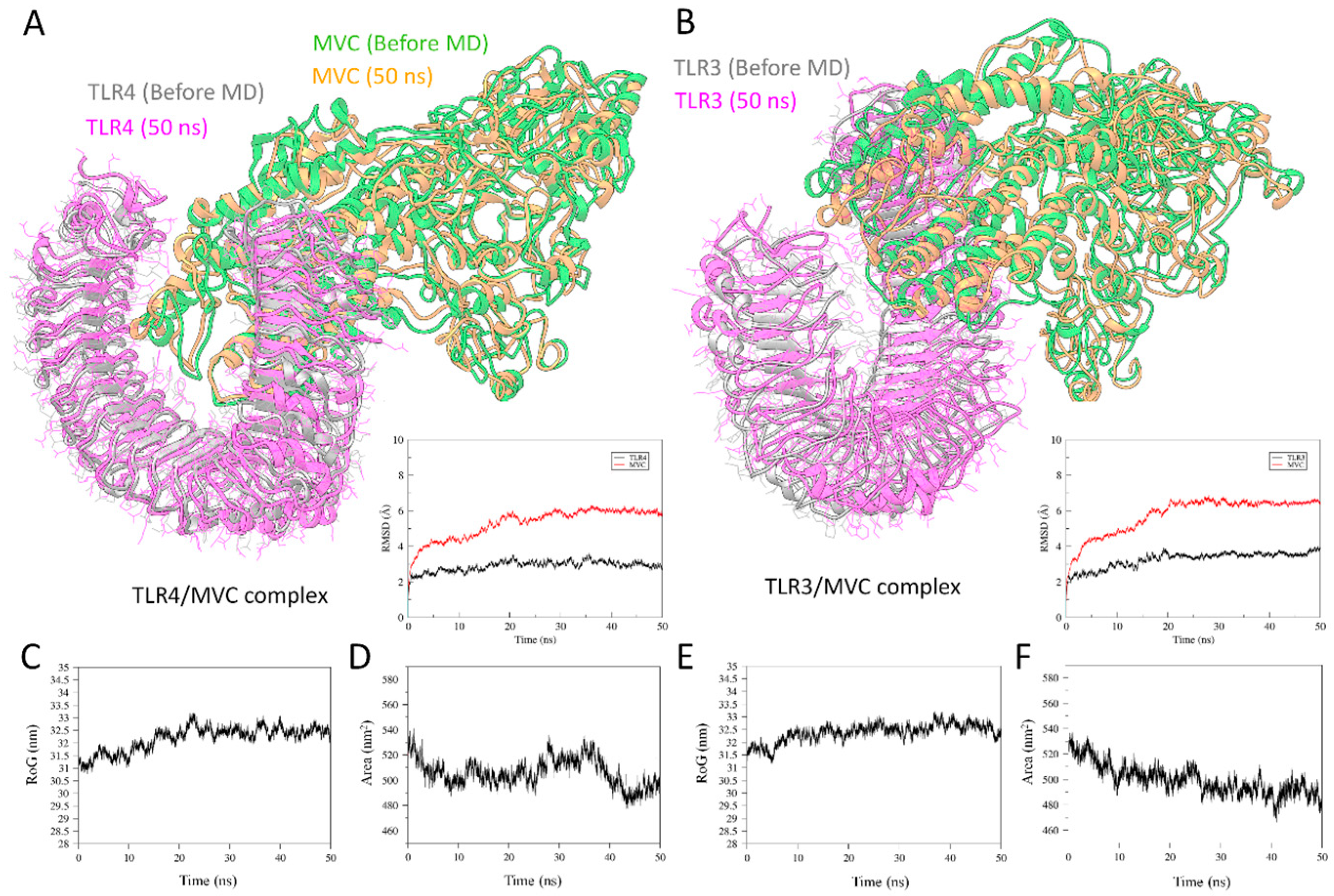
3.11. Molecular Dynamics Simulation for TLRs/MVC Complex
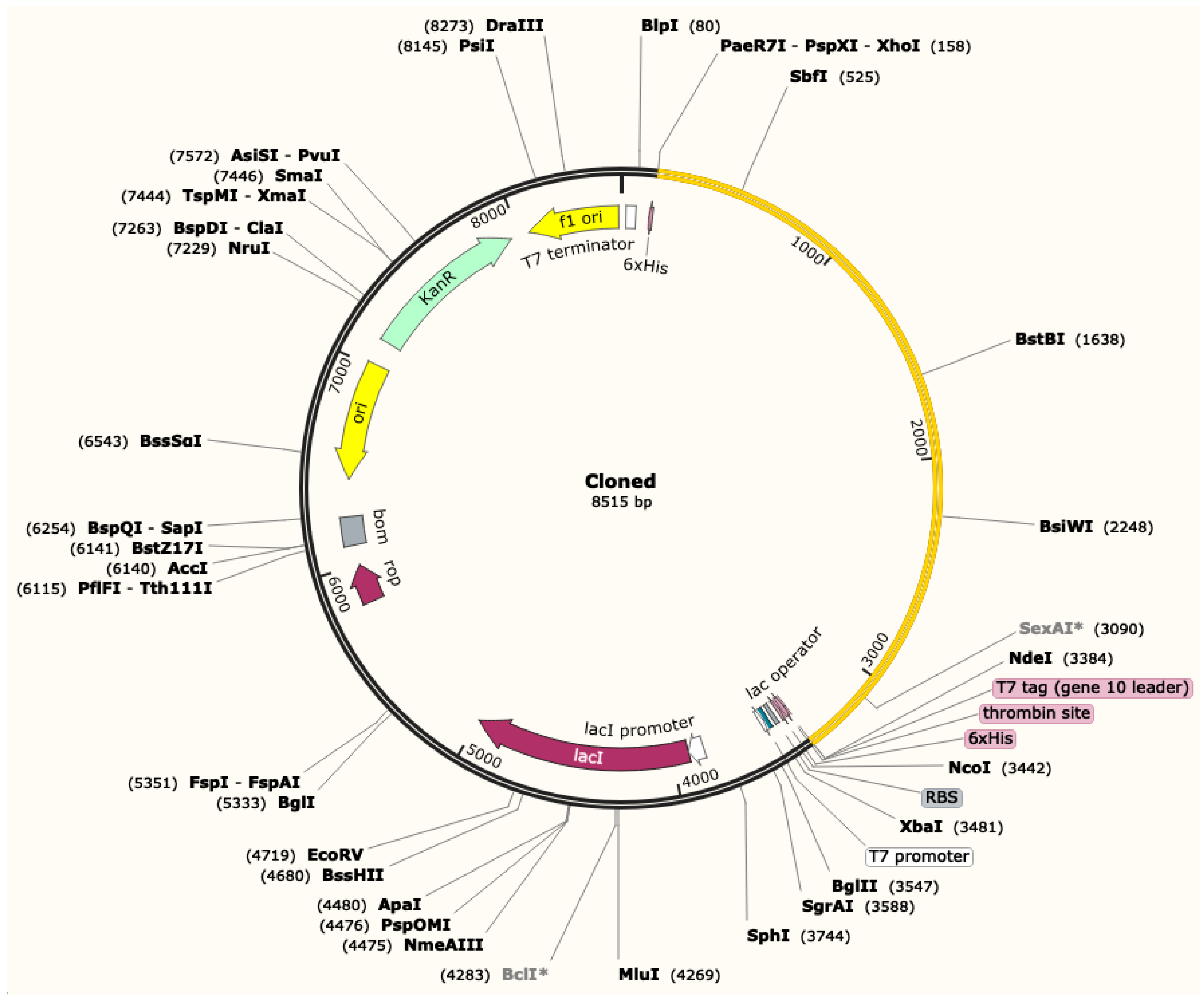
3.12. Codon Adaptation and In Silico Cloning of the MVC
3.13. Immune Simulation by MVC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parry, J. China coronavirus: Cases surge as official admits human to human transmission. BMJ 2020, 368, m236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benvenuto, D.; Giovanetti, M.; Ciccozzi, A.; Spoto, S.; Angeletti, S.; Ciccozzi, M. The 2019-new Coronavirus epidemic: Evidence for virus evolution. J. Med. Virol. 2020, 92, 455–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Perlman, S. Another Decade, Another Coronavirus. 2020, 760–762. [Google Scholar] [CrossRef]
- WHO. Coronavirus Disease 2019 (COVID-19) Situation Report-52. Available online: https://www.who.int/docs/default-source/coronaviruse/20200312-sitrep-52-covid-19.pdf?sfvrsn=e2bfc9c0_2 (accessed on 12 March 2020).
