Mass Spectrometry to Study Chromatin Compaction

 ,
, 
Abstract
1. Introduction
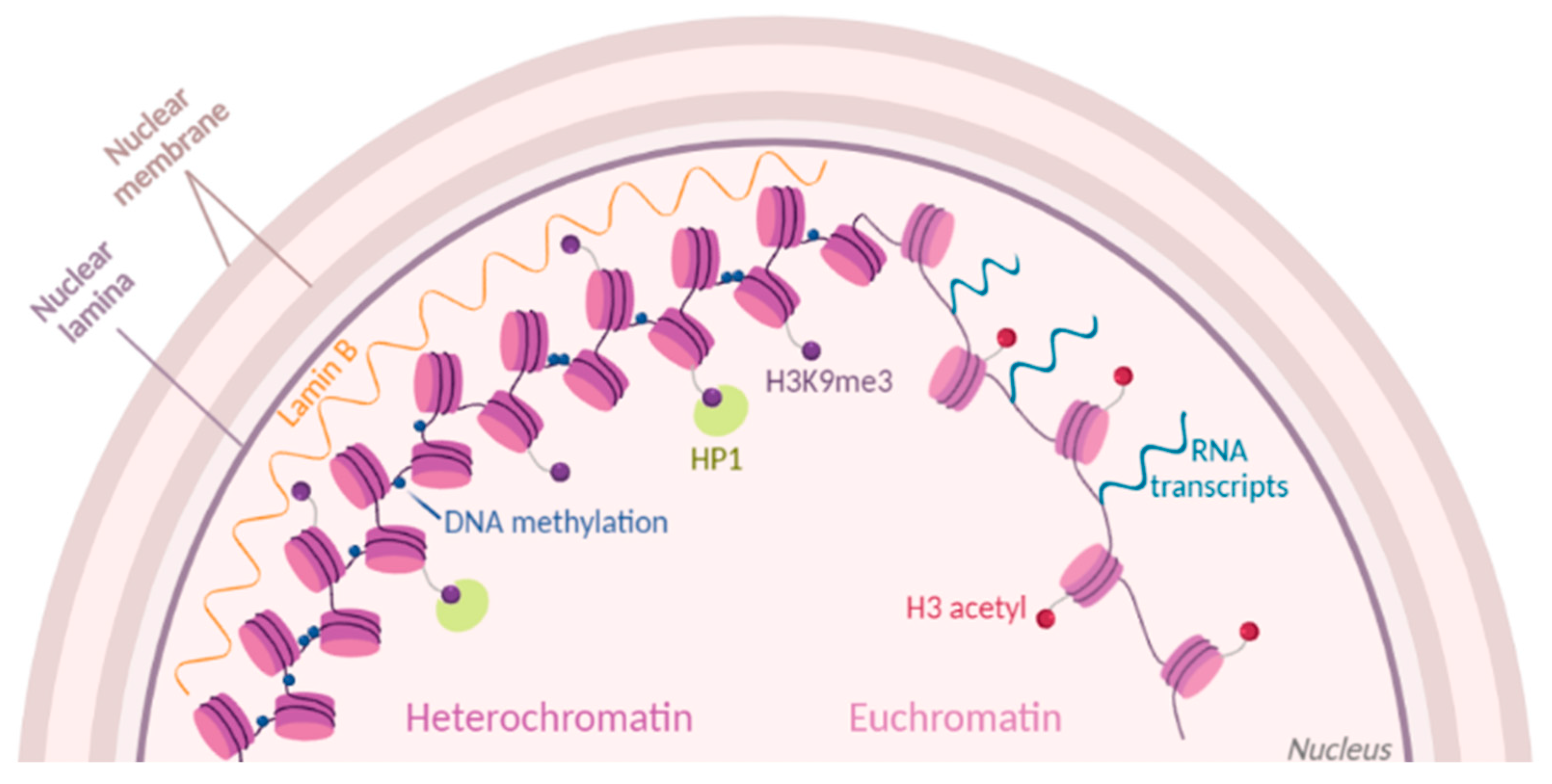
2. Popular Methods to Investigate Chromatin Accessibility
3. Mass Spectrometry to Study Chromatin State: First Steps with Nucleotide Modifications
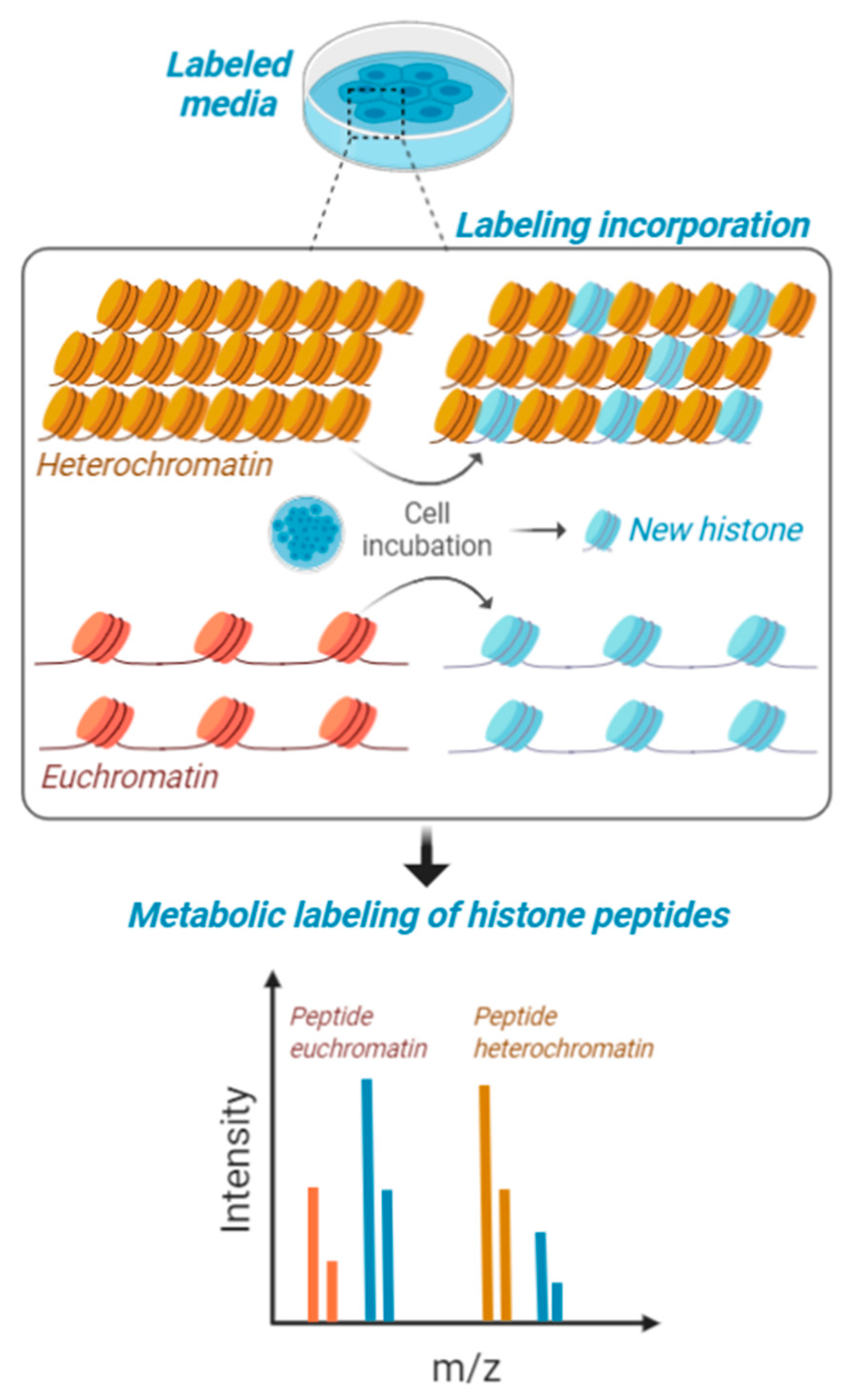
4. Multi-Dimensional Histone Modification Analysis Using Mass Spectrometry
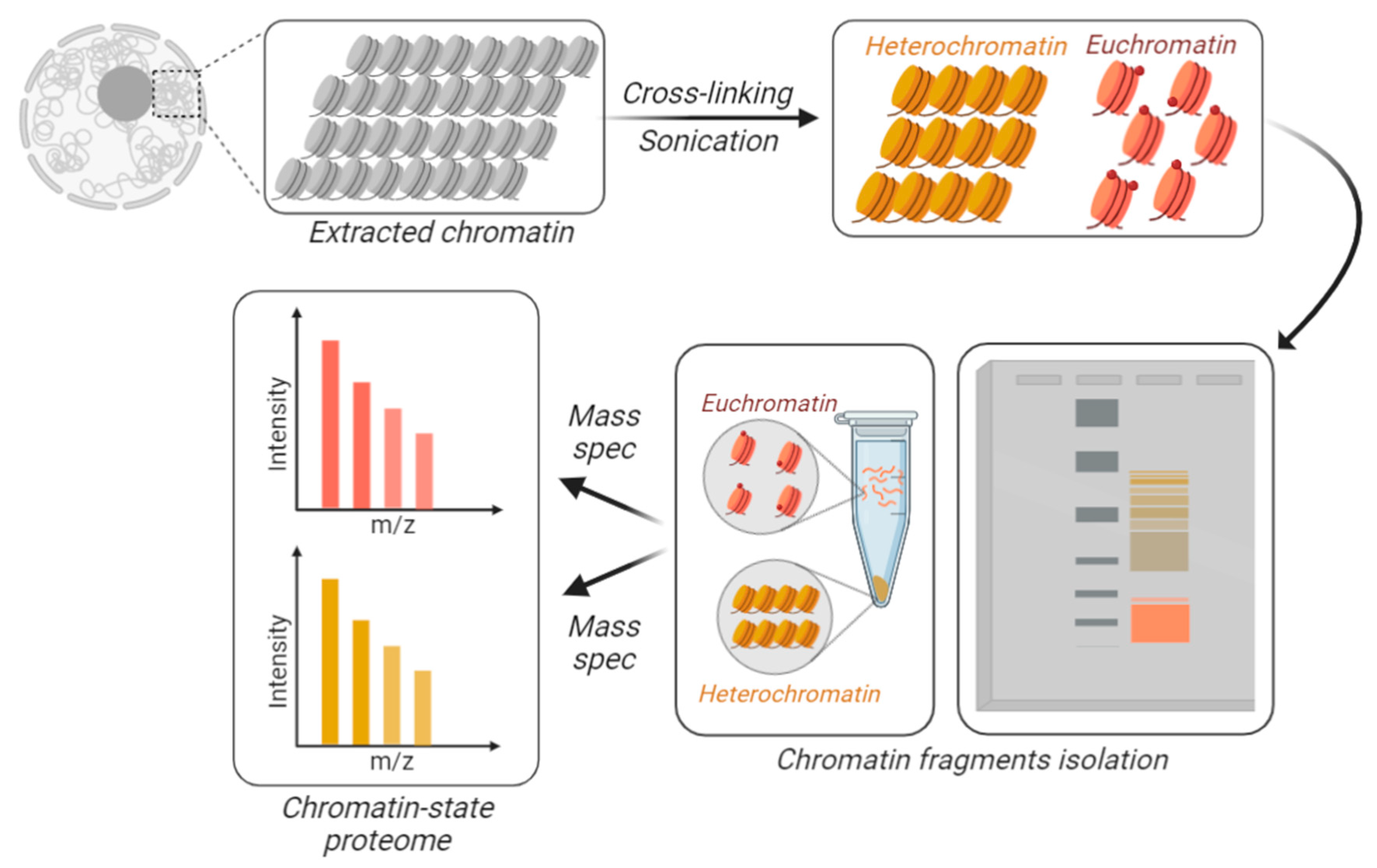
5. Quantifying the Chromatin-State Dependent Proteome with Mass Spectrometry
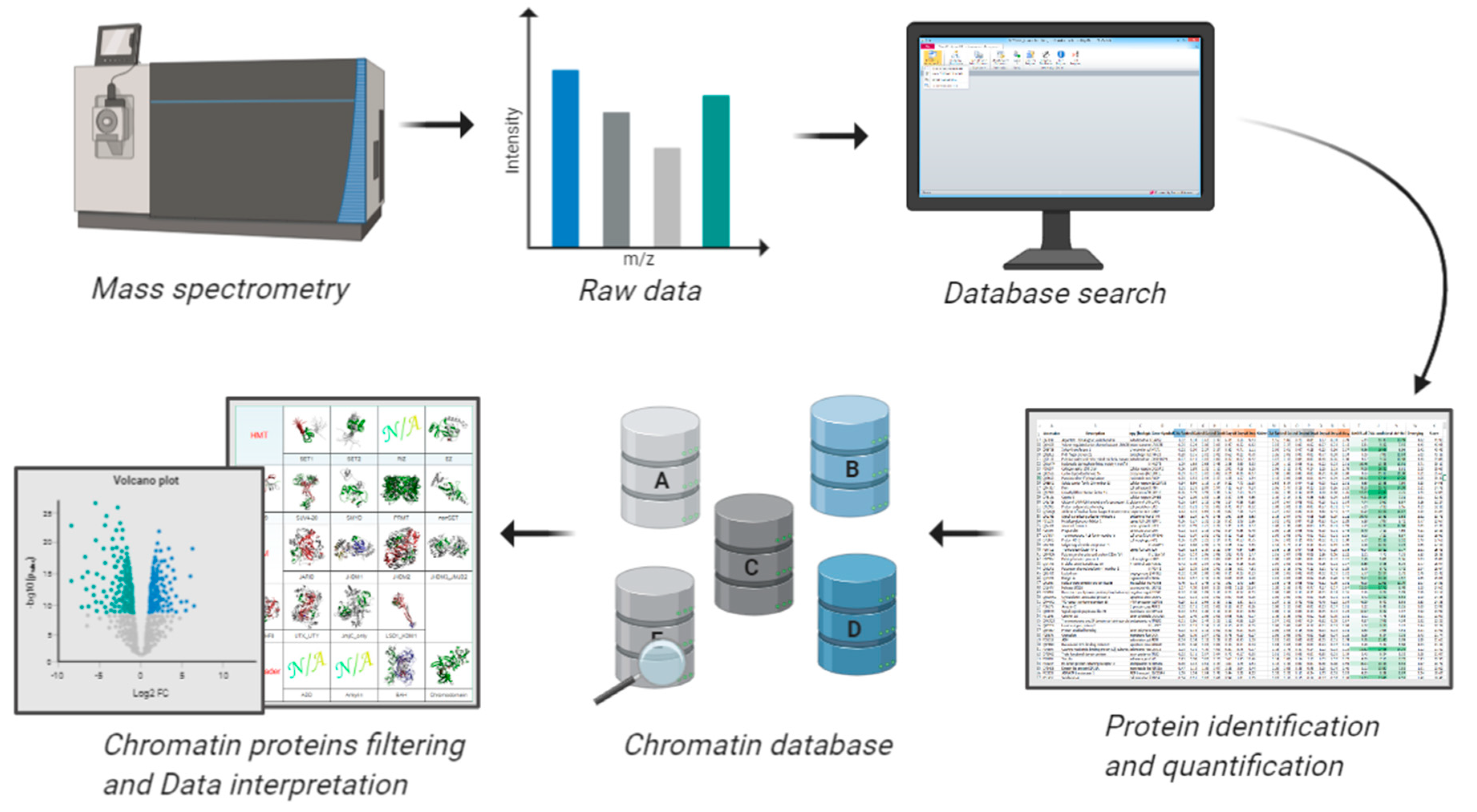
6. Bioinformatics Resources to Study the Chromatin Bound Proteome
7. Conclusive Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J.P. Targeting Epigenetic Readers in Cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Vickers, M.; Zhang, J.; Feng, X. Natural depletion of histone H1 in sex cells causes DNA demethylation, heterochromatin decondensation and transposon activation. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Maile, T.M.; Izrael-Tomasevic, A.; Cheung, T.; Guler, G.D.; Tindell, C.; Masselot, A.; Liang, J.; Zhao, F.; Trojer, P.; Classon, M.; et al. Mass spectrometric quantification of histone post-translational modifications by a hybrid chemical labeling method. Mol. Cell. Proteom. 2015, 14, 1148–1158. [Google Scholar] [CrossRef]
- Simithy, J.; Sidoli, S.; Yuan, Z.-F.; Coradin, M.; Bhanu, N.V.; Marchione, D.M.; Klein, B.J.; Bazilevsky, G.A.; McCullough, C.E.; Magin, R.; et al. Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 2017, 8, 1141. [Google Scholar] [CrossRef]
- Bueno, R.R.; Ruiz, P.D.L.C.; Artal-Sanz, M.; Askjaer, P.; Dobrzynska, A. Nuclear Organization in Stress and Aging. Cells 2019, 8, 664. [Google Scholar] [CrossRef]
- Quinn, J.; Fyrberg, A.M.; Ganster, R.W.; Schmidt, M.C.; Peterson, C.L. DNA-binding properties of the yeast SWI/SNF complex. Nature 1996, 379, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Boil. 2018, 34, 265–288. [Google Scholar] [CrossRef] [PubMed]
- Harr, J.; Luperchio, T.R.; Wong, X.; Cohen, E.; Wheelan, S.J.; Reddy, K. Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. J. Cell Boil. 2015, 208, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Poleshko, A.; Smith, C.L.; Nguyen, S.C.; Sivaramakrishnan, P.; Wong, K.G.; Murray, J.I.; Lakadamyali, M.; Joyce, E.F.; Jain, R.; Epstein, J.A.; et al. H3K9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Gozalo, A.; Duke, A.; Lan, Y.; Pascual-Garcia, P.; Talamas, J.A.; Nguyen, S.C.; Shah, P.P.; Jain, R.; Joyce, E.F.; Capelson, M. Core Components of the Nuclear Pore Bind Distinct States of Chromatin and Contribute to Polycomb Repression. Mol. Cell 2020, 77, 67–81. [Google Scholar] [CrossRef]
- Feinberg, A.P. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N. Engl. J. Med. 2018, 378, 1323–1334. [Google Scholar] [CrossRef]
- Bartlett, A.A.; Hunter, R.G. Transposons, stress and the functions of the deep genome. Front. Neuroendocr. 2018, 49, 170–174. [Google Scholar] [CrossRef]
