Skim-Sequencing Based Genotyping Reveals Genetic Divergence of the Wild and Domesticated Population of Black Tiger Shrimp (Penaeus monodon) in the Indo-Pacific Region

 ,
,
Abstract
1. Introduction
2. Materials and Methods
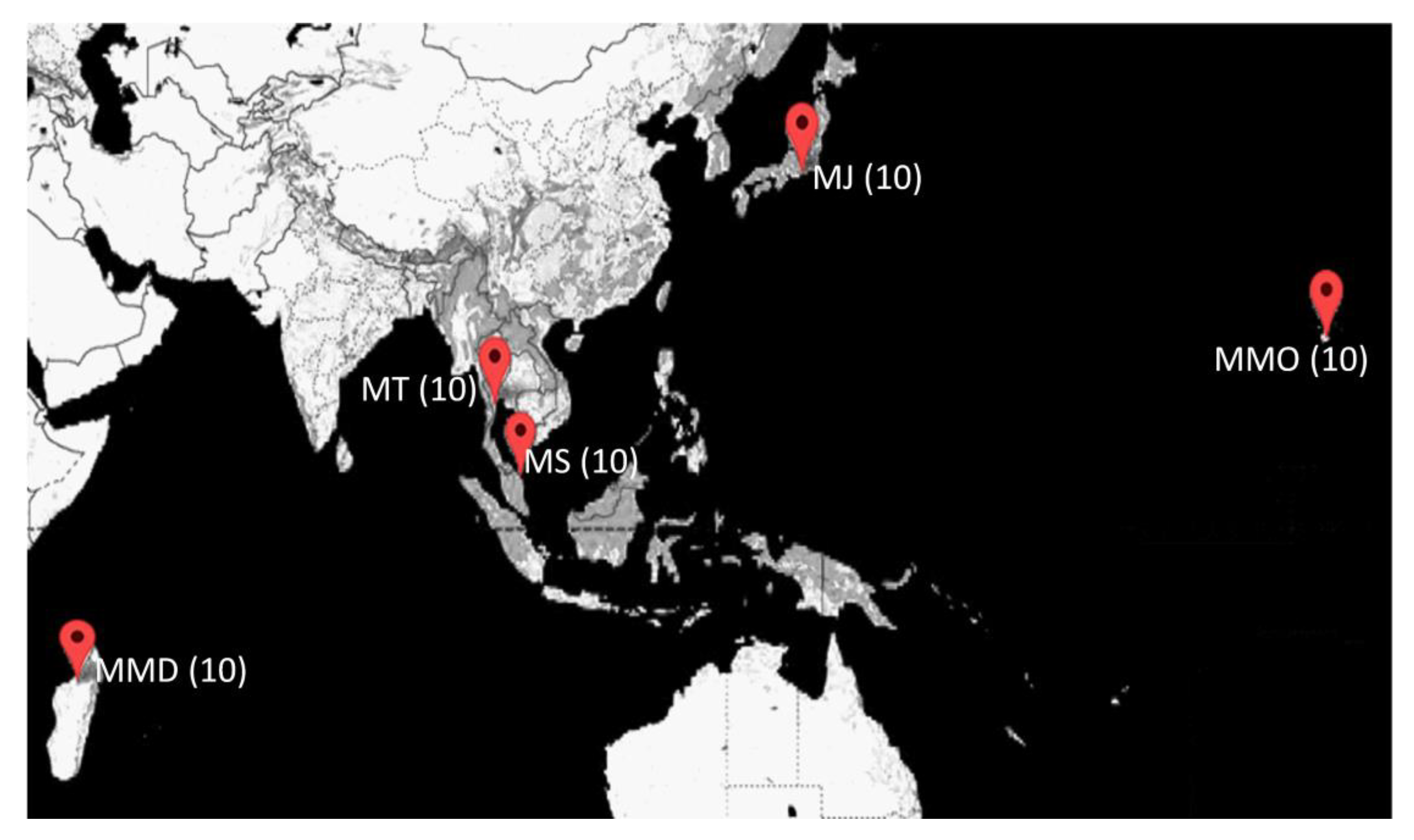
2.1. Sample Collection
2.2. DNA Extraction and Library Preparation
2.3. Sequence Assembly, Filtering and SNPs Discovery
2.4. Power Analysis
2.5. Genetic Variation Analysis
2.6. GO and KEGG Enrichment Analysis of Putatively Adaptive SNP Loci
2.7. Data Accessibility
3. Results
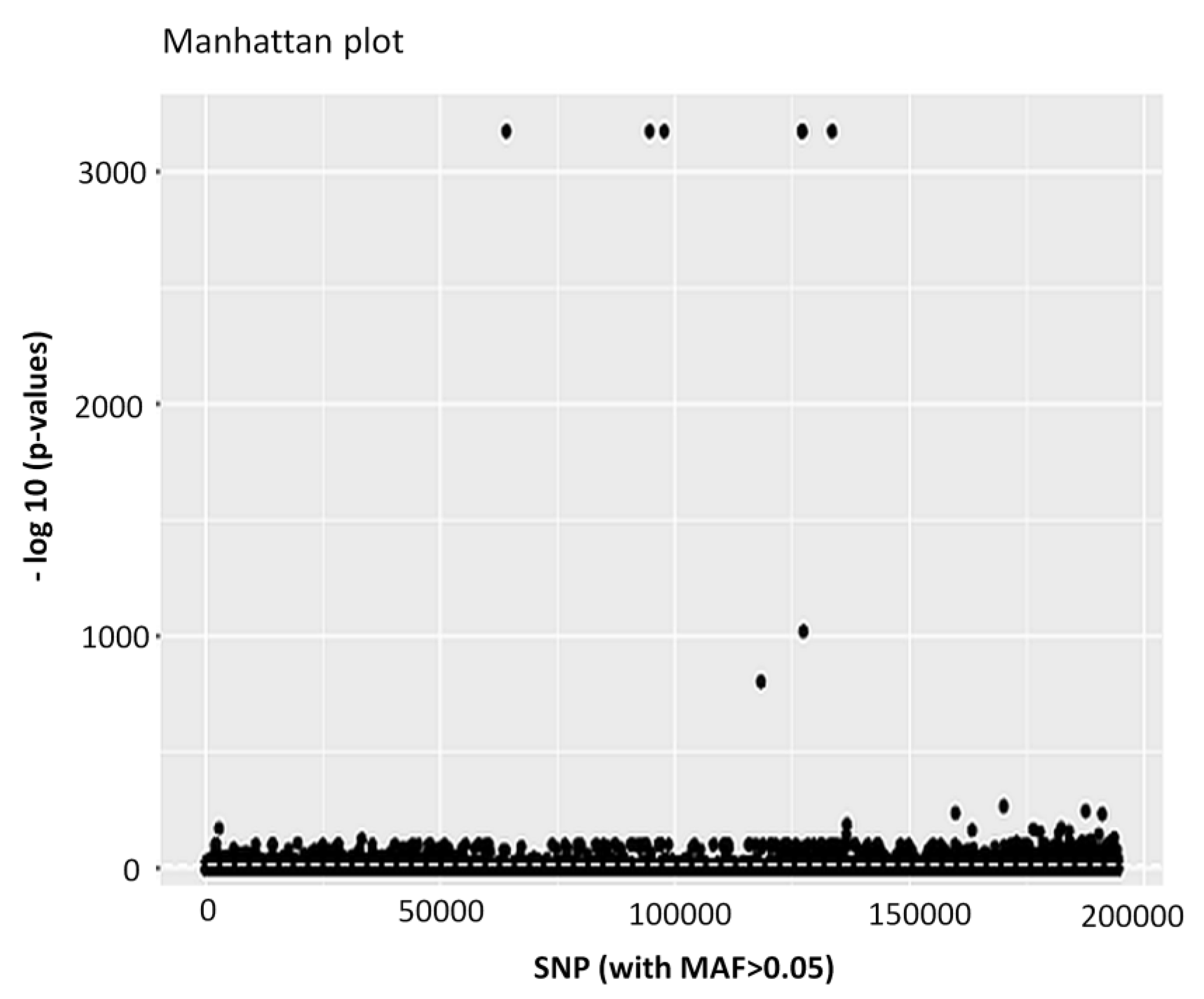
3.1. Genome Assembly, Annotation and Quality Filtering of SNP Loci
3.2. Power Analysis
3.3. Demographic Interpretations from FST Statistics and AMOVA Analysis
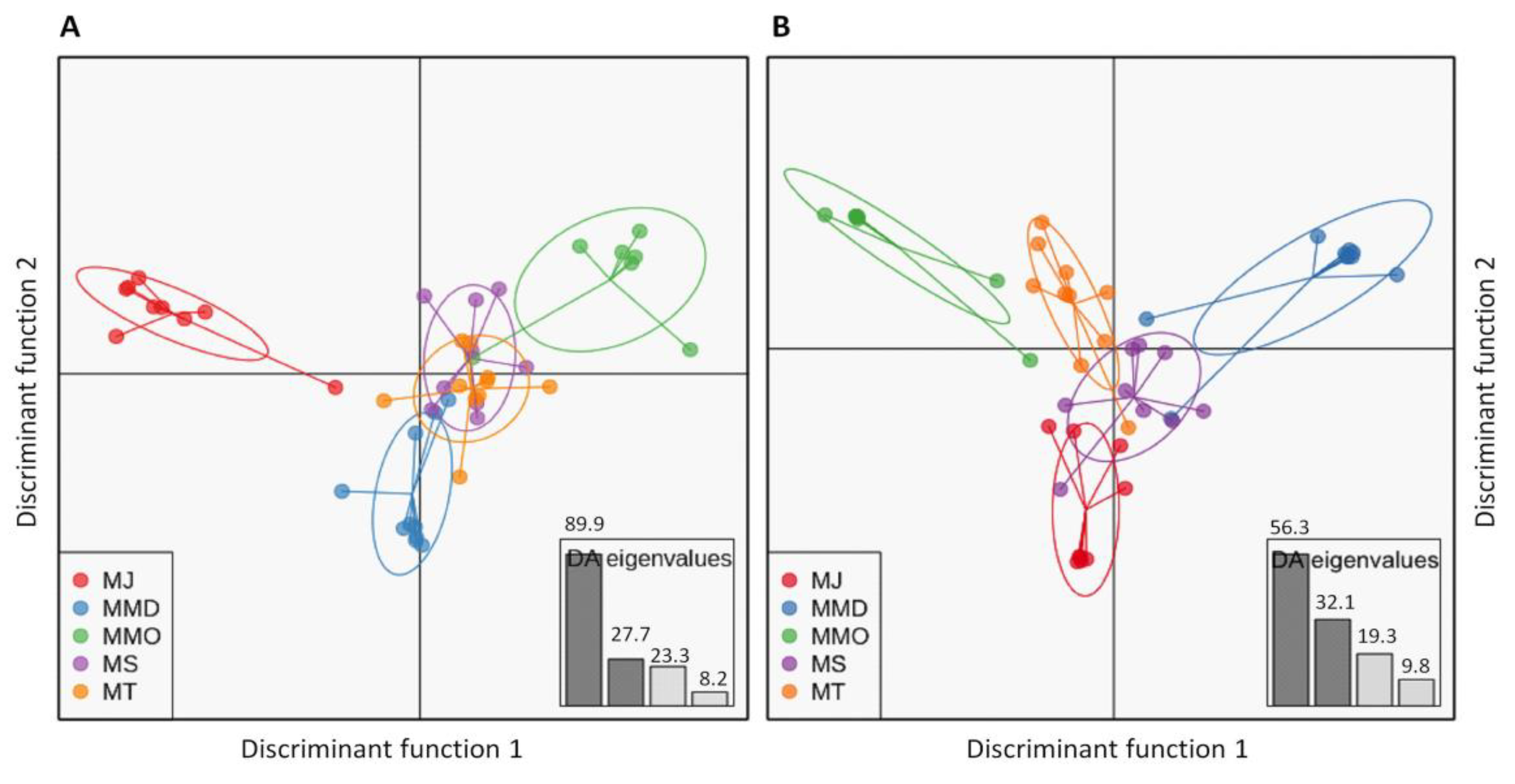
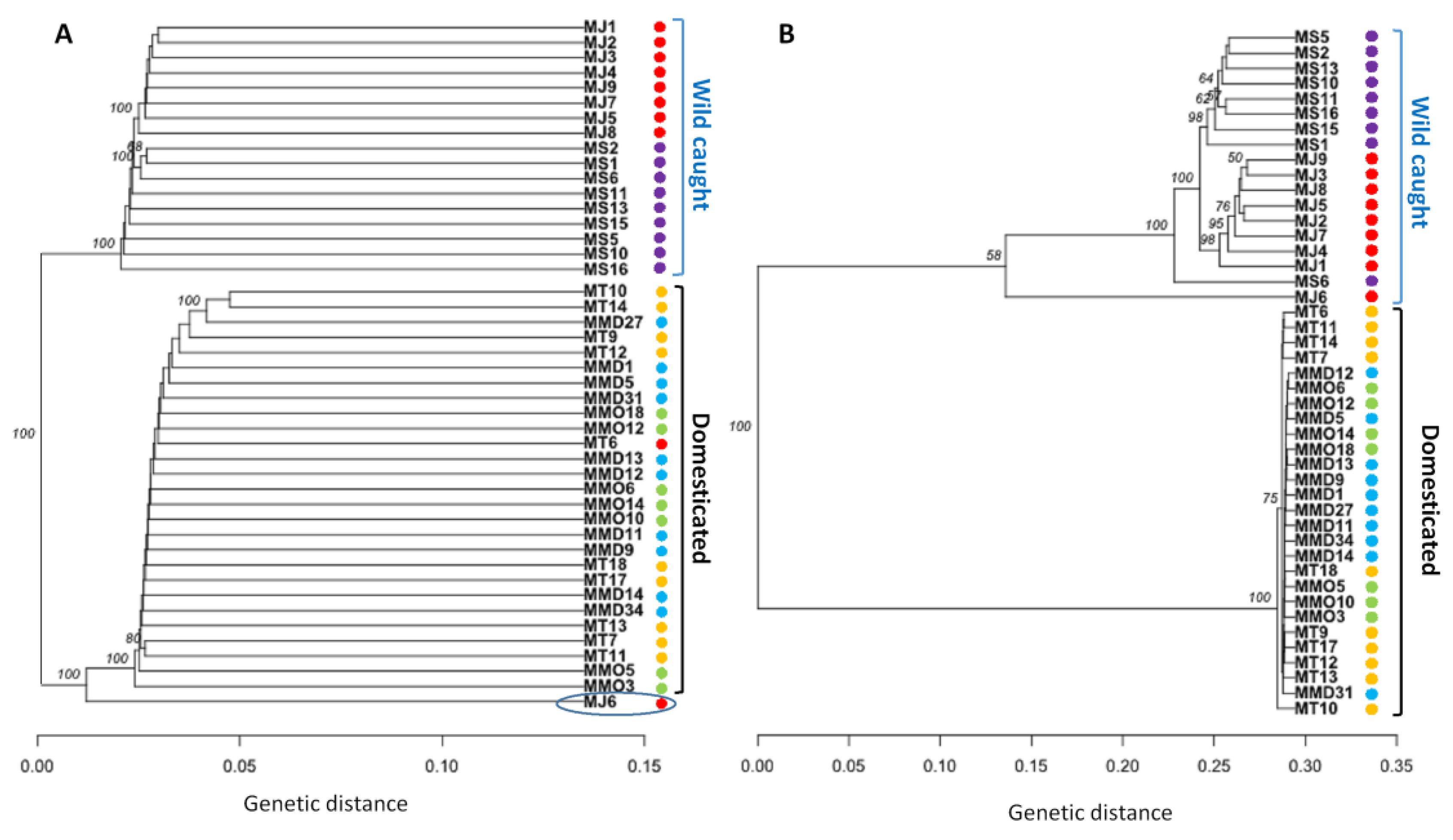
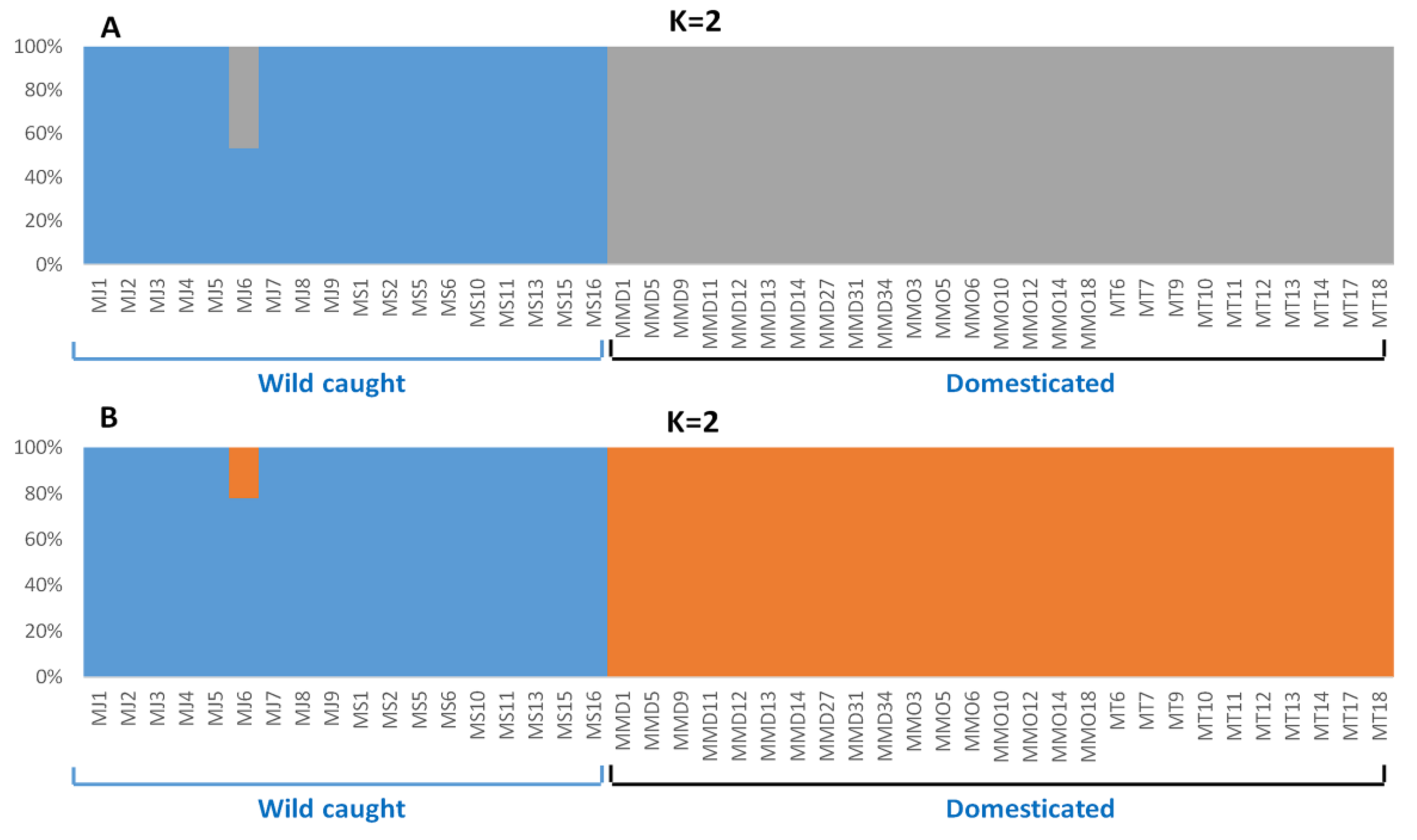
3.4. Genetic Structure Based on Clustering Analyses
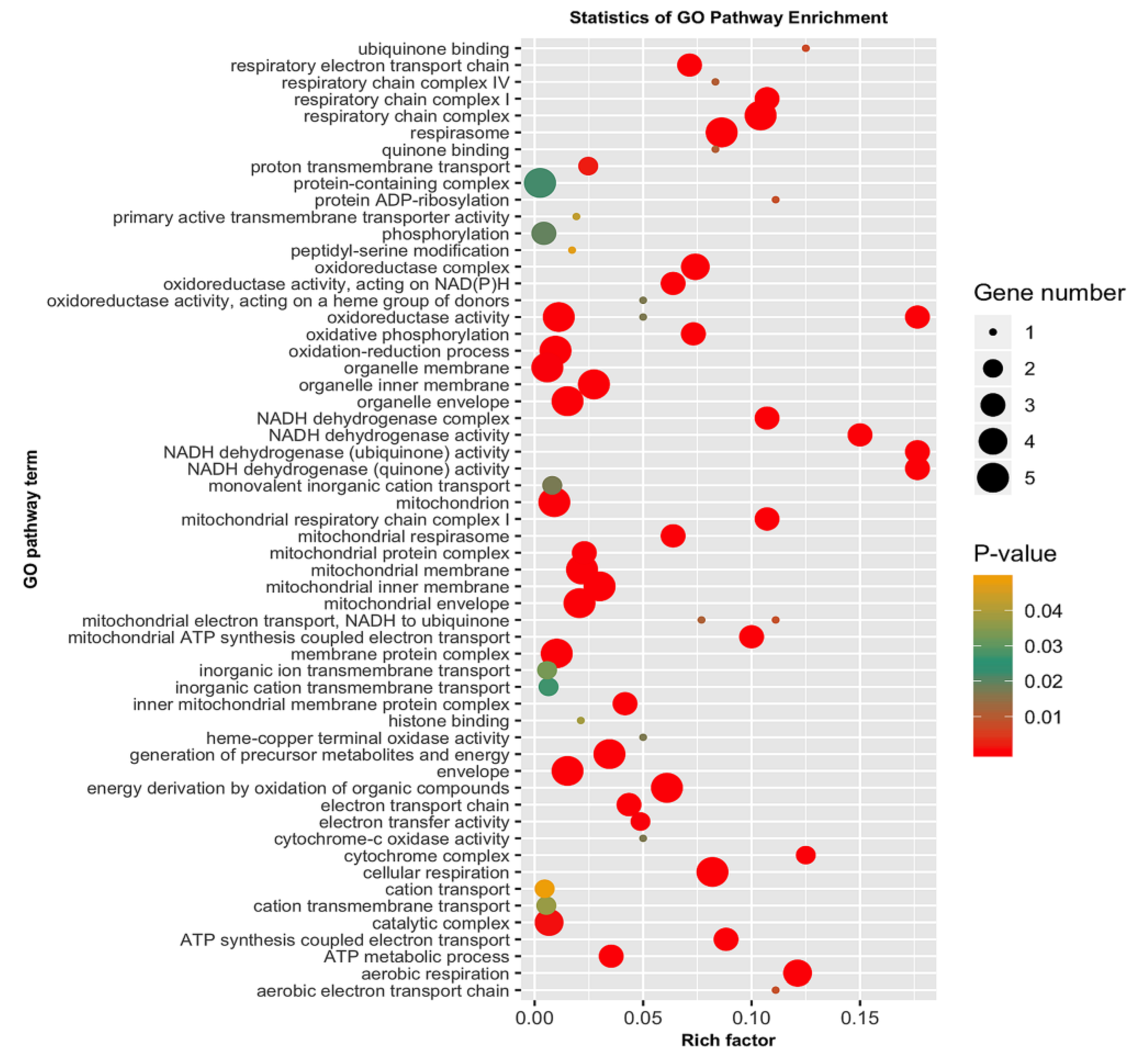
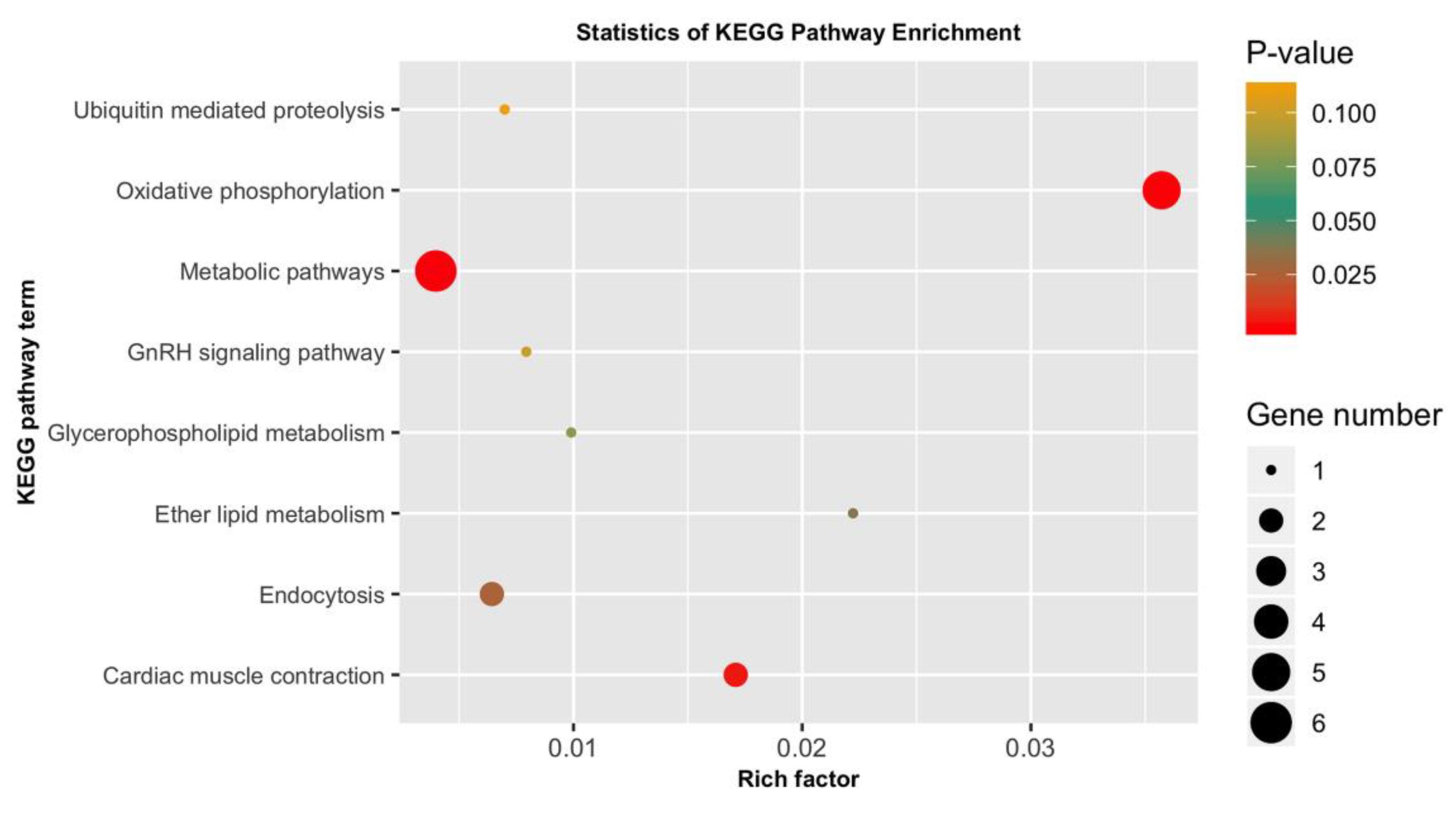
3.5. GO Categorization of the Encoding Genes of Putatively Adaptive SNP Loci
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- FAO (Food and Agriculture Organization of the United Nations). The State of World Fisheries and Aquaculture 2020; Sustainability in Action; FAO: Rome, Italy, 2020.
- FAO (Food and Agriculture Organization of the United Nations). GLOBEFISH—Information and Analysis on World Fish Trade; FAO: Italy, Rome, 2019.
- Venkateswarlu, V.; Venkatrayulu, C. Prevalence of disease problems affecting shrimp Litopenaeus vannamei farming in Andhra Pradesh, India. Int. J. Fish. Aquat. Stud. 2019, 7, 275–279. [Google Scholar]
- Wyban, J.; Martinez, G.; Sweeney, J.N. Adding paprika to Penaeus vannamei maturation diet improves nauplii quality. World Aquac. 1997, 28, 59–62. [Google Scholar]
- Klinbunga, S.; Siludjai, D.; Wuthijinda, W.; Tassanakajon, A.; Jarayabhand, P.; Menasveta, P. Genetic heterogeneity of the giant tiger shrimp (Penaeus monodon) in Thailand revealed by RAPD and mtDNA-RFLP analyses. Mar. Biotechnol. 2001, 3, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Nahavandi, R.; Hafezamini, P.; Shamsudin, M.N. Genetic diversity of intensive cultured and wild tiger shrimp Penaeus monodon (Fabricius) in Malaysia using microsatellite markers. Afr. J. Biotechnol. 2011, 10, 15501–15508. [Google Scholar] [CrossRef]
- FAO (Food and Agriculture Organization of the United Nations). Improving Penaeus Monodon Hatchery Practices; FAO Fisheries Technical Paper 446; FAO: Rome, Italy, 2007.
- Andam, C.P.; Challagundla, L.; Azarian, T.; Hanage, W.P.; Robinson, D.A. 3—Population Structure 2017, Pages 51–70 of Pathogenic Bacteria. In Genetics and Evolution of Infectious Diseases, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 51–70. [Google Scholar]
- Fowler, S.; Roush, R.; Wise, J. Concept of Biology. OpenStax, Pressbook. 2019. Available online: https://openstax.org/details/books/concepts-biology (accessed on 28 June 2020).
- De León, F.J.G.; Galván-Tirado, C.; Velasco, L.S.; Segundo, C.A.S.; Guzmán, H.R.; Barriga-Sosa, I.A.; Jaimes, P.D.; Canino, M.; Cruz-Hernandez, P. Role of oceanography in shaping the genetic structure in the North Pacific hake Merluccius productus. PLoS ONE 2018, 13, e0194646. [Google Scholar]
- Perina, A.; Mari-Mena, N.; Torrecilla, Z.; González-Tizón, A.M.; González-Castellano, I.; González-Ortegón, E.; Martínez-Lage, A. Assessment of genetic diversity and population structure of the common littoral shrimp Palaemon serratus (Pennant, 1777) by microsatellites: Towards a sustainable management. Aquatic Conservation. Mar. Freshw. Ecosyst. 2018, 29, 528–536. [Google Scholar] [CrossRef]
- Yu, L.; Zhu, X.; Liang, J.; Fan, J.; Chen, C. Analysis of genetic structure of wild and cultured Giant Freshwater Prawn (Macrobrachium rosenbergii) using newly developed microsatellite. Front. Mar. Sci. 2019, 6, 323. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, L.; Fu, J.; Xu, X.; Yue, G.H.; Li, J. Population structure, demographic history and local adaptation of the grass carp. BMC Genom. 2019, 20, 467. [Google Scholar] [CrossRef]