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Azhar, E.I.; Hui, D.S.; Memish, Z.A.; Drosten, C.; Zumla, A. The Middle East Respiratory Syndrome (MERS). Infect. Dis. Clin. 2019, 33, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging Coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Buchholz, U.J.; Bukreyev, A.; Yang, L.; Lamirande, E.W.; Murphy, B.R.; Subbarao, K.; Collins, P.L. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc. Nat. Acad. Sci. USA 2004, 101, 9804–9809. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Xie, J.; He, Y.; Fan, H.; Baril, L.; Qiu, Z.; Han, Y.; Xu, W.; Zhang, W.; You, H.; et al. Long-term persistence of robust antibody and cytotoxic T cell responses in recovered patients infected with SARS coronavirus. PLoS ONE 2006, 1, e24. [Google Scholar] [CrossRef] [Green Version]
- Zakhartchouk, A.N.; Sharon, C.; Satkunarajah, M.; Auperin, T.; Viswanathan, S.; Mutwiri, G.; Petric, M.; See, R.H.; Brunham, R.C.; Finlay, B.B.; et al. Immunogenicity of a receptor-binding domain of SARS coronavirus spike protein in mice: Implications for a subunit vaccine. Vaccine 2007, 25, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Y.; Kobinger, G.P.; Jordan, H.; Suchma, K.; Weiss, S.R.; Shen, H.; Schumer, G.; Gao, G.; Boyer, J.L.; Crystal, R.G.; et al. Identification of murine CD8 T cell epitopes in codon-optimized SARS-associated coronavirus spike protein. Virology 2005, 335, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. LoS Biol. 2005, 3. [Google Scholar]
- Bacha, U.; Barrila, J.; Velazquez-Campoy, A.; Leavitt, S.A.; Freire, E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry 2004, 43, 4906–4912. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Yan, L.; Ren, Z.; Wu, L.; Wang, J.; Guo, J.; Zheng, L.; Ming, Z.; Zhang, L.; Lou, Z.; et al. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019, 47, 6538–6550. [Google Scholar] [CrossRef] [Green Version]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhao, S.; Teng, T.; Abdalla, A.E.; Zhu, W.; Xie, L.; Wang, Y.; Guo, X. Systematic Comparison of Two Animal-to-Human Transmitted Human Coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 2020, 12, 244. [Google Scholar] [CrossRef] [Green Version]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. Chembiochem 2020, 21, 730–738. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 149–150. [Google Scholar] [CrossRef] [Green Version]
- Shang, W.; Yang, Y.; Rao, Y.; Rao, X. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. NPJ Vaccines 2020, 5, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andaleeb Sajid, S.; Singh, Y.; Shukla, P. Computational tools for modern vaccine development. Hum Vaccin Immunother. 2019. [Google Scholar]
- Havranek, B.; Islam, S.M. An in silico approach for identification of novel inhibitors as potential therapeutics targeting COVID-19 main protease. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Abu-Saleh, A.A.-A.A.; Awad, I.; Yadav, A.; Poirier, R.A. Computational Design of Potent Inhibitors for SARS-CoV-2′s Main Protease. ChemRxiv 2020. [Google Scholar]
- Arnon, R.; Ben-Yedidia, T. Old and new vaccine approaches. Int. Immunopharmacol. 2003, 3, 1195–1204. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Felipe, L.S.; Vercruysse, T.; Sharma, S.; Ma, J.; Lemmens, V.; van Looveren, D.; Javarappa, M.P.A.; Boudewijns, R.; Malengier-Devlies, B.; Kaptein, S.F.; et al. A single-dose live-attenuated YF17D-vectored SARS-CoV2 vaccine candidate. BioRxiv 2020. [Google Scholar] [CrossRef]
- Damfo, S.A.; Reche, P.; Gatherer, D.; Flower, D.R. In silico design of knowledge-based Plasmodium falciparum epitope ensemble vaccines. J. Mol. Graph. Model. 2017, 78, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Jabbar, B.; Rafique, S.; Salo-Ahen, O.M.; Ali, A.; Munir, M.; Idrees, M.; Mirza, M.U.; Vanmeert, M.; Shah, S.Z.; Jabbar, I. Antigenic Peptide Prediction From E6 and E7 Oncoproteins of HPV Types 16 and 18 for Therapeutic Vaccine Design Using Immunoinformatics and MD Simulation Analysis. Front. Immunol. 2018, 9, 3000. [Google Scholar] [CrossRef] [Green Version]
- Michel-Todó, L.; Reche, P.A.; Bigey, P.; Pinazo, M.-J.; Gascón, J.; Alonso-Padilla, J. In silico design of an epitope-based vaccine ensemble for Chagas disease. Front. Immunol. 2019, 10, 2698. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.U.; Rafique, S.; Ali, A.; Munir, M.; Ikram, N.; Manan, A.; Salo-Ahen, O.M.; Idrees, M. Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins. Sci. Rep. 2016, 6, 37313. [Google Scholar] [CrossRef] [PubMed]
- Sabetian, S.; Nezafat, N.; Dorosti, H.; Zarei, M.; Ghasemi, Y. Exploring dengue proteome to design an effective epitope-based vaccine against dengue virus. J. Biomol. Struct. Dyn. 2019, 37, 2546–2563. [Google Scholar] [CrossRef] [PubMed]
- Ul Qamar, M.T.; Saleem, S.; Ashfaq, U.A.; Bari, A.; Anwar, F.; Alqahtani, S. Epitope-based peptide vaccine design and target site depiction against Middle East Respiratory Syndrome Coronavirus: An immune-informatics study. J. Transl. Med. 2019, 17, 362. [Google Scholar] [CrossRef] [PubMed]
- Kathwate, G.H. In silico design and characterization of multiepitopes vaccine for SARS-CoV2 from its Spike proteins. BioRxiv 2020. [Google Scholar] [CrossRef]