- McMurchy, A.N.; Stempor, P.; Gaarenstroom, T.; Wysolmerski, B.; Dong, Y.; Aussianikava, D.; Appert, A.; Huang, N.; Kolasinska-Zwierz, P.; Sapetschnig, A.; et al. A team of heterochromatin factors collaborates with small RNA pathways to combat repetitive elements and germline stress. eLife 2017, 6. [Google Scholar] [CrossRef]
- Feng, J.X.; Riddle, N.C. Epigenetics and genome stability. Mamm. Genome 2020, 1–15. [Google Scholar] [CrossRef]
- Herman, J.G.; Baylin, S.B. Gene Silencing in Cancer in Association with Promoter Hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed]
- Feser, J.; Tyler, J.K. Chromatin structure as a mediator of aging. FEBS Lett. 2010, 585, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin A-Dependent Nuclear Defects in Human Aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem. Mol. Boil. 2019, 54, 61–83. [Google Scholar] [CrossRef]
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Desmarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free. Radic. Boil. Med. 2015, 82, 22–28. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Boil. 2013, 1, 8–16. [Google Scholar] [CrossRef]
- Zhang, Y.; Máté, G.; Müller, P.; Hillebrandt, S.; Krufczik, M.; Bach, M.; Kaufmann, R.; Hausmann, M.; Heermann, D.W. Radiation Induced Chromatin Conformation Changes Analysed by Fluorescent Localization Microscopy, Statistical Physics, and Graph Theory. PLoS ONE 2015, 10, e0128555. [Google Scholar] [CrossRef]
- Schick, S.; Fournier, D.; Thakurela, S.; Sahu, S.K.; Garding, A.; Tiwari, V.K. Dynamics of chromatin accessibility and epigenetic state in response to UV damage. J. Cell Sci. 2015, 128, 4380–4394. [Google Scholar] [CrossRef]
- Hamilton, C.; Hayward, R.L.; Gilbert, N. Global chromatin fibre compaction in response to DNA damage. Biochem. Biophys. Res. Commun. 2011, 414, 820–825. [Google Scholar] [CrossRef]
- Warnault, V.; Darcq, E.; Levine, A.; Barak, S.; Ron, D. Chromatin remodeling—A novel strategy to control excessive alcohol drinking. Transl. Psychiatry 2013, 3, e231. [Google Scholar] [CrossRef] [PubMed]
- Thu, K.L.; Vucic, E.; Kennett, J.Y.; Heryet, C.; Brown, C.J.; Lam, W.L.; Wilson, I.M. Methylated DNA Immunoprecipitation. J. Vis. Exp. 2009. [Google Scholar] [CrossRef] [PubMed]
- Pastor, W.A.; Pape, U.J.; Huang, Y.; Henderson, H.R.; Lister, R.; Ko, M.; McLoughlin, E.M.; Brudno, Y.; Mahapatra, S.; Kapranov, P.; et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 2011, 473, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, W.A.M.; Bártfai, R.; Visa, N.; Jordán-Pla, A. Characterization of the Nucleosome Landscape by Micrococcal Nuclease-Sequencing (MNase-seq). Adv. Struct. Saf. Stud. 2017, 1689, 83–101. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Boil. 2015, 109. [Google Scholar] [CrossRef]
- Trotter, K.W.; Archer, T.K. Assaying Chromatin Structure and Remodeling by Restriction Enzyme Accessibility. Breast Cancer 2012, 833, 89–102. [Google Scholar] [CrossRef]
- Shipony, Z.; Marinov, G.K.; Swaffer, M.P.; A Sinnott-Armstrong, N.; Skotheim, J.M.; Kundaje, A.; Greenleaf, W.J. Long-range single-molecule mapping of chromatin accessibility in eukaryotes. Nat. Methods 2020, 17, 319–327. [Google Scholar] [CrossRef]
