Essentiality and Transcriptome-Enriched Pathway Scores Predict Drug-Combination Synergy

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
2.1.1. Data Regarding the Synergy of Drug Combinations
2.1.2. Transcriptome Data
2.1.3. Essentiality Data
2.1.4. Training and Validation Data
2.2. Features
2.3. Logistic Regression Models in Selecting Features for Drug-Synergy Prediction
2.4. Model Training and Validation
3. Results
3.1. In Overall Trend Analysis, Pathway Features Showed Stronger Statistical Correlation Evidence Than Gene Features with Drug-Combination Synergy Scores
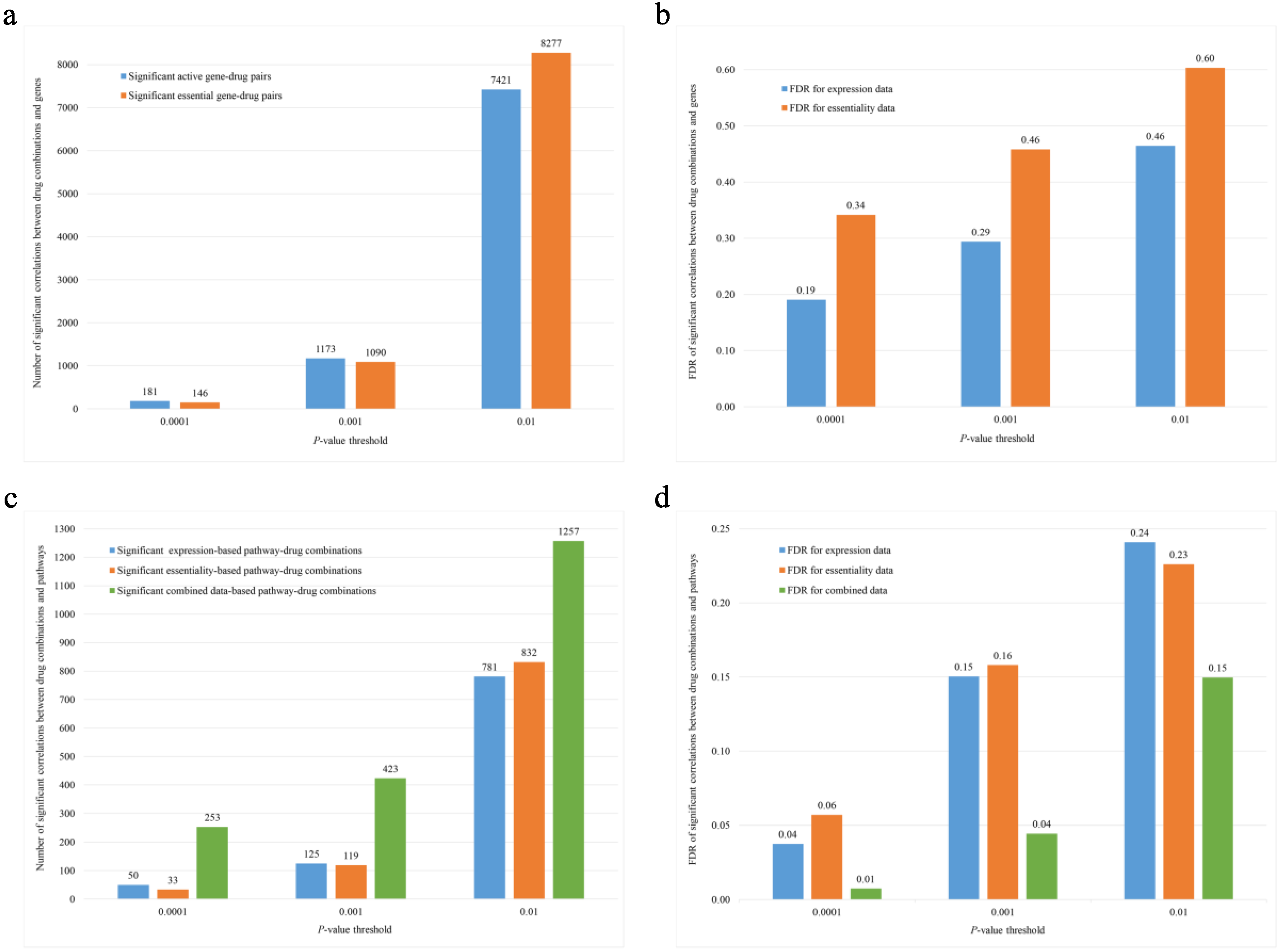
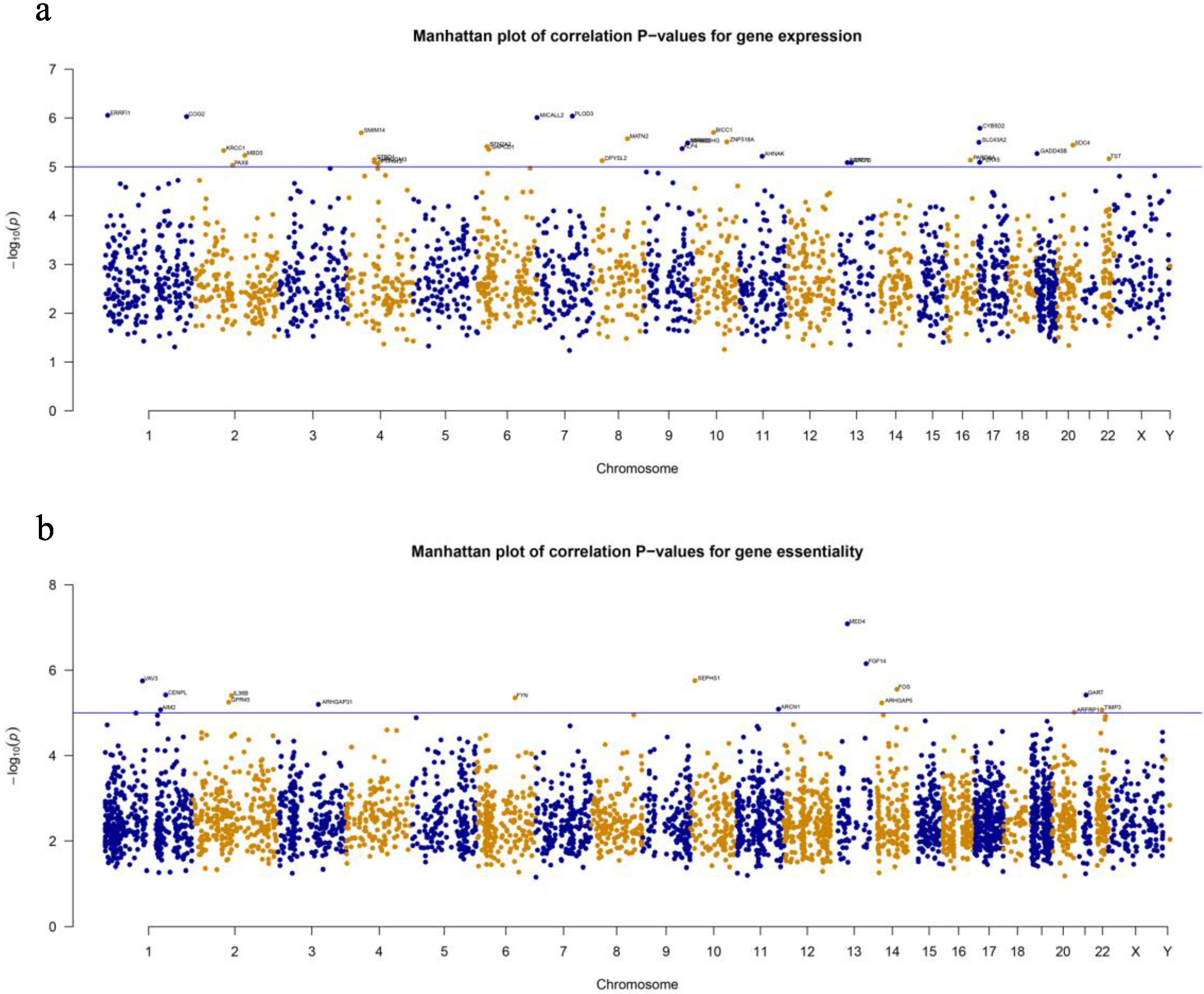
3.2. Genome-Wide Association Analyses Revealed Significant Associations of Genes and Pathways with Scores of Drug Synergy
3.3. Overlap Was Limited between Significant Gene Expressions and Essentialities in Predicting Drug Synergy
3.4. No Statistically Significant Evidence Supported Relationship between Target and Non-Target Genes and Pathways in the Prediction of Drug Synergy
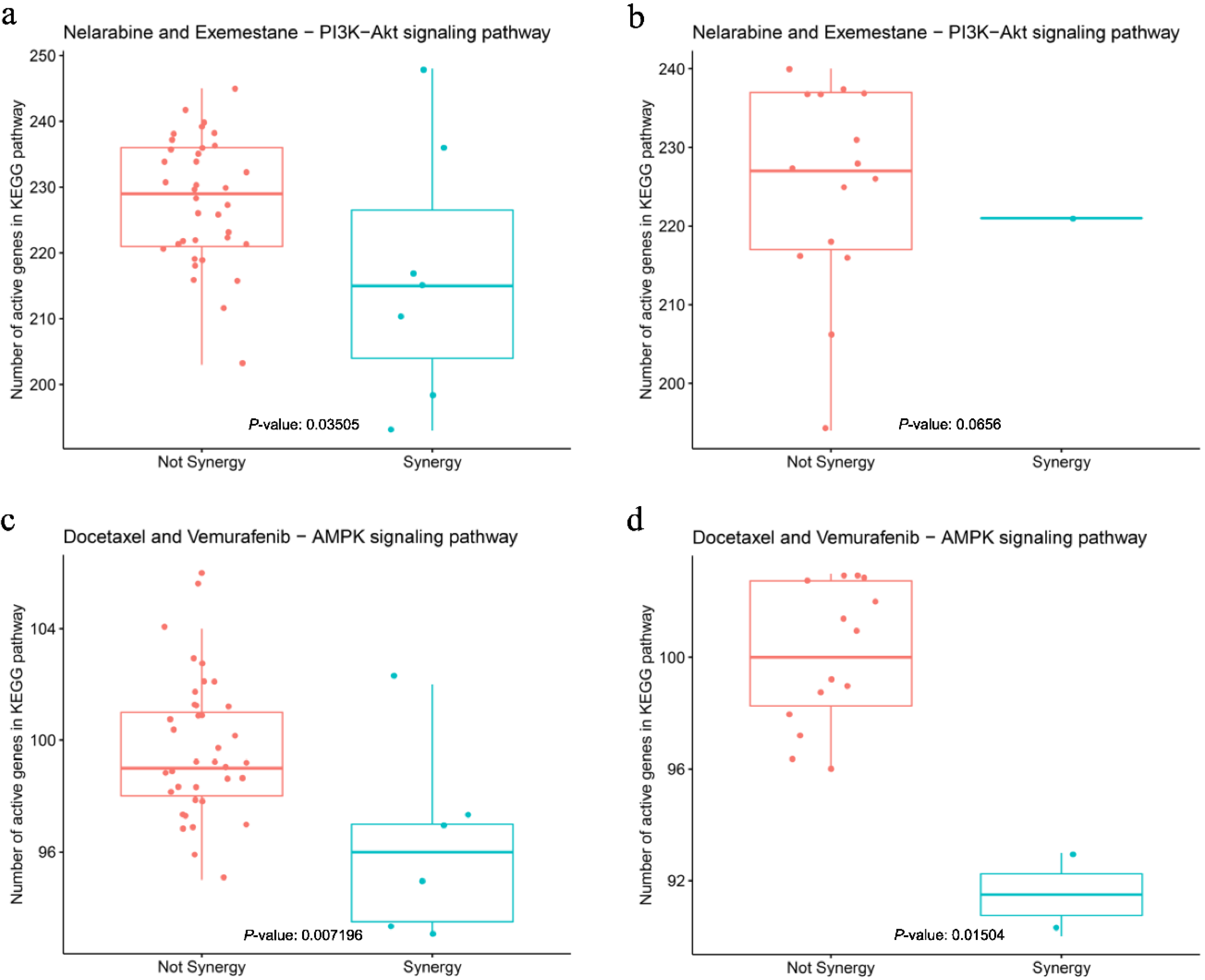
3.5. Feature Comparison in Pathway Analysis
3.6. Model Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Allen, E.M.; Wagle, N.; Stojanov, P.; Perrin, D.L.; Cibulskis, K.; Marlow, S.; Jane-Valbuena, J.; Friedrich, D.C.; Kryukov, G.; Carter, S.L.; et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 2014, 20, 682–688. [Google Scholar] [CrossRef]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef]
- Wagle, N.; Berger, M.F.; Davis, M.J.; Blumenstiel, B.; Defelice, M.; Pochanard, P.; Ducar, M.; Van Hummelen, P.; Macconaill, L.E.; Hahn, W.C.; et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012, 2, 82–93. [Google Scholar] [CrossRef]
- Garraway, L.A.; Jänne, P.A. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef]
- Dias-Santagata, D.; Akhavanfard, S.; David, S.S.; Vernovsky, K.; Kuhlmann, G.; Boisvert, S.L.; Stubbs, H.; McDermott, U.; Settleman, J.; Kwak, E.L.; et al. Rapid targeted mutational analysis of human tumours: A clinical platform to guide personalized cancer medicine. EMBO Mol. Med. 2010, 2, 146–158. [Google Scholar] [CrossRef]