- Bhatnager, R.; Bhasin, M.; Arora, J.; Dang, A.S. Epitope based peptide vaccine against SARS-COV2: An immune-informatics approach. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Green, D.R. SARS-CoV2 Vaccines: Slow is Fast. Am. Assoc. Adv. Sci. 2020. [Google Scholar] [CrossRef]
- de Oliveira Tosta, S.F.; Passos, M.S.; Kato, R.; Salgado, Á.; Xavier, J.; Jaiswal, A.K.; Soares, S.C.; Azevedo, V.; Giovanetti, M.; Tiwari, S.; et al. Multi-epitope based vaccine against Yellow fever virus applying immunoinformatics approaches. J. Biomol. Struct. Dyn. 2019, 1–28. [Google Scholar] [CrossRef]
- Srivastava, S.; Kamthania, M.; Kumar Pandey, R.; Kumar Saxena, A.; Saxena, V.; Kumar Singh, S.; Kumar Sharma, R.; Sharma, N. Design of novel multi-epitope vaccines against severe acute respiratory syndrome validated through multistage molecular interaction and dynamics. J. Biomol. Struct. Dyn. 2019, 37, 4345–4360. [Google Scholar] [CrossRef]
- Goodman, A.G.; Heinen, P.P.; Guerra, S.; Vijayan, A.; Sorzano, C.O.S.; Gomez, C.E.; Esteban, M. A human multi-epitope recombinant vaccinia virus as a universal T cell vaccine candidate against influenza virus. PLoS ONE 2011, 6, e25938. [Google Scholar] [CrossRef] [Green Version]
- Shahid, F.; Ashfaq, U.A.; Javaid, A.; Khalid, H. Immunoinformatics guided rational design of a next generation multi epitope based peptide (MEBP) vaccine by exploring Zika virus proteome. Infect. Genet. Evol. 2020, 80, 104199. [Google Scholar] [CrossRef]
- Nosrati, M.; Behbahani, M.; Mohabatkar, H. Towards the first multi-epitope recombinant vaccine against Crimean-Congo hemorrhagic fever virus: A computer-aided vaccine design approach. J Biomed Inform. 2019, 93, 103160. [Google Scholar] [CrossRef]
- Khatoon, N.; Pandey, R.K.; Ojha, R.; Aathmanathan, V.S.; Krishnan, M.; Prajapati, V.K. Exploratory algorithm to devise multi-epitope subunit vaccine by investigating Leishmania donovani membrane proteins. J. Biomol. Struct. Dyn. 2019, 37, 2381–2393. [Google Scholar] [CrossRef] [PubMed]
- Dorosti, H.; Eslami, M.; Negahdaripour, M.; Ghoshoon, M.B.; Gholami, A.; Heidari, R.; Dehshahri, A.; Erfani, N.; Nezafat, N.; Ghasemi, Y.; et al. Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. J. Biomol. Struct. Dyn. 2019, 37, 3524–3535. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yin, R.; Liu, K.; Lv, X.; Li, Y.; Duan, X.; Chu, Y.; Xi, T.; Xing, Y. Immunological features and efficacy of a multi-epitope vaccine CTB-UE against H. pylori in BALB/c mice model. Appl. Microbiol. Biotechnol. 2014, 98, 3495–3507. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.-Y.; Shi, Y.; Wu, C.; Zhang, W.-J.; Mao, X.-H.; Guo, G.; Li, H.-X.; Zou, Q.-M. Therapeutic efficacy of a multi-epitope vaccine against Helicobacter pylori infection in BALB/c mice model. Vaccine 2009, 27, 5013–5019. [Google Scholar] [CrossRef]
- Depla, E.; Van der Aa, A.; Livingston, B.D.; Crimi, C.; Allosery, K.; De Brabandere, V.; Krakover, J.; Murthy, S.; Huang, M.; Power, S.; et al. Rational design of a multiepitope vaccine encoding T-lymphocyte epitopes for treatment of chronic hepatitis B virus infections. J. Virol. 2008, 82, 435–450. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Li, D.; Fu, Y.; Bai, Q.; Chen, Y.; Bai, X.; Jing, Z.; Sun, P.; Bao, H.; Li, P.; et al. Rational design and efficacy of a multi-epitope recombinant protein vaccine against foot-and-mouth disease virus serotype A in pigs. Antiviral. Res. 2017, 140, 133–141. [Google Scholar] [CrossRef]
- Van Duin, D.; Medzhitov, R.; Shaw, A.C. Triggering TLR signaling in vaccination. Trends Immunol. 2006, 27, 49–55. [Google Scholar] [CrossRef]
- Mirza, M.U.; Froeyen, M. Structural Elucidation of SARS-CoV-2 Vital Proteins: Computational Methods Reveal Potential Drug Candidates against Main Protease, Nsp12 RNA-dependent RNA Polymerase and Nsp13 Helicase. J. Pharm. Anal. 2020, 10, 320–328. [Google Scholar] [CrossRef]
- Nair, D.T.; Singh, K.; Siddiqui, Z.; Nayak, B.P.; Rao, K.V.; Salunke, D.M. Epitope recognition by diverse antibodies suggests conformational convergence in an antibody response. J. Immunol. 2002, 168, 2371–2382. [Google Scholar] [CrossRef] [Green Version]
- Fieser, T.M.; Tainer, J.A.; Geysen, H.M.; Houghten, R.A.; Lerner, R.A. Influence of protein flexibility and peptide conformation on reactivity of monoclonal anti-peptide antibodies with a protein alpha-helix. Proc. Nat. Acad. Sci. USA 1987, 84, 8568–8572. [Google Scholar] [CrossRef] [Green Version]
- Emini, E.A.; Hughes, J.V.; Perlow, D.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolaskar, A.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef] [Green Version]
- Karplus, P.; Schulz, G. Prediction of chain flexibility in proteins. Naturwissenschaften 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [Green Version]
- Thornton, J.; Edwards, M.; Taylor, W.; Barlow, D. Location of ‘continuous’ antigenic determinants in the protruding regions of proteins. EMBO J. 1986, 5, 409–413. [Google Scholar] [CrossRef]
- Taylor, W.; Thornton, J.t.; Turnell, W. An ellipsoidal approximation of protein shape. J. Mol. Graph. Model. 1983, 1, 30–38. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lund, O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinform. 2009, 10, 296. [Google Scholar] [CrossRef] [Green Version]