- Dixon, T.J.; Coman, G.J.; Arnold, S.J.; Sellars, M.J.; Lyons, R.E.; Dierens, L.E.; Preston, N.G.; Li, Y. Shifts in genetic diversity during domestication of Black Tiger shrimp, Penaeus monodon, monitored using two multiplexed microsatellite systems. Aquaculture 2008, 283, 1–6. [Google Scholar] [CrossRef]
- Souza de Lima, A.P.; Lira dos, S.A.C.; Dantas, H.L.; Gomes, F.M.; Maggioni, R.; Moura, C.M.R. Genetic monitoring of broodstocks of the marine shrimp Litopenaeus vannamei in a closed rearing system in Pernambuco, Brazil. Aquac. Res. 2008, 39, 1461–1466. [Google Scholar] [CrossRef]
- Benzie, J.A.H. Use and exchange of genetic resources of penaeid shrimps for food and aquaculture. Rev. Aquac. 2009, 1, 232–250. [Google Scholar] [CrossRef]
- Perez-Enriquez, R.; Hernández-Martínez, F.; Cruz, P. Genetic diversity status of White shrimp Penaeus (Litopenaeus) vannamei broodstock in Mexico. Aquaculture 2009, 297, 44–50. [Google Scholar] [CrossRef]
- Zhang, T.; Kong, J.; Wang, W.; Wang, Q. Genetic variability assessed by microsatellites in the breeding populations of the shrimp Penaeus (Fenneropenaeus) chinensis in China. Aquaculture 2010, 310, 229–233. [Google Scholar] [CrossRef]
- Li, Z.-M.; Xie, L.; Ye, F.; Chen, G. SSR analysis of three species from primary parent and their first generation of Litopenaeus vannamei. Agric. Biotechnol. 2010, 11, 57–61. [Google Scholar]
- De Donato, M.; Manrique, R.; Ramirez, R.; Mayer, L.; Howell, C. Mass selection and inbreeding effects on a cultivated strain of Penaeus (Litopenaeus) vannamei in Venezuela. Aquaculture 2005, 247, 159–167. [Google Scholar] [CrossRef]
- Khedkar, G.D.; Reddy, A.C.; Ron, T.B.; Haymer, D. High levels of genetic diversity in Penaeus monodon populations from the east coast of India. Springer Plus 2013, 2, 671. [Google Scholar] [CrossRef]
- Ríos-Pérez, L.D.L.; Campos-Montes, G.R.; Martínez-Ortegad, A.; Castillo-Juárez, H.; Montaldo, H.H. Inbreeding effects on reproductive traits in a breeding population of Pacific white shrimp Penaeus (Litopenaeus) vannamei. Aquaculture 2017, 479, 442–446. [Google Scholar] [CrossRef]
- Lynch, M.; Walsh, B. Genetics and Analysis of Quantitative Traits; Sinauer Associates: Sunderland, MA, USA, 1998. [Google Scholar]
- Hedrick, P.W.; Kalinowski, S.T. Inbreeding depression in conservation biology. Annu. Rev. Ecol. Syst. 2000, 31, 139–162. [Google Scholar] [CrossRef]
- Ponzoni, R.W.; Khaw, H.L.; Nguyen, N.H.; Hamzah, A. Inbreeding and effective population size in the Malaysian nucleus of the GIFT strain of Nile tilapia (Oreochromis niloticus). Aquaculture 2010, 302, 42–48. [Google Scholar] [CrossRef]
- Francisco, A.K.; Galetti, P.M., Jr. Genetic distance between broodstock of the marine shrimp Litopenaeus vannamei (Decapoda, Penaeidae) by mtDNA analyses. Genet. Mol. Biol. 2005, 28, 258–261. [Google Scholar] [CrossRef]
- Rezaee, S.; Farahmand, H.; Nematollahi, M.A. Genetic diversity status of Pacific white shrimp (Litopenaeus vannamei) using SSR markers in Iran. Aquac. Int. 2015, 247, 159–167. [Google Scholar] [CrossRef]
- Gaiotto, F.A.; Bramucci, M.; Grattapaglia, D. Estimation of outcrossing rate in a breeding population of Eucalyptus urophylla with dominant RAPD and AFLP markers. Theor. Appl. Genet. 1997, 95, 842–849. [Google Scholar] [CrossRef]
- Benzie, J.A.H.; Ballment, E.; Forbes, A.T.; Demetriades, N.T.; Sugama, K.; Haryanti; Moria, S. Mitochondrial DNA variation in Indo-Pacific populations of the Giant Tiger prawn, Penaeus monodon. Mol. Ecol. 2002, 11, 2553–2569. [Google Scholar] [CrossRef] [PubMed]
- Waqairatu, S.S.; Dierens, L.; Cowley, J.A.; Dixon, T.J.; Johnson, K.N.; Barnes, A.C.; Li, Y. Genetic analysis of Black Tiger shrimp (Penaeus monodon) across its natural distribution range reveals more recent colonization of Fiji and other South Pacific Islands. Ecol. Evol. 2012, 2, 2057–2071. [Google Scholar] [CrossRef]
- Sugama, K.; Haryanti Benzie, J.A.H.; Ballment, E. Genetic variation and population structure of the giant tiger prawn, Penaeus monodon, in Indonesia. Aquaculture 2002, 205, 37–48. [Google Scholar] [CrossRef]
- Brooker, A.L.; Benzie, J.A.H.; Blair, D.; Versini, J.J. Population structure of the giant tiger prawn Penaeus monodon in Australian waters, determined using microsatellite markers. Mar. Biol. 2000, 136, 149–157. [Google Scholar] [CrossRef]