- Kerick, M.; Isau, M.; Timmermann, B.; Sültmann, H.; Herwig, R.; Krobitsch, S.; Schaefer, G.; Verdorfer, I.; Bartsch, G.; Klocker, H.; et al. Targeted high throughput sequencing in clinical cancer settings: Formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med. Genom. 2011, 4, 68. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, S.; Iyer, M.K.; Robinson, D.R.; Lonigro, R.J.; Wu, Y.M.; Cao, X.; Kalyana-Sundaram, S.; Sam, L.; Balbin, O.A.; Quist, M.J.; et al. Personalized oncology through integrative high-throughput sequencing: A pilot study. Sci. Transl. Med. 2011, 3, 111ra121. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Anagnostou, V.; Lytle, K.; Parpart-Li, S.; Nesselbush, M.; Riley, D.R.; Shukla, M.; Chesnick, B.; Kadan, M.; Papp, E.; et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci. Transl. Med. 2015, 7, 283ra53. [Google Scholar] [CrossRef]
- Carter, J.; Cheng, L.; Zucker, J.; Marshall, M.; Pollok, K.; Murray, M.; Li, L.; Renbarger, J. Use of Precision Medicine Molecular Profiling of Baseline Tumor Specimen May Not Benefit Outcomes in Children with Relapsed or Refractory Pediatric Sarcomas. Clin. Pharmacol. Ther. 2017, 101, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.; Yin, R.; Luo, J.; Kwoh, C.K. Ensemble Prediction of Synergistic Drug Combinations Incorporating Biological, Chemical, Pharmacological, and Network Knowledge. IEEE J. Biomed. Health Inform. 2019, 23, 1336–1345. [Google Scholar] [CrossRef]
- O′Neil, J.; Benita, Y.; Feldman, I.; Chenard, M.; Roberts, B.; Liu, Y.; Li, J.; Kral, A.; Lejnine, S.; Loboda, A.; et al. An Unbiased Oncology Compound Screen to Identify Novel Combination Strategies. Mol. Cancer Ther. 2016, 15, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Holbeck, S.L.; Camalier, R.; Crowell, J.A.; Govindharajulu, J.P.; Hollingshead, M.; Anderson, L.W.; Polley, E.; Rubinstein, L.; Srivastava, A.; Wilsker, D.; et al. The National Cancer Institute ALMANAC: A Comprehensive Screening Resource for the Detection of Anticancer Drug Pairs with Enhanced Therapeutic Activity. Cancer Res. 2017, 77, 3564–3576. [Google Scholar] [CrossRef] [PubMed]