- Kohlgraf, K.G.; Pingel, L.C.; Dietrich, D.E.; Brogden, K.A. Defensins as anti-inflammatory compounds and mucosal adjuvants. Future Microbiol. 2010, 5, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Park, M.S.; Kim, J.I.; Lee, I.; Park, S.; Bae, J.-Y.; Park, M.-S. Towards the application of human defensins as antivirals. Biomol. Ther. (Seoul) 2018, 26, 242. [Google Scholar] [CrossRef]
- Weinberg, A.; Jin, G.; Sieg, S.; McCormick, T.S. The yin and yang of human Beta-defensins in health and disease. Front. Immunol. 2012, 3, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulla, F.; Adhikari, U.K.; Uddin, M.K. Exploring T & B-cell epitopes and designing multi-epitope subunit vaccine targeting integration step of HIV-1 lifecycle using immunoinformatics approach. Microb. Pathog. 2019, 137, 103791. [Google Scholar] [PubMed]
- Saha, S.; Raghava, G. AlgPred: Prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2005; pp. 571–607. [Google Scholar]
- Ma, J.; Wang, S.; Zhao, F.; Xu, J. Protein threading using context-specific alignment potential. Bioinformatics 2013, 29, i257–i265. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall III, W.B.; Snoeyink, J.; Richardson, J.S. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [Green Version]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255. [Google Scholar] [CrossRef]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the C lus P ro server motivated by CAPRI. Proteins 2017, 85, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahbar, M.R.; Ghasemi, Y. Designing an efficient multi-epitope oral vaccine against Helicobacter pylori using immunoinformatics and structural vaccinology approaches. Mol. Syst. Biol. 2017, 13, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Castiglione, F. Immune system simulation online. Bioinformatics 2011, 27, 2013–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Nielsen, M.; Lundegaard, C.; Lund, O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinform. 2007, 8, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. Des. Sel. 2001, 14, 529–532. [Google Scholar] [CrossRef]
- Bergmann, C.C.; Yao, Q.; Ho, C.-K.; Buckwold, S.L. Flanking residues alter antigenicity and immunogenicity of multi-unit CTL epitopes. J. Immunol. 1996, 157, 3242–3249. [Google Scholar]
- Bachmair, A.; Finley, D.; Varshavsky, A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986, 234, 179–186. [Google Scholar] [CrossRef]
- Ikai, A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Brooks, C.L., III. Can molecular dynamics simulations provide high-resolution refinement of protein structure? Proteins 2007, 67, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Mirjalili, V.; Noyes, K.; Feig, M. Physics-based protein structure refinement through multiple molecular dynamics trajectories and structure averaging. Proteins 2014, 82, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Raval, A.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Refinement of protein structure homology models via long, all-atom molecular dynamics simulations. Proteins 2012, 80, 2071–2079. [Google Scholar] [CrossRef]
- Ikram, N.; Mirza, M.U.; Vanmeert, M.; Froeyen, M.; Salo-Ahen, O.M.; Tahir, M.; Qazi, A.; Ahmad, S. Inhibition of Oncogenic Kinases: An In Vitro Validated Computational Approach Identified Potential Multi-Target Anticancer Compounds. Biomolecules 2019, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Iman, K.; Mirza, M.U.; Mazhar, N.; Vanmeert, M.; Irshad, I.; Kamal, M.A. In silico Structure-based Identification of Novel Acetylcholinesterase Inhibitors Against Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2018, 17, 54–68. [Google Scholar] [CrossRef]
- Mirza, M.U.; Ikram, N. Integrated computational approach for virtual hit identification against ebola viral proteins VP35 and VP40. Int. J. Mol. Sci. 2016, 17, 1748. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.U.; Vanmeert, M.; Froeyen, M.; Ali, A.; Rafique, S.; Idrees, M. In silico structural elucidation of RNA-dependent RNA polymerase towards the identification of potential Crimean-Congo Hemorrhagic Fever Virus inhibitors. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: A fully automated algorithm for protein–protein docking. Nucleic Acids Res. 2004, 32, W96–W99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Tsai, C.-J.; Nussinov, R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. 1997, 10, 999–1012. [Google Scholar] [CrossRef] [Green Version]
- McGillewie, L.; Soliman, M.E. The binding landscape of plasmepsin V and the implications for flap dynamics. Mol. Syst. Biol. 2016, 12, 1457–1467. [Google Scholar]
- Sindhu, T.; Srinivasan, P. Exploring the binding properties of agonists interacting with human TGR5 using structural modeling, molecular docking and dynamics simulations. RSC Adv. 2015, 5, 14202–14213. [Google Scholar] [CrossRef]
- Richmond, T.J. Solvent accessible surface area and excluded volume in proteins: Analytical equations for overlapping spheres and implications for the hydrophobic effect. J. Mol. Biol. 1984, 178, 63–89. [Google Scholar] [CrossRef]
- Livingston, B.; Crimi, C.; Newman, M.; Higashimoto, Y.; Appella, E.; Sidney, J.; Sette, A. A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J. Immunol. 2002, 168, 5499–5506. [Google Scholar] [CrossRef] [PubMed]
- Saadi, M.; Karkhah, A.; Nouri, H.R. Development of a multi-epitope peptide vaccine inducing robust T cell responses against brucellosis using immunoinformatics based approaches. Infect. Genet. Evol. 2017, 51, 227–234. [Google Scholar] [CrossRef]