- Xu, Z.K.; Primavera, J.H.; de la Pena, L.D.; Belak, J.; Alcivar-Warren, A. Genetic diversity of wild and cultured Black tiger Shrimp (Penaeus monodon) in the Philippines using microsatellites. Aquaculture 2001, 199, 13–40. [Google Scholar] [CrossRef]
- You, E.M.; Chiu, T.S.; Liu, K.F.; Tassanakajon, A.; Klinbunga, S.; Triwitayakorn, K.; de la Peña, L.D.; Li, Y.; Yu, H.T. Microsatellite and mitochondrial haplotype diversity reveals population differentiation in the tiger shrimp (Penaeus monodon) in the Indo-Pacific region. Anim. Genet. 2008, 39, 267–277. [Google Scholar] [CrossRef]
- Walther, E.; Schöfl, G.; Mrotzek, G.; Sugama, K.; Saluz, H.P. Paralogous mitochondrial control region in the giant tiger shrimp, Penaeus monodon (F.) affects population genetics inference: A cautionary tale. Mol. Phylogenetics Evol. 2011, 58, 404–408. [Google Scholar] [CrossRef]
- Abdul-Aziz, M.A.; Sch€ofl, G.; Mrotzek, G.; Haryanti, H.; Sugama, K.; Saluz, H.P. Population structure of the Indonesian giant tiger shrimp Penaeus monodon: A window into evolutionary similarities between paralogous mitochondrial DNA sequences and their genomes. Ecol. Evol. 2015, 5, 3570–3584. [Google Scholar] [CrossRef]
- Duda, T.F.; Palumbi, S.R. Population structure of the black tiger prawn, Penaeus monodon, among western Indian Ocean and western Pacific populations. Mar. Biol. 1999, 134, 705–710. [Google Scholar] [CrossRef]
- Carreras, C.; Ordóñez, V.; Zane, L.; Kruschel, C.; Nasto, I.; Macpherson, E.; Pascual, M. Population genomics of an endemic Mediterranean fish: Differentiation by fine scale dispersal and adaptation. Sci. Rep. 2016, 7, 43417. [Google Scholar] [CrossRef] [PubMed]
- Vendrami, D.L.; Telesca, L.; Weigand, H.; Weiss, M.; Fawcett, K.; Lehman, K.; Clark, M.S.; Leese, F.; McMinn, C.; Moore, H.; et al. RAD sequencing resolves fine-scale population structure in a benthic invertebrate: Implications for understanding phenotypic plasticity. R. Soc. Open Sci. 2017, 4, 160548. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.O.; Jones, K.S.; Price, S.J.; Weisrock, D.W. A genomic assessment of population structure and gene flow in an aquatic salamander identifies the roles of spatial scale, barriers, and river architecture. Freshw. Biol. 2018, 63, 407–419. [Google Scholar] [CrossRef]
- Asaduzzaman, M.; Wahab, M.A.; Rahman, M.J.; Nahiduzzzaman, M.; Dickson, M.W.; Igarashi, Y.; Asakawa, S.; Wong, L.L. Fine-scale population structure and ecotypes of anadromous Hilsa shad (Tenualosa ilisha) across complex aquatic ecosystems revealed by NextRAD genotyping. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Zhang, B.D.; Xue, D.X.; Li, Y.L.; Liu, J.X. RAD genotyping reveals fine-scale population structure and provides evidence for adaptive divergence in a commercially important fish from the northwestern Pacific Ocean. PeerJ 2019, 7, e7242. [Google Scholar] [CrossRef]
- Sherman, K.D.; Paris, J.R.; King, R.A.; Moore, K.A.; Dahlgren, C.P.; Knowles, L.C.; Stump, K.; Tyler, C.R.; Stevens, J.R. RAD-Seq Analysis and in situ Monitoring of Nassau Grouper Reveal Fine-Scale Population Structure and Origins of Aggregating Fish. Front. Mar. Sci. 2020, 7, 157. [Google Scholar] [CrossRef]
- Gamboa, M.; Watanabe, K. Genome-wide signatures of local adaptation among seven stoneflies species along a nationwide latitudinal gradient in Japan. BMC Genom. 2019, 20, 84. [Google Scholar] [CrossRef]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef]
- Bayer, P.E.; Ruperao, P.; Mason, A.S.; Stiller, J.; Chan, C.K.K.; Hayashi, S.; Long, Y.; Meng, J.; Sutton, T.; Visendi, P.; et al. High-resolution skim genotyping by sequencing reveals the distribution of crossovers and gene conversions in Cicer arietinum and Brassica napus. Theor. Appl. Genet. 2015, 128, 1039–1045. [Google Scholar] [CrossRef]
- Golicz, A.A.; Bayer, P.E.; Edwards, D. Skim-based genotyping by sequencing. Methods Mol. Biol. 2015, 1245, 257–270. [Google Scholar] [PubMed]