- Li, Z.; Schulz, M.H.; Look, T.; Begemann, M.; Zenke, M.; Costa, I.G. Identification of transcription factor binding sites using ATAC-seq. Genome Boil. 2019, 20, 45. [Google Scholar] [CrossRef]
- Song, L.; Crawford, G.E. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010, 2010, 5384. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Bennink, M.L.; Leuba, S.; Leno, G.H.; Zlatanova, J.; De Grooth, B.G.; Greve, J. Unfolding individual nucleosomes by stretching single chromatin fibers with optical tweezers. Nat. Genet. 2001, 8, 606–610. [Google Scholar] [CrossRef]
- Boichenko, I.; Fierz, B. Chemical and biophysical methods to explore dynamic mechanisms of chromatin silencing. Curr. Opin. Chem. Boil. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ma, H.; Jin, J.; Uttam, S.; Fu, R.; Huang, Y.; Liu, Y. Super-Resolution Imaging of Higher-Order Chromatin Structures at Different Epigenomic States in Single Mammalian Cells. Cell Rep. 2018, 24, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Marr, L.T.; Clark, D.J.; Hayes, J.J. A method for assessing histone surface accessibility genome-wide. Methods 2019. [Google Scholar] [CrossRef]
- Oluwadare, O.; Highsmith, M.; Cheng, J. An Overview of Methods for Reconstructing 3-D Chromosome and Genome Structures from Hi-C Data. Boil. Proced. Online 2019, 21, 7. [Google Scholar] [CrossRef]
- Hainsworth, A.H.; Lee, S.; Foot, P.; Patel, A.; Poon, W.W.; Knight, A.E. Super-resolution imaging of subcortical white matter using stochastic optical reconstruction microscopy (STORM) and super-resolution optical fluctuation imaging (SOFI). Neuropathol. Appl. Neurobiol. 2017, 44, 417–426. [Google Scholar] [CrossRef]
- Būtaitė, U.G.; Gibson, G.M.; Ho, Y.-L.D.; Taverne, M.; Taylor, J.M.; Phillips, D.B. Indirect optical trapping using light driven micro-rotors for reconfigurable hydrodynamic manipulation. Nat. Commun. 2019, 10, 1215. [Google Scholar] [CrossRef]
- Mirsaidov, U.; Timp, W.; Timp, K.; Mir, M.; Matsudaira, P.; Timp, G. Optimal optical trap for bacterial viability. Phys. Rev. E 2008, 78, 021910. [Google Scholar] [CrossRef]
- Halstead, M.M.; Kern, C.; Saelao, P.; Chanthavixay, G.; Wang, Y.; Delany, M.E.; Zhou, H.; Ross, P.J. Systematic alteration of ATAC-seq for profiling open chromatin in cryopreserved nuclei preparations from livestock tissues. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Sidoli, S.; Garcia, B.A. Middle-down proteomics: A still unexploited resource for chromatin biology. Expert Rev. Proteom. 2017, 14, 617–626. [Google Scholar] [CrossRef]
- Önder, Ö.; Sidoli, S.; Carroll, M.; Garcia, B.A. Progress in epigenetic histone modification analysis by mass spectrometry for clinical investigations. Expert Rev. Proteom. 2015, 12, 499–517. [Google Scholar] [CrossRef] [PubMed]
- Janssen, K.A.; Sidoli, S.; Garcia, B.A. Recent Achievements in Characterizing the Histone Code and Approaches to Integrating Epigenomics and Systems Biology. Methods Enzym. 2017, 586, 359–378. [Google Scholar] [CrossRef]