- Mahmoodi, S.; Nezafat, N.; Barzegar, A.; Negahdaripour, M.; R Nikanfar, A.; Zarghami, N.; Ghasemi, Y. Harnessing bioinformatics for designing a novel multiepitope peptide vaccine against breast cancer. Curr Pharm. Biotechno. 2016, 17, 1100–1114. [Google Scholar] [CrossRef]
- Jiang, P.; Cai, Y.; Chen, J.; Ye, X.; Mao, S.; Zhu, S.; Xue, X.; Chen, S.; Zhang, L. Evaluation of tandem Chlamydia trachomatis MOMP multi-epitopes vaccine in BALB/c mice model. Vaccine 2017, 35, 3096–3103. [Google Scholar] [CrossRef]
- Lennerz, V.; Gross, S.; Gallerani, E.; Sessa, C.; Mach, N.; Boehm, S.; Hess, D.; Von Boehmer, L.; Knuth, A.; Ochsenbein, A.F.; et al. Immunologic response to the survivin-derived multi-epitope vaccine EMD640744 in patients with advanced solid tumors. Cancer Immunol. Immunother. 2014, 63, 381–394. [Google Scholar] [CrossRef]
- Zhu, S.; Feng, Y.; Rao, P.; Xue, X.; Chen, S.; Li, W.; Zhu, G.; Zhang, L. Hepatitis B virus surface antigen as delivery vector can enhance Chlamydia trachomatis MOMP multi-epitope immune response in mice. Appl. Microbiol. Biotechnol. 2014, 98, 4107–4117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Multi-epitope vaccines: A promising strategy against tumors and viral infections. Immunol. Cell Biol. 2018, 15, 182–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappuoli, R. Reverse vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Channappanavar, R.; Perlman, S. Evaluation of Activation and Inflammatory Activity of Myeloid Cells during Pathogenic Human Coronavirus Infection. In MERS Coronavirus: Methods and Protocols; Vijay, R., Ed.; Springer US: New York, NY, USA, 2020; pp. 195–204. [Google Scholar]
- Yin, D.; Li, L.; Song, X.; Li, H.; Wang, J.; Ju, W.; Qu, X.; Song, D.; Liu, Y.; Meng, X. A novel multi-epitope recombined protein for diagnosis of human brucellosis. BMC Infect. Dis. 2016, 16, 219. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.N.; Flower, D.R. Harnessing bioinformatics to discover new vaccines. Drug Discov. Today 2007, 12, 389–395. [Google Scholar] [CrossRef]
- Slingluff, C.L.; Lee, S.; Zhao, F.; Chianese-Bullock, K.A.; Olson, W.C.; Butterfield, L.H.; Whiteside, T.L.; Leming, P.D.; Kirkwood, J.M. A randomized phase II trial of multiepitope vaccination with melanoma peptides for cytotoxic T cells and helper T cells for patients with metastatic melanoma (E1602). Clin. Cancer Res. 2013, 19, 4228–4238. [Google Scholar] [CrossRef] [Green Version]
- Toledo, H.; Baly, A.; Castro, O.; Resik, S.; Laferté, J.; Rolo, F.; Navea, L.; Lobaina, L.; Cruz, O.; Míguez, J.; et al. A phase I clinical trial of a multi-epitope polypeptide TAB9 combined with Montanide ISA 720 adjuvant in non-HIV-1 infected human volunteers. Vaccine 2001, 19, 4328–4336. [Google Scholar] [CrossRef]
- Yu, K.; Liu, C.; Kim, B.-G.; Lee, D.-Y. Synthetic fusion protein design and applications. Biotechnol. Adv. 2015, 33, 155–164. [Google Scholar] [CrossRef]
- Yano, A.; Onozuka, A.; Asahi-Ozaki, Y.; Imai, S.; Hanada, N.; Miwa, Y.; Nisizawa, T. An ingenious design for peptide vaccines. Vaccine 2005, 23, 2322–2326. [Google Scholar] [CrossRef]
- Kavoosi, M.; Creagh, A.L.; Kilburn, D.G.; Haynes, C.A. Strategy for selecting and characterizing linker peptides for CBM9-tagged fusion proteins expressed in Escherichia coli. Biotechnol. Bioeng. 2007, 98, 599–610. [Google Scholar] [CrossRef]
- Scheiblhofer, S.; Laimer, J.; Machado, Y.; Weiss, R.; Thalhamer, J. Influence of protein fold stability on immunogenicity and its implications for vaccine design. Expert Rev. Vaccines 2017, 16, 479–489. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Start | End | Peptide | Length |
|---|---|---|---|---|
| 1 | 15 | 23 | GCMVQVTCG | 9 |
| 2 | 32 | 45 | LDDVVYCPRHVICT | 14 |
| 3 | 65 | 72 | NFLVQAGN | 8 |
| 4 | 83 | 91 | QNCVLKLKV | 9 |
| 5 | 101 | 107 | YKFVRIQ | 7 |
| 6 | 111 | 120 | TFSVLACYNG | 10 |
| 7 | 123 | 129 | SGVYQCA | 7 |
| 8 | 153 | 162 | DYDCVSFCYM | 10 |
| 9 | 201 | 212 | TVNVLAWLYAAV | 12 |
| 10 | 244 | 253 | QDHVDILGPL | 10 |
| 11 * | 258 | 271 | GIAVLDMCASLKEL | 14 |
| No. | Start | End | Peptide | Length |
|---|---|---|---|---|
| 17 | 395 | 400 | CFSVAA | 6 |
| 3 | 50 | 56 | KTNCCRF | 7 |
| 8 | 171 | 177 | ILRVYAN | 7 |
| 10 | 201 | 207 | IVGVLTL | 7 |
| 13 | 327 | 333 | GPLVRKI | 7 |
| 20 | 557 | 563 | VAGVSIC | 7 |
| 21 | 573 | 579 | QKLLKSI | 7 |
| 22 | 585 | 591 | ATVVIGT | 7 |
| 26 | 670 | 676 | GGSLYVK | 7 |
| 28 | 725 | 731 | HRLYECL | 7 |
| 31 | 773 | 779 | QGLVASI | 7 |
| 2 | 28 | 35 | TDVVYRAF | 8 |
| 6 | 125 | 132 | ADLVYALR | 8 |
| 15 | 350 | 357 | ELGVVHNQ | 8 |
| 16 | 369 | 376 | KELLVYAA | 8 |
| 18 | 435 | 442 | VELKHFFF | 8 |
| 23 | 633 | 640 | MASLVLAR | 8 |
| 29 | 744 | 751 | EFYAYLRK | 8 |
| 32 | 783 | 790 | KSVLYYQN | 8 |
| 34 | 825 | 832 | DYVYLPYP | 8 |
| 4 | 67 | 75 | DSYFVVKRH | 9 |
| 25 | 658 | 666 | ECAQVLSEM | 9 |
| 30 | 760 | 768 | DDAVVCFNS | 9 |
| 35 | 839 | 847 | GAGCFVDDI | 9 |
| 36 | 859 | 867 | FVSLAIDAY | 9 |
| 1 | 8 | 17 | LNRVCGVSAA | 10 |
| 27 | 694 | 703 | FNICQAVTAN | 10 |
| 33 | 810 | 819 | HEFCSQHTML | 10 |
| 7 | 144 | 154 | EILVTYNCCDD | 11 |
| 9 | 183 | 193 | RQALLKTVQFC | 11 |
| 14 | 335 | 345 | VDGVPFVVSTG | 11 |
| 24 | 643 | 653 | TTCCSLSHRFY | 11 |
| 5 | 87 | 99 | YNLLKDCPAVAKH | 13 |
| 37 * | 878 | 890 | ADVFHLYLQYIRK | 13 |