- Scheben, A.; Batley, J.; Edwards, D. Genotyping-by-sequencing approaches to characterize crop genomes: Choosing the right tool for the right application. Plant Biotechnol. J. 2017, 15, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Scheben, A.; Batley, J.; Edwards, D. Revolution in Genotyping Platforms for Crop Improvement. In Advances in Biochemical Engineering Biotechnology; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Anderson, J.; Kantar, M.; Bock, D.; Grubbs, K.C.; Schilling, E.; Rieseberg, L. Skim-Sequencing Reveals the Likely Origin of the Enigmatic Endangered Sunflower Helianthus schweinitzii. Genes 2019, 10, 1040. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.E. Skim-Based Genotyping by Sequencing Using a Double Haploid Population to Call SNPs, Infer Gene Conversions, and Improve Genome Assemblies. In Plant Bioinformatics; Humana Press: New York, NY, USA, 2016; pp. 285–292. [Google Scholar]
- Bayer, P.E.; Hurgobin, B.; Golicz, A.A.; Chan, C.K.K.; Yuan, Y.; Lee, H.; Renton, M.; Meng, J.; Li, R.; Long, Y.; et al. Assembly and comparison of two closely related Brassica napus genomes. Plant Biotechnol. J. 2017, 15, 1602–1610. [Google Scholar] [CrossRef]
- Gacek, K.; Bayer, P.E.; Bartkowiak-Broda, I.; Szala, L.; Bocianowski, J.; Edwards, D.; Batley, J. Genome-wide association study of genetic control of seed fatty acid biosynthesis in Brassica napus. Front. Plant Sci. 2017, 7, 2062. [Google Scholar] [CrossRef] [PubMed]
- Blotta, I.; Prestinaci, F.; Mirante, S.; Cantafora, A. Quantitative assay of total dsDNA with PicoGreen reagent and real-time fluorescent detection. Ann. Ist Super Sanita 2005, 41, 119–123. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzburg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, Y.L.; Weng, J.C.; Hsiao, C.C.; Chou, M.T.; Tseng, C.W.; Hung, J.H. PEAT: An intelligent and efficient paired-end sequencing adapter trimming algorithm. BMC Bioinform. 2015, 16, S2. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient shortread de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Boeckmann, B.; Ferro, S.; Gasteiger, E. Swiss-Prot: Juggling between evolution and stability. Brief. Bioinform. 2004, 5, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Pedersen, B.S.; Layer, R.M.; Quinlan, A.R. Vcfanno: Fast, flexible annotation of genetic variants. Genome Biol. 2016, 17, 118. [Google Scholar] [CrossRef]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Duforet-Frebourg, N.; Luu, K.; Laval, G.; Bazin, E.; Blum, M.G. Detecting genomic signatures of natural selection with principal component analysis: Application to the 1000 Genomes data. Mol. Biol. Evol. 2015, 334, 1082–1093. [Google Scholar] [CrossRef]
- Luu, K.; Bazin, E.; Blum, M.G.B. pcadapt: An R package to perform genome scans for selection based on principal component analysis. Mol. Ecol. Resour. 2017, 17, 67–77. [Google Scholar] [CrossRef]
- Ryman, N.; Palm, S. POWSIM: A computer program for assessing statistical power when testing for genetic differentiation. Mol. Ecol. Notes 2006, 6, 600–602. [Google Scholar] [CrossRef]
- Jombart, T. adegenet: An R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Meirmans, P.G.; van Tienderen, P.H. GENOTYPE and GENODIVE: Two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes 2004, 4, 792–794. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Hollenbeck, C.M.; Portnoy, D.S.; Gold, J.R. Evolution of population structure in an estuarine-dependent marine fish. Ecol. Evol. 2019, 9, 3141–3152. [Google Scholar] [CrossRef]
- Milano, I.; Babbucci, M.; Cariani, A.; Atanassova, M.; Bekkevold, D.; Carvalho, G.R.; Espiñeira, M.; Fiorentino, F.; Garofalo, G.; Geffen, A.J.; et al. Outlier SNP markers reveal fine-scale genetic structuring across European hake populations (Merluccius merluccius). Mol. Ecol. 2014, 23, 118–135. [Google Scholar] [CrossRef]
- Purves, W.K.; Sadava, D.; Orians, G.H.; Heller, H.C. Genetic drift may cause large changes in small populations. In Life: The Science of Biology, 7th ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 2003; pp. 468–469. [Google Scholar]
- Nazareno, A.G.; Bemmels, J.B.; Dick, C.W.; Lohmann, L.G. Minimum sample sizes for population genomics: An empirical study from an Amazonian plant species. Mol. Ecol. Resour. 2017, 17, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, D.L.; Copp, G.H.; Lawson Handley, L.; Olsén, K.H.; Sayer, C.D.; Hänfling, B. Comparing RAD seq and microsatellites to infer complex phylogeographic patterns, an empirical perspective in the Crucian carp, Carassius carassius, L. Mol. Ecol. 2016, 25, 2997–3018. [Google Scholar] [CrossRef] [PubMed]