- Nguyen, T.; Pappireddi, N.; Wühr, M. Proteomics of nucleocytoplasmic partitioning. Curr. Opin. Chem. Boil. 2019, 48, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Mittler, G.; Butter, F.; Mann, M. A SILAC-based DNA protein interaction screen that identifies candidate binding proteins to functional DNA elements. Genome Res. 2008, 19, 284–293. [Google Scholar] [CrossRef]
- Casas-Vila, N.; Scheibe, M.; Freiwald, A.; Kappei, D.; Butter, F. Identification of TTAGGG-binding proteins in Neurospora crassa, a fungus with vertebrate-like telomere repeats. BMC Genom. 2015, 16, 965. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R.; Pugh, J. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef]
- Riggs, A. X inactivation, differentiation, and DNA methylation. Cytogenet. Genome Res. 1975, 14, 9–25. [Google Scholar] [CrossRef]
- Harrison, A.; Parle-McDermott, A. DNA Methylation: A Timeline of Methods and Applications. Front. Genet. 2011, 2, 74. [Google Scholar] [CrossRef]
- Chowdhury, B.; Cho, I.-H.; Irudayaraj, J. Technical advances in global DNA methylation analysis in human cancers. J. Boil. Eng. 2017, 11, 10. [Google Scholar] [CrossRef]
- Kuo, K.C.; McCune, R.A.; Gehrke, C.W.; Midgett, R.; Ehrlich, M. Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in DNA. Nucleic Acids Res. 1980, 8, 4763–4776. [Google Scholar] [CrossRef]
- Annan, R.S.; Kresbach, G.M.; Giese, R.W.; Vouros, P. Trace detection of modified dna bases via moving-belt liquid chromatography—Mass spectrometry using electrophoric derivatization and negative chemica. J. Chromatogr. A 1989, 465, 285–296. [Google Scholar] [CrossRef]
- Del Gaudio, R.; Di Giaimo, R.; Geraci, G. Genome methylation of the marine annelid worm Chaetopterus variopedatus: Methylation of a CpG in an expressed H1 histone gene. FEBS Lett. 1997, 417, 48–52. [Google Scholar] [CrossRef]
- Zambonin, C.G.; Palmisano, F. Electrospray ionization mass spectrometry of 5-methyl-2’-deoxycytidine and its determination in urine by liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid. Commun. Mass. Spectrom. 1999, 13, 2160–2165. [Google Scholar] [CrossRef]
- Friso, S.; Choi, S.-W.; Dolnikowski, G.; Selhub, J. A Method to Assess Genomic DNA Methylation Using High-Performance Liquid Chromatography/Electrospray Ionization Mass Spectrometry. Anal. Chem. 2002, 74, 4526–4531. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; James, S.R.; Kazim, L.; Karpf, A.R. Specific Method for the Determination of Genomic DNA Methylation by Liquid Chromatography-Electrospray Ionization Tandem Mass Spectrometry. Anal. Chem. 2005, 77, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Kim, K.-P.; Fan, G.; Faull, K.F. A sensitive mass spectrometry method for simultaneous quantification of DNA methylation and hydroxymethylation levels in biological samples. Anal. Biochem. 2011, 412, 203–209. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Tang, Y.; Zheng, S.-J.; Qi, C.-B.; Feng, Y.-Q.; Yuan, B.-F. Sensitive and Simultaneous Determination of 5-Methylcytosine and Its Oxidation Products in Genomic DNA by Chemical Derivatization Coupled with Liquid Chromatography-Tandem Mass Spectrometry Analysis. Anal. Chem. 2015, 87, 3445–3452. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Chen, G.; Huang, Q.; Zhang, J.; Wen, J.; Ye, X.; Cai, C. Validation and quantification of genomic 5-carboxylcytosine (5caC) in mouse brain tissue by liquid chromatography-tandem mass spectrometry. Anal. Methods 2016, 8, 5812–5817. [Google Scholar] [CrossRef]