| 19 | 466 | 482 | IRQLLFVVEVVDKYFDC | 17 |
| 11 | 230 | 248 | GVPVVDSYYSLLMPILTLT | 19 |
| 12 | 295 | 323 | HPNCVNCLDDRCILHCANFNVLFSTVFPP | 29 |
| No. | Start | End | Peptide | Length |
|---|---|---|---|---|
| 3 | 70 | 75 | YYCKSH | 6 |
| 5 | 207 | 212 | DAVVYR | 6 |
| 10 | 369 | 374 | DIVVFD | 6 |
| 11 | 384 | 389 | LSVVNA | 6 |
| 13 | 423 | 428 | NSVCRL | 6 |
| 15 | 493 | 498 | IGVVRE | 6 |
| 17 | 542 | 547 | DYVIFT | 6 |
| 12 | 394 | 400 | KHYVYIG | 7 |
| 16 | 522 | 528 | ASKILGL | 7 |
| 18 * | 570 | 576 | VGILCIM | 7 |
| 1 | 4 | 11 | ACVLCNSQ | 8 |
| 6 | 222 | 230 | GDYFVLTSH | 9 |
| 9 | 353 | 361 | EQYVFCTVN | 9 |
| 4 | 78 | 87 | PISFPLCANG | 10 |
| 14 | 449 | 458 | VDTVSALVYD | 10 |
| 7 | 237 | 250 | APTLVPQEHYVRIT | 14 |
| 8 | 292 | 325 | AIGLALYYPSARIVYTACSHAAVDALCEKALKYL | 34 |
| 2 | 21 | 58 | RRPFLCCKCCYDHVISTSHKLVLSVNPYVCNAPGCDVT | 38 |
| No. | Start | End | Peptide | Length |
|---|---|---|---|---|
| 1 | 4 | 18 | FLVLLPLVSSQCVNL | 15 |
| 2 | 34 | 41 | RGVYYPDK | 8 |
| 3 | 44 | 51 | RSSVLHST | 8 |
| 4 | 53 | 60 | DLFLPFFS | 8 |
| 5 | 65 | 70 | FHAIHV | 6 |
| 6 | 81 | 87 | NPVLPFN | 7 |
| 7 | 115 | 121 | QSLLIVN | 7 |
| 8 | 125 | 134 | NVVIKVCEFQ | 10 |
| 9 | 136 | 146 | CNDPFLGVYYH | 11 |
| 10 | 168 | 174 | FEYVSQP | 7 |
| 11 | 210 | 216 | INLVRDL | 7 |
| 12 | 223 | 230 | LEPLVDLP | 8 |
| 13 | 239 | 248 | QTLLALHRSY | 10 |
| 14 | 263 | 270 | AAYYVGYL | 8 |
| 15 | 272 | 278 | PRTFLLK | 7 |
| 16 | 288 | 295 | AVDCALDP | 8 |
| 17 | 333 | 339 | TNLCPFG | 7 |
| 18 | 359 | 371 | SNCVADYSVLYNS | 13 |
| 19 | 376 | 385 | TFKCYGVSPT | 10 |
| 20 | 430 | 435 | TGCVIA | 6 |
| 21 | 488 | 495 | CYFPLQSY | 8 |
| 22 | 505 | 527 | YQPYRVVVLSFELLHAPATVCGP | 23 |
| 23 | 592 | 599 | FGGVSVIT | 8 |
| 24 | 607 | 615 | QVAVLYQDV | 9 |
| 25 | 617 | 627 | CTEVPVAIHAD | 11 |
| 26 | 647 | 653 | AGCLIGA | 7 |
| 27 | 667 | 674 | GAGICASY | 8 |
| 28 | 687 | 693 | VASQSII | 7 |
| 29 | 723 | 730 | TTEILPVS | 8 |
| 30 | 735 | 741 | SVDCTMY | 7 |
| 31 | 750 | 763 | SNLLLQYGSFCTQL | 14 |
| 32 | 781 | 788 | VFAQVKQI | 8 |
| 33 | 803 | 808 | SQILPD | 6 |
| 34 | 837 | 843 | YGDCLGD | 7 |
| 35 | 847 | 853 | RDLICAQ | 7 |
| 36 | 858 | 864 | LTVLPPL | 7 |
| 37 | 873 | 880 | YTSALLAG | 8 |
| 38 | 959 | 966 | LNTLVKQL | 8 |
| 39 | 973 | 979 | ISSVLND | 7 |
| 40 | 1003 | 1011 | SLQTYVTQQ | 9 |
| 41 | 1030 | 1037 | SECVLGQS | 8 |
| 42 | 1057 | 1070 | PHGVVFLHVTYVPA | 14 |
| 43 | 1079 | 1085 | PAICHDG | 7 |
| 44 | 1123 | 1132 | SGNCDVVIGI | 10 |
| 45 | 1174 | 1179 | ASVVNI | 6 |
| 46 * | 1221 | 1256 | IAGLIAIVMVTIMLCCMTSCCSCLKGCCSCGSCCKF | 36 |
| Residue Number | Peptide Sequence | Predicted MHC Binding Affinity | Rescale Binding Affinity | C-Terminal Cleavage Affinity | TAP Transport | Prediction Score | MHC |
|---|---|---|---|---|---|---|---|
| Efficiency | Ligand | ||||||
| 604 | TSNQVAVLY | 0.6559 | 2.7847 | 0.944 | 2.991 | 3.0758 | yes |
| 361 | CVADYSVLY | 0.5348 | 2.2705 | 0.9764 | 3.18 | 2.5759 | yes |
| 733 | KTSVDCTMY | 0.4908 | 2.084 | 0.9649 | 3.016 | 2.3795 | yes |
| 687 | VASQSIIAY | 0.3529 | 1.4986 | 0.9656 | 3.089 | 1.7978136 | yes |
| 136 | CNDPFLGVY | 0.2613 | 1.1095 | 0.69 | 2.45 | 1.3355 | yes |
| 261 | GAAAYYVGY | 0.2253 | 0.9568 | 0.7608 | 2.969 | 1.2194 | yes |
| 357 | RISNCVADY | 0.2106 | 0.8941 | 0.9292 | 3.394 | 1.2032 | yes |
| 285 | ITDAVDCAL | 0.235 | 0.9979 | 0.8708 | 0.79 | 1.168 | yes |
| 1237 | MTSCCSCLK | 0.226 | 0.9595 | 0.7525 | 0.479 | 1.0963 | yes |
| 50 | STQDLFLPF | 0.1974 | 0.8383 | 0.553 | 2.511 | 1.0468 | yes |
| 748 | ECSNLLLQY | 0.1413 | 0.6 | 0.5316 | 2.747 | 0.8171 | yes |
| Residue Number | Peptide Sequence | Predicted MHC Binding Affinity | Rescale Binding Affinity | C-Terminal Cleavage Affinity | TAP Transport Efficiency | Prediction Score | MHC Ligand |
|---|---|---|---|---|---|---|---|
| 201 | TVNVLAWLY | 0.6255 | 2.6559 | 0.8852 | 2.957 | 2.9365 | yes |
| 110 | QTFSVLACY | 0.2625 | 1.1146 | 0.9725 | 2.998 | 1.4104 | yes |
| 153 | DYDCVSFCY | 0.2097 | 0.8905 | 0.9722 | 0.9722 | 1.1717 | yes |
| 93 | TANPKTPKY | 0.1676 | 0.7118 | 0.9755 | 2.676 | 0.9088 | yes |
| Residue Number | Peptide Sequence | Predicted MHC Binding Affinity | Rescale Binding Affinity | C-Terminal Cleavage Affinity | TAP Transport Efficiency | Prediction Score | MHC Ligand |
|---|---|---|---|---|---|---|---|
| 738 | DTDFVNEFY | 0.7922 | 3.3634 | 0.8873 | 2.458 | 3.6194 | yes |
| 336 | LSFKELLVY | 0.3898 | 1.6552 | 0.9676 | 3.213 | 1.961 | yes |
| 27 | STDVVYRAF | 0.4019 | 1.7065 | 0.6174 | 2.4 | 1.9191 | yes |
| 859 | FVSLAIDAY | 0.3709 | 1.5746 | 0.7669 | 3.096 | 1.8444 | yes |
| 666 | MVMCGGSLY | 0.3637 | 1.5441 | 0.9482 | 3.008 | 1.8368 | yes |
| 758 | LSDDAVVCF | 0.3143 | 1.3345 | 0.9556 | 2.412 | 1.5985 | yes |
| 686 | TTAYANSVF | 0.2963 | 1.258 | 0.4772 | 2.663 | 1.4627 | yes |
| 762 | AVVCFNSTY | 0.2435 | 1.0339 | 0.9754 | 3.146 | 1.3375 | yes |
| 463 | MCDIRQLLF | 0.2518 | 1.0691 | 0.1005 | 2.436 | 1.206 | yes |
| 233 | VVDSYYSLL | 0.2332 | 0.9901 | 0.7134 | 0.834 | 1.1388 | yes |
| 700 | VTANVNALL | 0.2007 | 0.8523 | 0.9705 | 1.166 | 1.0562 | yes |
| 818 | MLVKQGDDY | 0.1793 | 0.7614 | 0.8328 | 3.079 | 1.0403 | yes |
| 823 | GDDYVYLPY | 0.1821 | 0.7733 | 0.8456 | 2.213 | 1.0108 | yes |
| 879 | DVFHLYLQY | 0.1677 | 0.7119 | 0.9529 | 3.013 | 1.0055 | yes |
| 876 | EYADVFHLY | 0.1624 | 0.6894 | 0.9603 | 2.953 | 0.9811 | yes |
| 230 | GVPVVDSYY | 0.1504 | 0.6386 | 0.9521 | 2.923 | 0.9276 | yes |