- Puckett, E.E.; Eggert, L.S. Comparison of SNP and microsatellite genotyping panels for spatial assignment of individuals to natal range: A case study using the American black bear (Ursus americanus). Biol. Conserv. 2016, 193, 86–93. [Google Scholar] [CrossRef]
- Qu, W.M.; Liang, N.; Wu, Z.K.; Zhao, Y.G.; Chu, D. Minimum sample sizes for invasion genomics: Empirical investigation in an invasive whitefly. Ecol. Evol. 2020, 10, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qu, W.; Obrycki, J.J.; Meng, L.; Zhou, X.; Chu, D.; Li, B. Optimizing Sample Size for Population Genomic Study in a Global Invasive Lady Beetle, Harmonia Axyridis. Insects 2020, 11, 290. [Google Scholar] [CrossRef]
- Flesch, E.P.; Rotella, J.J.; Thomson, J.M.; Graves, T.A.; Garrott, R.A. Evaluating sample size to estimate genetic management metrics in the genomics era. Mol. Ecol. Resour. 2018, 18, 1077–1091. [Google Scholar] [CrossRef]
- Thanh, H.N.; Liu, Q.; Zhao, L.; Zhang, H.; Liu, J. Genetic diversity of the cultured giant freshwater prawn (Macrobrachium rosenbergii) in China based on microsatellite markers. Biochem. Syst. Ecol. 2015, 59, 144–154. [Google Scholar] [CrossRef]
- Miao, W.M.; Ge, X.P. Freshwater prawn culture in China: An overview. Aquaculture Asia 2002, VII, 9–12. [Google Scholar]
- Rhode, C.; Hepple, J.; Jansen, S.; Davis, T.; Vervalle, J.; Bester-van der Merwe, A.E.; Roodt-Wilding, R. A population genetic analysis of abalone domestication events in South Africa: Implications for the management of the abalone resource. Aquaculture 2012, 356–357, 235–242. [Google Scholar] [CrossRef]
- An, H.A.; Wook, J.; Kim, H.C.; Myeong, J. Genetic characterization of five hatchery populations of pacific abalone (Haliotis discus hannai) using microsatellite markers. Int. J. Mol. Sci. 2011, 12, 4836–4849. [Google Scholar] [CrossRef]
- Norris, A.T.; Bradley, D.G.; Cunningham, E.P. Microsatellite genetic variation between and within farmed and wild Atlantic salmon (Salmo salar) populations. Aquaculture 1999, 180, 247–264. [Google Scholar] [CrossRef]
- Zhang, D.C.; Yu, L.N.; Fang, Y.L. A study on genetic diversity of wild and cultured grass carp populations using RAPD. Freshw. Fish 2004, 4, 5–7. [Google Scholar]
- Liu, F.; Xia, J.; Bai, Z.; Fu, J.; Li, J.; Yue, G.H. High genetic diversity and substantial population differentiation in grass carp (Ctenopharyngodon idella) revealed by microsatellite analysis. Aquaculture 2009, 297, 51–56. [Google Scholar] [CrossRef]
- Yue, G.H.; Zhu, Z.Y.; Lo, L.C.; Wang, C.M.; Lin, G.; Feng, F.; Pang, H.Y.; Li, J.; Gong, P.; Liu, H.M.; et al. Genetic variation and population structure of Asian seabass (Lates calcarifer) in the Asia-Pacific region. Aquaculture 2009, 293, 22–28. [Google Scholar] [CrossRef]
- Li, L.; Lin, H.; Tang, W.; Liu, D.; Bao, B.; Yang, J. Population genetic structure in wild and aquaculture populations of Hemibarbus maculates inferred from microsatellites markers. Aquac. Fish. 2017, 2, 78–83. [Google Scholar] [CrossRef]
- Tassanakajon, A.; Pongsomboon, S.; Jarayabhand, P.; Klinbunga, S.; Boonsaeng, V. Genetic structure in wild populations of black tiger shrimp Penaeus monodon using randomly amplified polymorphic DNA analysis. J. Mar. Biotechnol. 1998, 6, 249–254. [Google Scholar]
- Benzie, J.A.H.; Frusher, S.; Ballment, E. Geographical variation in allozyme frequencies of populations of Penaeus monodon Crustacea: Decapoda in Australia. Aust. J. Mar. Freshw. Res. 1992, 43, 715–725. [Google Scholar] [CrossRef]
- Sekino, M.; Hara, M.; Taniguchi, N. Loss of microsatellite and mitochondrial DNA variation in hatchery strains of Japanese flounder Paralichthys olivaceus. Aquaculture 2002, 213, 101–122. [Google Scholar] [CrossRef]
- Lundrigan, T.A.; Reist, J.D.; Ferguson, M.M. Microsatellite genetic variation within and among Arctic charr (Salvelinus alpinus) from aquaculture and natural populations in North America. Aquaculture 2005, 244, 63–75. [Google Scholar] [CrossRef]