- Menden, M.P.; Wang, D.; Mason, M.J.; Szalai, B.; Bulusu, K.C.; Guan, Y.; Yu, T.; Kang, J.; Jeon, M.; Wolfinger, R.; et al. Community assessment to advance computational prediction of cancer drug combinations in a pharmacogenomic screen. Nat. Commun. 2019, 10, 2674. [Google Scholar] [CrossRef] [PubMed]
- Preuer, K.; Lewis, R.P.I.; Hochreiter, S.; Bender, A.; Bulusu, K.C.; Klambauer, G. DeepSynergy: Predicting anti-cancer drug synergy with Deep Learning. Bioinformatics 2018, 34, 1538–1546. [Google Scholar] [CrossRef]
- Sidorov, P.; Naulaerts, S.; Ariey-Bonnet, J.; Pasquier, E.; Ballester, P.J. Predicting Synergism of Cancer Drug Combinations Using NCI-ALMANAC Data. Front. Chem. 2019, 7, 509. [Google Scholar] [CrossRef]
- Jiang, P.; Huang, S.; Fu, Z.; Sun, Z.; Lakowski, T.M.; Hu, P. Deep graph embedding for prioritizing synergistic anticancer drug combinations. Comput. Struct. Biotechnol. J. 2020, 18, 427–438. [Google Scholar] [CrossRef]
- Huang, L.; Brunell, D.; Stephan, C.; Mancuso, J.; Yu, X.; He, B.; Thompson, T.C.; Zinner, R.; Kim, J.; Davies, P.; et al. Driver network as a biomarker: Systematic integration and network modeling of multi-omics data to derive driver signaling pathways for drug combination prediction. Bioinformatics 2019, 35, 3709–3717. [Google Scholar] [CrossRef]
- Li, H.; Li, T.; Quang, D.; Guan, Y. Network Propagation Predicts Drug Synergy in Cancers. Cancer Res. 2018, 78, 5446–5457. [Google Scholar] [CrossRef]
- Chen, D.; Liu, X.; Yang, Y.; Yang, H.; Lu, P. Systematic synergy modeling: Understanding drug synergy from a systems biology perspective. BMC Syst. Biol. 2015, 9, 56. [Google Scholar] [CrossRef]
- Zhang, C.; Yan, G. Synergistic drug combinations prediction by integrating pharmacological data. Synth. Syst. Biotechnol. 2019, 4, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ren, B.; Chen, M.; Wang, Q.; Zhang, L.; Yan, G. NLLSS: Predicting Synergistic Drug Combinations Based on Semi-supervised Learning. PLoS Comput. Biol. 2016, 12, e1004975. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.M.; Iskar, M.; Zeller, G.; Kuhn, M.; van Noort, V.; Bork, P. Prediction of drug combinations by integrating molecular and pharmacological data. PLoS Comput. Biol. 2011, 7, e1002323. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sheng, Z.; Ma, C.; Tang, K.; Zhu, R.; Wu, Z.; Shen, R.; Feng, J.; Wu, D.; Huang, D.; et al. Combining genomic and network characteristics for extended capability in predicting synergistic drugs for cancer. Nat. Commun. 2015, 6, 8481. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, Y.; Liu, F.; Luo, F.; Tian, G.; Li, X. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data. BMC Bioinform. 2017, 18, 18. [Google Scholar] [CrossRef]
- Yang, J.; Tang, H.; Li, Y.; Zhong, R.; Wang, T.; Wong, S.; Xiao, G.; Xie, Y. DIGRE: Drug-Induced Genomic Residual Effect Model for Successful Prediction of Multidrug Effects. CPT Pharmacomet. Syst. Pharmacol. 2015, 4, 91–97. [Google Scholar] [CrossRef]
- Jaeger, S.; Igea, A.; Arroyo, R.; Alcalde, V.; Canovas, B.; Orozco, M.; Nebreda, A.R.; Aloy, P. Quantification of Pathway Cross-talk Reveals Novel Synergistic Drug Combinations for Breast Cancer. Cancer Res. 2017, 77, 459–469. [Google Scholar] [CrossRef]
- Cheng, F.; Kovacs, I.A.; Barabási, A.L. Network-based prediction of drug combinations. Nat. Commun. 2019, 10, 1197. [Google Scholar] [CrossRef]
- Cheng, F.; Lu, W.; Liu, C.; Fang, J.; Hou, Y.; Handy, D.E.; Wang, R.; Zhao, Y.; Yang, Y.; Huang, J.; et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun. 2019, 10, 3476. [Google Scholar] [CrossRef]
- Reinhold, W.C.; Sunshine, M.; Liu, H.; Varma, S.; Kohn, K.W.; Morris, J.; Doroshow, J.; Pommier, Y. CellMiner: A web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012, 72, 3499–3511. [Google Scholar] [CrossRef]
- Reinhold, W.C.; Mergny, J.L.; Liu, H.; Ryan, M.; Pfister, T.D.; Kinders, R.; Parchment, R.; Doroshow, J.; Weinstein, J.N.; Pommier, Y. Exon array analyses across the NCI-60 reveal potential regulation of TOP1 by transcription pausing at guanosine quartets in the first intron. Cancer Res. 2010, 70, 2191–2203. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Li, F.; Zhou, Y.; Zhang, Y.; Wang, Z.; Zhang, R.; Zhu, J.; Ren, Y.; Tan, Y.; et al. Therapeutic target database 2020: Enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2020, 48, D1031–D1041. [Google Scholar] [CrossRef]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer. Res. 2015, 5, 1602–1609. [Google Scholar]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell. Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.; Miller, D.D.; Li, W. Synergistic combination of novel tubulin inhibitor ABI-274 and vemurafenib overcome vemurafenib acquired resistance in BRAFV600E melanoma. Mol. Cancer Ther. 2014, 13, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- El-Masry, O.S.; Al-Sakkaf, K.; Brown, B.L.; Dobson, P.R. Differential crosstalk between the AMPK and PI3K/Akt pathways in breast cancer cells of differing genotypes: Leptin inhibits the effectiveness of AMPK activation. Oncol. Rep. 2015, 34, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Gong, J.; Luo, X.; Zang, M.; Guo, W.; Wen, R.; Luo, Z. AMPK exerts dual regulatory effects on the PI3K pathway. J. Mol. Signal. 2010, 5, 1. [Google Scholar] [CrossRef]
- He, G.; Zhang, Y.W.; Lee, J.H.; Zeng, S.X.; Wang, Y.V.; Luo, Z.; Dong, X.C.; Viollet, B.; Wahl, G.M.; Lu, H. AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol. Cell. Biol. 2014, 34, 148–157. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data | Feature | Feature Description | Type |
|---|---|---|---|
| Drug-combination targets | nD_union_ab | Number of total drug targets for Drug A or Drug B | |
| nD_intersection_ab | Number of total drug targets for both Drug A and Drug B | ||
| KEGG pathways | nk | Number of genes in KEGG Pathway K | |
| Cell lines | ncell_c_expression | Number of active genes in Cell Line C | 1 |
| ncell_c_essentiality | Number of essential genes in Cell Line C | 1 | |
| Drug-combination targets in cell lines based on gene expression | ncell_c_expression_D_union_ab | Number of total drug targets for Drug A or Drug B that are active in Cell Line C | 1 |
| ncell_c_expression_D_intersection_ab | Number of total drug targets for both Drug A and Drug B that are active in Cell Line C | ||
| ncell_c_expression_D_union_ab/ncell_c_expression | Ratio of the number of active drug targets for Drug A or Drug B to all active genes in Cell Line C | ||