- Giessing, A.M.B.; Scott, L.G.; Kirpekar, F. A Nano-Chip-LC/MS n Based Strategy for Characterization of Modified Nucleosides Using Reduced Porous Graphitic Carbon as a Stationary Phase. J. Am. Soc. Mass Spectrom. 2011, 22, 1242–1251. [Google Scholar] [CrossRef][Green Version]
- Sidoli, S.; Bhanu, N.V.; Karch, K.; Wang, X.; Garcia, B.A. Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J. Vis. Exp. 2016, e54112. [Google Scholar] [CrossRef] [PubMed]
- Egelhofer, T.A.; Minoda, A.; Klugman, S.; Lee, K.; Kolasinska-Zwierz, P.; Alekseyenko, A.A.; Cheung, M.-S.; Day, D.S.; Gadel, S.; Gorchakov, A.A.; et al. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Boil. 2010, 18, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Dickson, B.M.; Raab, J.R.; Grzybowski, A.; Krajewski, K.; Guo, A.H.; Shanle, E.K.; Josefowicz, S.Z.; Fuchs, S.M.; Allis, C.D.; et al. An interactive database for the assessment of histone antibody specificity. Mol. Cell 2015, 59, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Sidoli, S.; Cheng, L.; Jensen, O.N. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J. Proteom. 2012, 75, 3419–3433. [Google Scholar] [CrossRef]
- Alabert, C.; Barth, T.K.; Reverón-Gómez, N.; Sidoli, S.; Schmidt, A.; Jensen, O.N.; Imhof, A.; Groth, A. Two distinct modes for propagation of histone PTMs across the cell cycle. Genome Res. 2015, 29, 585–590. [Google Scholar] [CrossRef]
- Bonaldi, T.; Imhof, A.; Regula, J.T. A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics 2004, 4, 1382–1396. [Google Scholar] [CrossRef]
- Garcia, B.A.; Mollah, S.; Ueberheide, B.; Busby, S.A.; Muratore, T.L.; Shabanowitz, J.; Hunt, N.F. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007, 2, 933–938. [Google Scholar] [CrossRef]
- Liao, R.; Wu, H.; Deng, H.; Yu, Y.; Hu, M.; Zhai, H.; Yang, P.; Zhou, S.; Yi, W. Specific and Efficient N-Propionylation of Histones with Propionic Acid N-Hydroxysuccinimide Ester for Histone Marks Characterization by LC-MS. Anal. Chem. 2013, 85, 2253–2259. [Google Scholar] [CrossRef]
- Sidoli, S.; Yuan, Z.-F.; Lin, S.; Karch, K.; Wang, X.; Bhanu, N.; Arnaudo, A.M.; Britton, L.-M.; Cao, X.-J.; Gonzales-Cope, M.; et al. Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Proteomics 2015, 15, 1459–1469. [Google Scholar] [CrossRef]
- Soldi, M.; Cuomo, A.; Bonaldi, T. Quantitative assessment of chemical artefacts produced by propionylation of histones prior to mass spectrometry analysis. Proteomics 2016, 16, 1952–1954. [Google Scholar] [CrossRef]
- Meert, P.; Dierickx, S.; Govaert, E.; De Clerck, L.; Willems, S.; Dhaenens, M.; Deforce, D. Tackling aspecific side reactions during histone propionylation: The promise of reversing overpropionylation. Proteomics 2016, 16, 1970–1974. [Google Scholar] [CrossRef] [PubMed]