- Kohlmann, K.; Kersten, P.; Flajshans, M. Microsatellite-based genetic variability and differentiation of domesticated, wild and feral common carp (Cyprinus carpio L.) populations. Aquaculture 2005, 247, 253–266. [Google Scholar] [CrossRef]
- Koljonen, M.L.; Tähtinen, J.; Säisä, M.; Koskiniemi, J. Maintenance of genetic diversity of Atlantic salmon (Salmo salar) by captive breeding programmes and the geographic distribution of microsatellite variation. Aquaculture 2002, 212, 69–92. [Google Scholar] [CrossRef]
- Wang, L.; Meng, Z.; Liu, X.; Zhang, Y.; Lin, H. Genetic Diversity and differentiation of the Orange Spotted Grouper (Epinephelus coioides) between and bithin cultured stocks and wild populations Inferred from Microsatellite DNA analysis. Int. J. Mol. Sci. 2011, 12, 4378–4394. [Google Scholar] [CrossRef] [PubMed]
- Bester-van der Merwe, A.E.; Roodt-Wilding, R.; Volckaert, F.A.M.; D’Amato, M.E. Historical isolation and hydrodynamically constrained gene flow in declining population of the South-African abalone, Haliotis midae. Conserv. Genet. 2011, 12, 543–555. [Google Scholar] [CrossRef]
- Alejandrino, A.; Puslednik, L.; Serb, J.M. Convergent and parallel evolution in life habit of the scallops (Bivalvia: Pectinidae). BMC Evol. Biol. 2011, 11, 164. [Google Scholar] [CrossRef]
- Lester, L.J. Population genetics of penaeid shrimp from the Gulf of Mexico. J. Hered. 1979, 70, 175–180. [Google Scholar] [CrossRef]
- Benzie, J.A.H.; Ballment, E.; Frusher, S. Genetic structure of Penaeus monodon in Australia: Concordant results from mtDNA and allozymes. Aquaculture 1993, 111, 89–93. [Google Scholar] [CrossRef]
- Wade, M.J. Extinction and recolonization: Their effects on the genetic differentiation of local population. Evolution 1988, 42, 995–1005. [Google Scholar] [CrossRef]
- Li, J.; Wang, G.; Bai, Z. Genetic variability in four wild and two farmed stocks of the Chinese freshwater pearl mussel (Hyriopsis cumingii) estimated by microsatellite DNA Markers. Aquaculture 2009, 287, 286–291. [Google Scholar] [CrossRef]
- Qi, P.; Qin, J.; Xie, C. Determination of genetic diversity of wild and cultured topmouth culter (Culter alburnus) inhabiting China using mitochondrial DNA and microsatellites. Biochem. Syst. Ecol. 2015, 61, 232–239. [Google Scholar] [CrossRef]
- Hemmer-Hansen, J.; Nielsen, E.E.; Frydenberg, J.; Loeschcke, V. Adaptive divergence in a high gene flow environment: Hsc70 variation in the European flounder (Platichthys flesus L.). Heredity 2007, 99, 592–600. [Google Scholar] [CrossRef]
- Nielsen, E.E.; Hemmer-Jansen, J.; Larsen, P.F.; Bekkevold, D. Population genomics of marine fishes: Identifying adaptive variation in space and time. Mol. Ecol. 2009, 18, 3128–3150. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.S.; Serrão, E.A.; Pearson, G.A.; Riginos, C.; Rawson, P.D.; Hilbish, T.J.; Brawley, S.H.; Trussell, G.C.; Carrington, E.; Wethey, D.S.; et al. Ecological genetics in the North Atlantic: Environmental gradients and adaptation at specific loci. Ecology 2008, 89, S91–S107. [Google Scholar] [CrossRef] [PubMed]
- Asaduzzaman, M.; Igarashi, Y.; Wahab, M.A.; Nahiduzzzaman, M.; Rahman, M.J.; Phillips, M.J.; Huang, S.; Asakawa, S.; Rahman, M.M.; Wong, L.L. Population Genomics of an Anadromous Hilsa Shad Tenualosa ilisha Species across its diverse migratory habitats: Discrimination by fine-scale local adaptation. Genes 2020, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Asaduzzaman, M.; Wahab, M.A.; Rahman, M.M.; Mariom; Nahiduzzaman, M.; Rahman, M.J.; Roy, B.K.; Phillips, M.J.; Wong, L.L. Morpho-Genetic Divergence and Adaptation of Anadromous Hilsa shad (Tenualosa ilisha) Along Their Heterogenic Migratory Habitats. Front. Mar. Sci. 2020, 7, 554. [Google Scholar] [CrossRef]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Evol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Moritz, C.; Dowling, T.E.; Brown, W.M. Evolution of animal mitochondrial DNA: Relevance for population biology and systematics. Annu. Rev. Ecol. Evol. Syst. 1987, 18, 269–292. [Google Scholar] [CrossRef]
- Ballard, J.W.O.; Kreitman, M. Is mitochondrial DNA a strictly neutral marker? Trends Ecol. Evol. 1995, 10, 485–488. [Google Scholar] [CrossRef]