| ncell_c_expression_D_intersection_ab/ncell_c_expression | Ratio of the number of active drug targets for Drug A and Drug B to all active genes in Cell Line C | ||
| Drug-combination targets and cell lines based on gene essentiality | ncell_c_ essentiality _D_union_ab | Number of total drug targets for Drug A or Drug B that are essential in Cell Line C | 1 |
| ncell_c_essentiality _D_intersection_ab | Number of total drug targets for both Drug A and Drug B that are essential in Cell Line C | ||
| ncell_c_essentiality_D_union_ab/ncell_c_essentiality | Ratio of the number of essential drug targets for Drug A or Drug B relative to all essential genes in Cell Line C | ||
| ncell_c_essentiality_D_intersection_ab/ncell_c_essentiality | Ratio of the number of essential drug targets for Drug A and Drug B relative to all essential genes in Cell Line C | ||
| KEGG pathways and cell lines based on gene expression | ncell_c_expression_kegg_k | Number of active genes in KEGG Pathway K for Cell Line C | 2 |
| ncell_c_expression_kegg_k/ncell_c_expression | Ratio of the number of active genes in KEGG Pathway K relative to all active genes in Cell Line C | ||
| KEGG pathways and cell lines Based on gene essentiality | ncell_c_essentiality_kegg_k | Number of essential genes in KEGG Pathway K for Cell Line C | 2 |
| ncell_c_essentiality_kegg_k/ncell_c_essentiality | Ratio of the number of essential genes in KEGG Pathway K to all essential genes in Cell Line C |
| Group | Gene Expression | Gene Essentiality | Combined Expression and Essentiality | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Threshold | Number Observed | Number Expected | FDR | Threshold | Number Observed | Number Expected | FDR | Threshold | Number Observed | Number Expected | FDR | ||
| 1 | 0.0001 | 0 | 0.01 | NA | 0.0001 | 1 | 0.01 | 0.01 | 0.0001 | 1 | 0.01 | 0.01 | |
| 0.001 | 0 | 0.11 | NA | 0.001 | 1 | 0.11 | 0.11 | 0.001 | 1 | 0.11 | 0.11 | ||
| 0.01 | 4 | 1.14 | 0.29 | 0.01 | 2 | 1.14 | 0.57 | 0.01 | 7 | 1.14 | 0.16 | ||
| 2 | DD * | 0.0001 | 7 | 0.01 | 1.43 × 10−3 | 0.0001 | 2 | 0.01 | 0.005 | 0.0001 | 11 | 0.01 | 9.09 × 10−4 |