- Paternoster, V.; Edhager, A.V.; Sibbersen, C.; Nielsen, A.L.; Børglum, A.D.; Christensen, J.H.; Palmfeldt, J. Quantitative assessment of methyl-esterification and other side reactions in a standard propionylation protocol for detection of histone modifications. Proteomics 2016, 16, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Meert, P.; Govaert, E.; Scheerlinck, E.; Dhaenens, M.; Deforce, D. Pitfalls in histone propionylation during bottom-up mass spectrometry analysis. Proteomics 2015, 15, 2966–2971. [Google Scholar] [CrossRef]
- Zhang, R.; Erler, J.; Langowski, J. Histone Acetylation Regulates Chromatin Accessibility: Role of H4K16 in Inter-nucleosome Interaction. Biophys. J. 2016, 112, 450–459. [Google Scholar] [CrossRef]
- Chase, K.A.; Gavin, D.P.; Guidotti, A.; Sharma, R.P. Histone methylation at H3K9: Evidence for a restrictive epigenome in schizophrenia. Schizophr. Res. 2013, 149, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Shema, E.; Jones, D.; Shoresh, N.; Donohue, L.; Ram, O.; Bernstein, B.E. Single-molecule decoding of combinatorially modified nucleosomes. Science 2016, 352, 717–721. [Google Scholar] [CrossRef]
- Sadeh, R.; Launer-Wachs, R.; Wandel, H.; Rahat, A.; Friedman, N. Elucidating Combinatorial Chromatin States at Single-Nucleosome Resolution. Mol. Cell 2016, 63, 1080–1088. [Google Scholar] [CrossRef][Green Version]
- Sidoli, S.; Schwämmle, V.; Ruminowicz, C.; Hansen, T.A.; Wu, X.; Helin, K.; Jensen, O.N. Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics 2014, 14, 2200–2211. [Google Scholar] [CrossRef]
- Deal, R.B.; Henikoff, J.G.; Henikoff, S. Genome-Wide Kinetics of Nucleosome Turnover Determined by Metabolic Labeling of Histones. Science 2010, 328, 1161–1164. [Google Scholar] [CrossRef]
- Dion, M.; Kaplan, T.; Kim, M.; Buratowski, S.; Friedman, N.; Rando, O.J. Dynamics of Replication-Independent Histone Turnover in Budding Yeast. Science 2007, 315, 1405–1408. [Google Scholar] [CrossRef]
- Radman-Livaja, M.; Verzijlbergen, K.F.; Weiner, A.; Van Welsem, T.; Friedman, N.; Rando, O.J.; Van Leeuwen, F. Patterns and Mechanisms of Ancestral Histone Protein Inheritance in Budding Yeast. PLoS Boil. 2011, 9, e1001075. [Google Scholar] [CrossRef] [PubMed]
- Zee, B.M.; Levin, R.S.; DiMaggio, P.A.; Garcia, B.A. Global turnover of histone post-translational modifications and variants in human cells. Epigenetics Chromatin 2010, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Sidoli, S.; Lopes, M.; Lund, P.J.; Goldman, N.; Fasolino, M.; Coradin, M.; Kulej, K.; Bhanu, N.V.; Vahedi, G.; Garcia, B.A. A mass spectrometry-based assay using metabolic labeling to rapidly monitor chromatin accessibility of modified histone proteins. Sci. Rep. 2019, 9, 13613–13615. [Google Scholar] [CrossRef]
- Sidoli, S.; Lu, C.; Coradin, M.; Wang, X.; Karch, K.; Ruminowicz, C.; Garcia, B.A. Metabolic labeling in middle-down proteomics allows for investigation of the dynamics of the histone code. Epigenetics Chromatin 2017, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Shiio, Y.; Eisenman, R.N.; Yi, E.C.; Donohoe, S.; Goodlett, D.R.; Aebersold, R. Quantitative proteomic analysis of chromatin-associated factors. J. Am. Soc. Mass Spectrom. 2003, 14, 696–703. [Google Scholar] [CrossRef]