| 434 | SVELKHFFF | 0.1454 | 0.6176 | 0.9285 | 2.636 | 0.8886 | yes |
| 334 | FVDGVPFVV | 0.1739 | 0.7382 | 0.8437 | 0.191 | 0.8743 | yes |
| 645 | CCSLSHRFY | 0.1586 | 0.6732 | 0.274 | 2.91 | 0.8598 | yes |
| Residue Number | Peptide Sequence | Predicted MHC Binding Affinity | Rescale Binding Affinity | C-Terminal Cleavage Affinity | TAP Transport Efficiency | Prediction Score | MHC Ligand |
|---|---|---|---|---|---|---|---|
| 57 | VTDVTQLYL | 0.4708 | 1.9988 | 0.6073 | 0.68 | 2.1239 | yes |
| 56 | DVTDVTQLY | 0.289 | 1.2271 | 0.9651 | 2.704 | 1.5071 | yes |
| 535 | SSQGSEYDY | 0.2761 | 1.1724 | 0.8149 | 2.847 | 1.437 | yes |
| 238 | PTLVPQEHY | 0.1794 | 0.7617 | 0.8719 | 2.595 | 1.0222 | yes |
| 448 | IVDTVSALV | 0.1991 | 0.8453 | 0.8977 | 0.133 | 0.9866 | yes |
| 574 | CIMSDRDLY | 0.1634 | 0.6937 | 0.1836 | 3.125 | 0.8775 | yes |
| 347 | KVNSTLEQY | 0.1391 | 0.5907 | 0.8156 | 2.971 | 0.8616 | yes |
| 245 | HYVRITGLY | 0.1102 | 0.4678 | 0.9598 | 3.009 | 0.7622 | yes |
| 85 | ANGQVFGLY | 0.1141 | 0.4845 | 0.9132 | 2.746 | 0.7588 | yes |
| 538 | GSEYDYVIF | 0.1401 | 0.5947 | 0.3528 | 2.203 | 0.7578 | yes |
| No. | Residues | Number of Residues | Score |
|---|---|---|---|
| 1 | A:S1, A:G2, A:F3, A:A211, A:V212, A:I213, A:N214, A:G215, A:D216, A:R217, A:W218, A:F219, A:L220, A:N221, A:R222, A:F223, A:T224, A:T225, A:T226, A:L227, A:N228, A:D229, A:F230, A:N231, A:L232, A:V233, A:A234, A:M235, A:K236, A:Y237, A:Y239, A:E240, A:P241, A:L242, A:T243, A:Q244, A:D245, A:V247, A:D248, A:L250, A:G251, A:P252, A:S254, A:A255, A:Q256, A:T257, A:G258, A:I259, A:A260, A:V261, A:L262, A:D263, A:A266, A:S267, A:K269, A:E270, A:L271, A:L272, A:Q273, A:N274, A:G275, A:M276, A:N277, A:G278, A:R279, A:T280, A:I281, A:L282, A:G283, A:S284, A:A285, A:L286, A:C300, A:S301, A:G302 | 75 | 0.716 |
| No. | Residues | Number of Residues | Score |
|---|---|---|---|
| 1 | A:L119, A:T120, A:K121, A:Y122, A:T123, A:D126, A:D135, A:E136, A:G137, A:N138, A:C139, A:D140, A:T141, A:K143, A:E144, A:I145, A:L146, A:V147, A:T148, A:Y149, A:N150, A:C151, A:C152, A:D153, A:D154, A:D155, A:Y156, A:F157, A:N158, A:K159, A:W162, A:Y163, A:N168, A:P169, A:D170, A:R173, A:V174, A:N177, A:L178, A:E180, A:R181, A:R183, A:Q184, A:A185, A:L187, A:K188, A:T189, A:V190, A:Q191, A:F192, A:C193, A:D194, A:A195, A:M196, A:R197, A:N198, A:A199, A:G200, A:I201, A:V202, A:G203, A:V204, A:L205, A:T206, A:D208, A:N209, A:Q210, A:D211, A:L212, A:N213, A:G214, A:N215, A:W216, A:Y217, A:D218, A:F219, A:G220, A:D221, A:F222, A:I223, A:Q224, A:T225, A:T226, A:P227, A:G228, A:S229, A:G230, A:V231, A:P232, A:V233, A:V234, A:A250, A:D284, A:K288, A:Y289 | 95 | 0.728 |
| 2 | A:D269, A:L270, A:L271, A:K272, A:Y273, A:D274, A:F275, A:E277, A:E278, A:K281, A:T324, A:L329, A:V330, A:R331, A:K332, A:I333, A:F334, A:V335, A:D336, A:G337, A:V338, A:P339, A:F340, A:V341, A:V342, A:S343, A:T344, A:H355, A:N356, A:Q357, A:D358, A:V359, A:N360, A:L361, A:H362, A:S363, A:S364, A:R365, A:L366, A:S367, A:F368, A:K369, A:E370, A:L371, A:L372, A:V373, A:Y374, A:D377, A:P378, A:A379, A:M380, A:H381, A:A382, A:A383, A:S384, A:G385, A:N386, A:L387, A:L388, A:L389, A:D390, A:K391, A:R392, A:T393, A:A399, A:A400, A:L401, A:T402, A:N403, A:N404, A:V405, A:A406, A:F407, A:Q408, A:T409, A:V410, A:K411, A:P412, A:G413, A:N414, A:F415, A:N416, A:K417, A:D418, A:F419, A:Y420, A:D421, A:F422, A:A423, A:V424, A:S425, A:K426, A:G427, A:F428, A:F429, A:K430, A:E431, A:G432, A:S433, A:S434, A:V435, A:E436, A:L437, A:K438, A:H439, A:F440, A:F441, A:F442, A:A443, A:Q444, A:D445, A:G446, A:N447, A:C487, A:I488, A:N489, A:A490, A:N491, A:Q492, A:V493, A:D517, A:S518, A:M519, A:S520, A:Y521, A:E522, A:D523, A:Q524, A:D525, A:A526, A:L527, A:A529, A:Y530, A:T531, A:K532, A:R533, A:N534, A:V535, A:I536, A:Y546, A:A550, A:F594, A:Y595, A:G596, A:H599, A:N600, A:K603, A:S607, A:D608, A:V609, A:E610, A:N611, A:P612, A:H613, A:H642, A:T643, A:T644, A:C645, A:C646, A:S647, A:H650, A:G670, A:G671, A:T710, A:D711, A:G712, A:N713, A:K714, A:I715, A:A716, A:D717, A:K718, A:Y719, A:V720, A:R721, A:N722, A:L723, A:R726, A:C730, A:V737, A:D738, A:T739, A:D740, A:F741, A:N743, A:E744, A:K751, A:H752, A:N767, A:S768, A:T769, A:Y770, A:S772, A:Q773, A:G774, A:L775, A:V776, A:T801, A:E802, A:T803, A:D804, A:L805, A:T806, A:K807, A:G808, A:M818, A:L819, A:V820, A:K821, A:Q822, A:G823, A:D824, A:D825, A:Y826, A:V827, A:Y828, A:L829, A:P832, A:D833, A:P834, A:L838, A:G839, A:G841, A:C842, A:F843, A:V844, A:D845, A:D846, A:I847, A:V848, A:K849, A:T850, A:D851, A:G852, A:T853, A:L854, A:M855, A:I856, A:E857, A:F859, A:V860, A:A863, A:I864, A:A866, A:Y867, A:P868, A:L869, A:T870, A:K871, A:H872, A:P873, A:N874, A:Q875, A:E876, A:Y877, A:A878, A:D879, A:V880, A:F881, A:H882, A:L883, A:Y884, A:L885, A:Q886, A:Y887, A:I888, A:R889, A:K890, A:L891, A:H892, A:D893, A:E894, A:L895, A:T896, A:G897, A:H898, A:M899, A:L900, A:D901, A:M902, A:Y903, A:S904, A:V905, A:M906, A:L907, A:T908, A:N909, A:D910, A:N911, A:T912, A:S913, A:R914, A:Y915, A:W916, A:E917, A:P918, A:E919 | 297 | 0.719 |
| No. | Residues | Number of Residues | Score |
|---|---|---|---|
| 1 | A:D1139, A:P1140, A:L1141, A:Q1142, A:P1143, A:E1144, A:L1145, A:D1146 | 8 | 0.975 |