| 0.001 | 25 | 0.11 | 4.4 × 10−3 | 0.001 | 15 | 0.11 | 7.33 × 10−3 | 0.001 | 35 | 0.11 | 3.14 × 10−3 | ||
| 0.01 | 72 | 1.14 | 0.016 | 0.01 | 77 | 1.14 | 0.015 | 0.01 | 85 | 1.14 | 0.013 | ||
| DDP # | 0.0001 | 8 | 1.88 | 0.24 | 0.0001 | 3 | 1.88 | 0.63 | 0.0001 | 16 | 1.88 | 0.12 | |
| 0.001 | 43 | 18.81 | 0.44 | 0.001 | 25 | 18.81 | 0.75 | 0.001 | 78 | 18.81 | 0.24 | ||
| 0.01 | 443 | 188.1 | 0.42 | 0.01 | 311 | 188.1 | 0.60 | 0.01 | 519 | 188.1 | 0.36 | ||
| 3 | DD * | 0.0001 | 23 | 0.01 | 4.35 × 10−4 | 0.0001 | 13 | 0.01 | 7.69 × 10−4 | 0.0001 | 37 | 0.01 | 2.72 × 10−4 |
| 0.001 | 36 | 0.11 | 3.06 × 10−3 | 0.001 | 26 | 0.11 | 4.23 × 10−3 | 0.001 | 62 | 0.11 | 1.77 × 10−3 | ||
| 0.01 | 86 | 1.14 | 0.013 | 0.01 | 61 | 1.14 | 0.019 | 0.01 | 98 | 1.14 | 0.012 | ||
| DDP # | 0.0001 | 50 | 1.88 | 0.038 | 0.0001 | 33 | 1.88 | 0.057 | 0.0001 | 253 | 1.88 | 7.43 × 10−3 | |
| 0.001 | 125 | 18.81 | 0.15 | 0.001 | 119 | 18.81 | 0.16 | 0.001 | 423 | 18.81 | 0.044 | ||
| 0.01 | 781 | 188.1 | 0.24 | 0.01 | 832 | 188.1 | 0.23 | 0.01 | 1257 | 188.1 | 0.15 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Huo, Y.; Wu, X.; Liu, E.; Zeng, Z.; Tian, Z.; Fan, K.; Stover, D.; Cheng, L.; Li, L. Essentiality and Transcriptome-Enriched Pathway Scores Predict Drug-Combination Synergy. Biology 2020, 9, 278. https://doi.org/10.3390/biology9090278
Li J, Huo Y, Wu X, Liu E, Zeng Z, Tian Z, Fan K, Stover D, Cheng L, Li L. Essentiality and Transcriptome-Enriched Pathway Scores Predict Drug-Combination Synergy. Biology. 2020; 9(9):278. https://doi.org/10.3390/biology9090278
Chicago/Turabian StyleLi, Jin, Yang Huo, Xue Wu, Enze Liu, Zhi Zeng, Zhen Tian, Kunjie Fan, Daniel Stover, Lijun Cheng, and Lang Li. 2020. "Essentiality and Transcriptome-Enriched Pathway Scores Predict Drug-Combination Synergy" Biology 9, no. 9: 278. https://doi.org/10.3390/biology9090278
APA StyleLi, J., Huo, Y., Wu, X., Liu, E., Zeng, Z., Tian, Z., Fan, K., Stover, D., Cheng, L., & Li, L. (2020). Essentiality and Transcriptome-Enriched Pathway Scores Predict Drug-Combination Synergy. Biology, 9(9), 278. https://doi.org/10.3390/biology9090278