- Becker, J.S.; McCarthy, R.L.; Sidoli, S.; Donahue, G.; Kaeding, K.; He, Z.; Lin, S.; Garcia, B.A.; Zaret, K.S. Genomic and Proteomic Resolution of Heterochromatin and its Restriction of Alternate Fate Genes. Mol. Cell 2017, 68, 1023–1037.e15. [Google Scholar] [CrossRef]
- Buxton, K.E.; Kennedy-Darling, J.; Shortreed, M.R.; Zaidan, N.Z.; Olivier, M.; Scalf, M.; Sridharan, R.; Smith, L.M. Elucidating Protein–DNA Interactions in Human Alphoid Chromatin via Hybridization Capture and Mass Spectrometry. J. Proteome Res. 2017, 16, 3433–3442. [Google Scholar] [CrossRef]
- Dejardin, J.; Kingston, R.E. Purification of Proteins Associated with Specific Genomic Loci. Cell 2009, 136, 175–186. [Google Scholar] [CrossRef]
- Fujita, T.; Fujii, H. Direct Identification of Insulator Components by Insertional Chromatin Immunoprecipitation. PLoS ONE 2011, 6, e26109. [Google Scholar] [CrossRef]
- Nikolov, M.; Stützer, A.; Mosch, K.; Krasauskas, A.; Soeroes, S.; Stark, H.; Urlaub, H.; Fischle, W. Chromatin Affinity Purification and Quantitative Mass Spectrometry Defining the Interactome of Histone Modification Patterns. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G.; Sakai, A.; Loeb, G.B.; Ahmad, K. Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res. 2008, 19, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Teves, S.S.; Henikoff, S. Salt Fractionation of Nucleosomes for Genome-Wide Profiling. Adv. Struct. Saf. Stud. 2011, 833, 421–432. [Google Scholar] [CrossRef]
- Federation, A.J.; Nandakumar, V.; Searle, B.C.; Stergachis, A.; Wang, H.; Pino, L.K.; Merrihew, G.; Ting, Y.S.; Howard, N.; Kutyavin, T.; et al. Highly Parallel Quantification and Compartment Localization of Transcription Factors and Nuclear Proteins. Cell Rep. 2020, 30, 2463–2471. [Google Scholar] [CrossRef] [PubMed]
- Kulej, K.; Avgousti, D.C.; Sidoli, S.; Herrmann, C.; Della Fera, A.N.; Kim, E.T.; Garcia, B.A.; Weitzman, M.D. Time-resolved Global and Chromatin Proteomics during Herpes Simplex Virus Type 1 (HSV-1) Infection. Mol. Cell. Proteom. 2017, 16, S92–S107. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.; Avgousti, D.C.; Weitzman, M.D. Differential Salt Fractionation of Nuclei to Analyze Chromatin-associated Proteins from Cultured Mammalian Cells. BIO-PROTOCOL 2017, 7, 7. [Google Scholar] [CrossRef]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef]
- Soldi, M.; Bonaldi, T. The Proteomic Investigation of Chromatin Functional Domains Reveals Novel Synergisms among Distinct Heterochromatin Components. Mol. Cell. Proteom. 2013, 12, 764–780. [Google Scholar] [CrossRef]
- Ji, X.; Dadon, D.B.; Abraham, B.J.; Lee, T.I.; Jaenisch, R.; Bradner, J.E.; Young, R.A. Chromatin proteomic profiling reveals novel proteins associated with histone-marked genomic regions. Proc. Natl. Acad. Sci. USA 2015, 112, 3841–3846. [Google Scholar] [CrossRef]