- Ciborowski, K.L.; Consuegra, S.; García de Leániz, C.; Wang, J.; Beaumont, M.A.; Jordan, W.C. Stocking may increase mitochondrial DNA diversity but fails to halt the decline of endangered Atlantic salmon populations. Conserv. Genet. 2007, 8, 1355–1367. [Google Scholar] [CrossRef]
- Blier, P.U.; Dufresne, F.; Burton, R.S. Natural selection and the evolution of mtDNA-encoded peptides: Evidence for intergenomic co-adaptation. Trends Genet. 2001, 17, 400–406. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Hogue, B.A.; Hansford, R.G. Influence of NAD-linked dehydrogenase activity on flux through oxidative phosphorylation. Biochem. J. 1990, 268, 421–428. [Google Scholar] [CrossRef]
- Bai, Y.; Shakeley, R.M.; Attardi, G. Tight control of respiration by NADH Dehydrogenase ND5 subunit gene expression in mouse mitochondria. Mol. Cell Biol. 2000, 20, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Meiklejohn, C.D.; Montooth, K.L.; Ran, D.M. Positive and negative selection on the mitochondrial genome. Trends Genet. 2007, 23, 259–263. [Google Scholar] [CrossRef]
- Blier, P.U.; Guderley, H.E. Mitochondrial activity in rainbow trout red muscle: The effect of temperature on the ADP-dependence of ATP synthesis. J. Exp. Biol. 1993, 176, 145–158. [Google Scholar]
- Brown, K.H.; Lee, R.W.; Thorgaard, G.H. Use of androgenesis for estimating maternal and mitochondrial genome effects on development and oxygen consumption in rainbow trout, Oncorhynchus mykiss. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2006, 143, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Kivisild, T. Maternal ancestry and population history from whole mitochondrial genomes. Investig. Genet. 2015, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes/Metab. Res. Rev. 2002, 51, 2944–2950. [Google Scholar] [CrossRef]
- Einum, S.; Fleming, I.A. Highly fecund mothers sacrifice offspring survival to maximize fitness. Nature 2000, 405, 565–567. [Google Scholar] [CrossRef]
- Rand, D.M.; Haney, R.A.; Fry, A.J. Cytonuclear coevolution: The genomics of cooperation. Trends Ecol. Evol. 2004, 19, 645–653. [Google Scholar] [CrossRef]
- Das, J. The role of mitochondrial respiration in physiological and evolutionary adaptation. BioEssays 2006, 28, 890–901. [Google Scholar] [CrossRef]
- Zhao, X.; Gou, X.; Qin, Z.; Li, D.; Wang, Y.; Ma, E.; Li, S.; Zhang, J. Identification and expression of cuticular protein genes based on Locusta migratoria transcriptome. Sci. Rep. 2017, 7, 45462. [Google Scholar] [CrossRef]
- McKay, S.E.; Purcell, A.L.; Carew, T.J. Regulation of synaptic function by neurotrophic factors in vertebrates and invertebrates: Implications for development and learning. Learn. Mem. 1999, 6, 193–215. [Google Scholar] [PubMed]
- Saleem, M.; Qadir, M.I.; Perveen, N.; Ahmad, B.; Saleem, U.; Irshad, T.; Ahmad, B. Inhibitors of apoptotic proteins: New targets for anticancer therapy. Chem. Biol. Drug. Des. 2013, 82, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Twigg, M.; Brockbank, S.; Lowry, P.; FitzGerald, S.P.; Taggart, C.; Weldon, S. The role of serine proteases and antiproteases in the cystic fibrosis lung. Mediat. Inflamm. 2015, 293053, 10. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locations | Location Abbreviation | Broodstock Source | Latitudes | Longitudes | Year |
|---|---|---|---|---|---|
| Mahajamba, Madagascar | MMD | Domesticated | 16°02′52.8″ | 47°11′38.0″ | 2018 |
| Hawaii, HI, USA | MMO | Domesticated | 19°42′55.9″ | 156°02′34.6″ | 2018 |
| Petchaburi Province, Thailand | MT | Domesticated | 12°58′06.5″ | 99°37′48.0″ | 2019 |
| Setiu Wetland, Malaysia | MS | Wild | 5°40′38.3″ | 102°42′36.8″ | 2019 |
| Shizuoka, Japan | MJ | Wild | 34°56′25.9″ | 138°02′17.9″ | 2018 |
| Genome Assembly Statistics | |
| Number of scaffolds | 6,425,442 |
| Size of largest scaffolds (bp) | 15,300 |
| Size of smallest scaffolds (bp) | 100 |
| Total scaffold size (bp) | 1,531,786,734 |
| Scaffold N50 | 306 |
| Genome annotation statistics | |
| Number of Scaffolds with at least one protein sequence match | 440,707 |
| Number of Decapoda (order) protein sequences with match to scaffold | 107,063 |