| 2 | A:Y707, A:S708, A:N709, A:N710, A:S711, A:I712, A:A713, A:I714, A:P715, A:T716, A:N717, A:Q1071, A:K1073, A:N1074, A:F1075, A:T1076, A:T1077, A:A1078, A:P1079, A:A1080, A:I1081, A:C1082, A:H1083, A:D1084, A:G1085, A:K1086, A:A1087, A:H1088, A:F1089, A:P1090, A:R1091, A:E1092, A:G1093, A:V1094, A:F1095, A:V1096, A:S1097, A:N1098, A:G1099, A:T1100, A:H1101, A:W1102, A:F1103, A:V1104, A:T1105, A:Q1106, A:R1107, A:F1109, A:Y1110, A:E1111, A:P1112, A:Q1113, A:I1114, A:I1115, A:T1116, A:T1117, A:D1118, A:N1119, A:T1120, A:F1121, A:V1122, A:S1123, A:G1124, A:N1125, A:C1126, A:D1127, A:V1128, A:V1129, A:I1130, A:G1131, A:I1132, A:V1133, A:N1134, A:N1135, A:T1136, A:V1137, A:Y1138 | 77 | 0.845 |
| 3 | A:L335, A:C336, A:P337, A:F338, A:G339, A:E340, A:V341, A:F342, A:N343, A:A344, A:T345, A:R346, A:F347, A:A348, A:S349, A:V350, A:Y351, A:A352, A:W353, A:N354, A:R355, A:K356, A:R357, A:I358, A:S359, A:N360, A:C361, A:V362, A:A363, A:D364, A:Y365, A:S366, A:V367, A:L368, A:Y369, A:N370, A:S371, A:A372, A:S373, A:F374, A:S375, A:T376, A:F377, A:K378, A:C379, A:Y380, A:L390, A:C391, A:F392, A:T393, A:N394, A:V395, A:Y396, A:A397, A:D398, A:S399, A:F400, A:V401, A:I402, A:R403, A:G404, A:D405, A:E406, A:V407, A:R408, A:Q409, A:I410, A:A411, A:P412, A:G413, A:Q414, A:T415, A:G416, A:K417, A:I418, A:A419, A:D420, A:Y421, A:N422, A:Y423, A:K424, A:L425, A:P426, A:D427, A:D428, A:F429, A:T430, A:G431, A:C432, A:V433, A:I434, A:A435, A:W436, A:N437, A:S438, A:N439, A:N440, A:L441, A:D442, A:S443, A:Y449, A:N450, A:Y451, A:L452, A:Y453, A:R454, A:P491, A:L492, A:Q493, A:S494, A:Y495, A:G496, A:F497, A:Q498, A:P499, A:T500, A:V503, A:G504, A:Y505, A:Q506, A:P507, A:Y508, A:R509, A:V510, A:V511, A:V512, A:L513, A:S514, A:F515, A:E516, A:L517, A:L518, A:H519, A:A520, A:P521, A:A522, A:T523, A:V524, A:C525, A:G526, A:P527, A:K528 | 142 | 0.799 |
| 4 | A:F559, A:L560, A:P561, A:F562, A:Q563 | 5 | 0.789 |
| 5 | A:F79, A:D80, A:N81, A:P82, A:V83, A:L84, A:P85, A:I100, A:I101, A:R102, A:G103, A:W104, A:I105, A:T108, A:T109, A:L110, A:D111, A:S112, A:K113, A:T114, A:Q115, A:S116, A:L117, A:L118, A:I119, A:V120, A:N121, A:N122, A:A123, A:T124, A:N125, A:V126, A:V127, A:I128, A:K129, A:V130, A:C131, A:E132, A:F133, A:Q134, A:F135, A:C136, A:N137, A:D138, A:P139, A:F140, A:L141, A:G142, A:E156, A:F157, A:R158, A:V159, A:Y160, A:S161, A:S162, A:A163, A:N164, A:N165, A:C166, A:T167, A:F168, A:E169, A:Y170, A:V171, A:S172, A:Q173, A:P174, A:F175, A:L176, A:T236, A:R237, A:F238, A:Q239, A:T240, A:L241, A:L242, A:A243, A:L244, A:H245, A:R246 | 80 | 0.756 |
| No. | Residues | Number of Residues | Score |
|---|---|---|---|
| 1 | A:A1, A:V2, A:G3, A:A4, A:C5, A:L7, A:C8, A:N9, A:S10, A:Q11, A:T12, A:S13, A:L14, A:R15, A:C16, A:G17, A:F24, A:L25, A:C26, A:C27, A:K28, A:C29, A:C30, A:Y31, A:D32, A:V34, A:I35, A:S36, A:T37, A:S38, A:H39, A:K40, A:L41, A:V42, A:L43, A:S44, A:V45, A:N46, A:P47, A:Y48, A:V49, A:C50, A:N51, A:A52, A:P53, A:G54, A:C55, A:D56, A:V57, A:T58, A:D59, A:V60, A:T61, A:Q62, A:L63, A:Y64, A:L65, A:G66, A:G67, A:M68, A:S69, A:Y70, A:Y71, A:C72, A:K73, A:S74, A:H75, A:K76, A:P77, A:P78, A:I79, A:S80, A:F81, A:P82, A:L83, A:C84, A:A85, A:N86, A:G87, A:Q88, A:V89, A:F90, A:G91, A:L92, A:Y93, A:K94, A:N95, A:T96, A:C97, A:V98, A:G99, A:S100, A:D101, A:N102, A:V103, A:T104 | 96 | 0.761 |
| 2 | A:D344, A:K345, A:F346 | 3 | 0.74 |
| 3 | A:G150, A:I151, A:A152, A:T153, A:V154, A:R155, A:E156, A:V157, A:L158, A:S159, A:D160, A:R161, A:E162, A:L163, A:H164, A:L165, A:S166, A:W167, A:E168, A:V169, A:G170, A:K171, A:P172, A:R173, A:G184, A:Y185, A:R186, A:V187, A:T188, A:K189, A:N190, A:S191, A:K192, A:V193, A:Q194, A:I195, A:G203, A:D204, A:Y205, A:G206, A:D207, A:A208, A:V209, A:Y217, A:K218, A:L219, A:N220, A:V221, A:G222, A:D223, A:Y224, A:F225 | 52 | 0.738 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, H.M.; Mirza, M.U.; Ahmad, M.A.; Saleem, M.; Froeyen, M.; Ahmad, S.; Gul, R.; Alghamdi, H.A.; Aslam, M.S.; Sajjad, M.; et al. A Putative Prophylactic Solution for COVID-19: Development of Novel Multiepitope Vaccine Candidate against SARS-COV-2 by Comprehensive Immunoinformatic and Molecular Modelling Approach. Biology 2020, 9, 296. https://doi.org/10.3390/biology9090296
Rehman HM, Mirza MU, Ahmad MA, Saleem M, Froeyen M, Ahmad S, Gul R, Alghamdi HA, Aslam MS, Sajjad M, et al. A Putative Prophylactic Solution for COVID-19: Development of Novel Multiepitope Vaccine Candidate against SARS-COV-2 by Comprehensive Immunoinformatic and Molecular Modelling Approach. Biology. 2020; 9(9):296. https://doi.org/10.3390/biology9090296
Chicago/Turabian StyleRehman, Hafiz Muzzammel, Muhammad Usman Mirza, Mian Azhar Ahmad, Mahjabeen Saleem, Matheus Froeyen, Sarfraz Ahmad, Roquyya Gul, Huda Ahmed Alghamdi, Muhammad Shahbaz Aslam, Muhammad Sajjad, and et al. 2020. "A Putative Prophylactic Solution for COVID-19: Development of Novel Multiepitope Vaccine Candidate against SARS-COV-2 by Comprehensive Immunoinformatic and Molecular Modelling Approach" Biology 9, no. 9: 296. https://doi.org/10.3390/biology9090296
APA StyleRehman, H. M., Mirza, M. U., Ahmad, M. A., Saleem, M., Froeyen, M., Ahmad, S., Gul, R., Alghamdi, H. A., Aslam, M. S., Sajjad, M., & Bhinder, M. A. (2020). A Putative Prophylactic Solution for COVID-19: Development of Novel Multiepitope Vaccine Candidate against SARS-COV-2 by Comprehensive Immunoinformatic and Molecular Modelling Approach. Biology, 9(9), 296. https://doi.org/10.3390/biology9090296