| Number of SwissProt protein sequences with match to scaffold | 42,351 |
| Variant identification results | |
| Number of Scaffolds with at least one variant site | 3,651,235 |
| Total number of variants | 17,226,908 |
| Variant annotation results | |
| Total number of variants with annotations | 1,347,070 |
| Number of variants annotated with Decapoda proteins | 1,339,785 |
| Number of variants annotated with SwissProt proteins | 142,710 |
| Filtering Steps | Number of Loci |
|---|---|
| Total number of raw variants loci | 17,226,908 |
| Remaining SNP loci after excluded indels and MNP sites | 13,530,393 |
| Remaining SNP loci after excluded sites with MAF < 0.05 | 10,212,187 |
| Remaining SNP loci after excluded sites with missing genotypes in >80% of the samples in any population | 417,048 |
| Remaining SNP loci after excluded sites with genotypes not in Hardy-Weinberg equilibrium in any population (PHWE < 0.001) | 328,028 |
| SNP loci remained after all quality filtering | 194,259 |
| Number of putatively adaptive SNP loci | 4983 |
| All 194,259 SNP Loci | 4983 Putatively Adaptive SNP Loci | ||||
|---|---|---|---|---|---|
| t | Ne | Power | t | Ne | Power |
| 10 | 1000 | 0.945 | 10 | 1000 | 1.000 |
| 20 | 1000 | 1.000 | 20 | 1000 | 1.000 |
| 10 | 2000 | 0.884 | 10 | 2000 | 1.000 |
| 20 | 2000 | 0.912 | 20 | 2000 | 0.994 |
| 10 | 3000 | 0.824 | 10 | 3000 | 0.996 |
| 20 | 3000 | 0.902 | 20 | 3000 | 0.985 |
| Wild | Domesticated | ||||
|---|---|---|---|---|---|
| MJ | MS | MMO | MMD | MT | |
| All SNP loci | |||||
| MJ | -- | 0.001 | 0.001 | 0.001 | 0.001 |
| MS | 0.008 | -- | 0.001 | 0.001 | 0.001 |
| MMO | 0.151 | 0.147 | -- | 0.023 | 0.001 |
| MMD | 0.150 | 0.145 | 0.003 | -- | 0.001 |
| MT | 0.153 | 0.148 | 0.010 | 0.008 | -- |
| Putatively adaptive SNP loci | |||||
| MJ | -- | 0.000 | 0.000 | 0.000 | 0.000 |
| MS | 0.106 | -- | 0.000 | 0.000 | 0.000 |
| MMO | 0.836 | 0.856 | -- | 0.105 | 0.002 |
| MMD | 0.853 | 0.870 | 0.003 | -- | 0.000 |
| MT | 0.850 | 0.868 | 0.035 | 0.042 | -- |
| Source of Variation | Sum of Square | Variance Components | % of Variation | Statistics | p-Value |
|---|---|---|---|---|---|
| All SNP loci | |||||
| Within individuals | 1,056,256 | 25,309.931 | 74.1 | F_it = 0.259 | - |
| Among individuals | 1,286,226.1 | 5502.952 | 16.1 | F_is = 0.179 | 0.000 |
| Among populations | 355,088.8 | 3342.094 | 9.8 | F_st = 0.098 | 0.000 |
| Putatively adaptive SNP loci | |||||
| Within individuals | 10,205.5 | 238.184 | 19.2 | F_it = 0.808 | - |
| Among individuals | 8192.9 | −0.083 | 0.0 | F_is = 0.000 | 0.483 |
| Among populations | 63029.16 | 1004.217 | 80.8 | F_st = 0.808 | 0.000 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, L.L.; Deris, Z.M.; Igarashi, Y.; Huang, S.; Asakawa, S.; Ayub, Q.; Lim, S.Y.; Ikhwanuddin, M.; Iehata, S.; Okamoto, K.; et al. Skim-Sequencing Based Genotyping Reveals Genetic Divergence of the Wild and Domesticated Population of Black Tiger Shrimp (Penaeus monodon) in the Indo-Pacific Region. Biology 2020, 9, 277. https://doi.org/10.3390/biology9090277
Wong LL, Deris ZM, Igarashi Y, Huang S, Asakawa S, Ayub Q, Lim SY, Ikhwanuddin M, Iehata S, Okamoto K, et al. Skim-Sequencing Based Genotyping Reveals Genetic Divergence of the Wild and Domesticated Population of Black Tiger Shrimp (Penaeus monodon) in the Indo-Pacific Region. Biology. 2020; 9(9):277. https://doi.org/10.3390/biology9090277
Chicago/Turabian StyleWong, Li Lian, Zulaikha Mat Deris, Yoji Igarashi, Songqian Huang, Shuichi Asakawa, Qasim Ayub, Shu Yong Lim, Mhd Ikhwanuddin, Shumpei Iehata, Kazutoshi Okamoto, and et al. 2020. "Skim-Sequencing Based Genotyping Reveals Genetic Divergence of the Wild and Domesticated Population of Black Tiger Shrimp (Penaeus monodon) in the Indo-Pacific Region" Biology 9, no. 9: 277. https://doi.org/10.3390/biology9090277
APA StyleWong, L. L., Deris, Z. M., Igarashi, Y., Huang, S., Asakawa, S., Ayub, Q., Lim, S. Y., Ikhwanuddin, M., Iehata, S., Okamoto, K., Mariom, & Asaduzzaman, M. (2020). Skim-Sequencing Based Genotyping Reveals Genetic Divergence of the Wild and Domesticated Population of Black Tiger Shrimp (Penaeus monodon) in the Indo-Pacific Region. Biology, 9(9), 277. https://doi.org/10.3390/biology9090277