- Iglesias, N.; Paulo, J.A.; Tatarakis, A.; Wang, X.; Edwards, A.L.; Bhanu, N.V.; Garcia, B.A.; Haas, W.; Gygi, S.P.; Moazed, D. Native Chromatin Proteomics Reveals a Role for Specific Nucleoporins in Heterochromatin Organization and Maintenance. Mol. Cell 2020, 77, 51–66. [Google Scholar] [CrossRef]
- Zukowski, A.; Phillips, J.; Park, S.; Wu, R.; Gygi, S.P.; Johnson, A.M. Proteomic profiling of yeast heterochromatin connects direct physical and genetic interactions. Curr. Genet. 2018, 65, 495–505. [Google Scholar] [CrossRef]
- Tsui, C.; Inouye, C.; Levy, M.; Lu, A.; Florens, L.; Washburn, M.P.; Tjian, R. dCas9-targeted locus-specific protein isolation method identifies histone gene regulators. Proc. Natl. Acad. Sci. USA 2018, 115, E2734–E2741. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.A.; Wright, J.; Peckner, R.; Kalish, B.T.; Zhang, F.; Carr, S.A. Discovery of proteins associated with a predefined genomic locus via dCas9-APEX-mediated proximity labeling. Nat. Methods 2018, 15, 437–439. [Google Scholar] [CrossRef] [PubMed]
- Escobar, T.M.; Oksuz, O.; Saldaña-Meyer, R.; Descostes, N.; Bonasio, R.; Reinberg, D. Active and Repressed Chromatin Domains Exhibit Distinct Nucleosome Segregation during DNA Replication. Cell 2019, 179, 953–963.e11. [Google Scholar] [CrossRef]
- Dutta, B.; Adav, S.S.; Koh, C.-G.; Lim, S.K.; Meshorer, E.; Sze, S.K. Elucidating the temporal dynamics of chromatin-associated protein release upon DNA digestion by quantitative proteomic approach. J. Proteomics 2012, 75, 5493–5506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Smith, D.L. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 2008, 2, 522–531. [Google Scholar] [CrossRef]
- Xu, G.; Chance, M.R. Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chem. Rev. 2007, 107, 3514–3543. [Google Scholar] [CrossRef]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Kong, A.T.; Vos, R.A.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef]
- Chalabi, M.H.; Tsiamis, V.; Käll, L.; Vandin, F.; Schwämmle, V. CoExpresso: Assess the quantitative behavior of protein complexes in human cells. BMC Bioinform. 2019, 20, 17. [Google Scholar] [CrossRef]
- Xue, Y.; Ren, J.; Gao, X.; Jin, C.; Wen, L.; Yao, X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteom. 2008, 7, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-F.; Sidoli, S.; Marchione, D.M.; Simithy, J.; Janssen, K.A.; Szurgot, M.; Garcia, B.A. EpiProfile 2.0: A Computational Platform for Processing Epi-Proteomics Mass Spectrometry Data. J. Proteome Res. 2018, 17, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Baliban, R.C.; DiMaggio, P.A.; Plazas-Mayorca, M.D.; Young, N.L.; Garcia, B.A.; Floudas, C.A. A novel approach for untargeted post-translational modification identification using integer linear optimization and tandem mass spectrometry. Mol. Cell. Proteom. 2010, 9, 764–779. [Google Scholar] [CrossRef]
- Khare, S.; Habib, F.; Sharma, R.; Gadewal, N.; Gupta, S.; Galande, S. HIstome--a relational knowledgebase of human histone proteins and histone modifying enzymes. Nucleic Acids Res. 2011, 40, D337–D342. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, S.; Lin, S.; Guo, Y.; Deng, W.; Zhang, Y.; Xue, Y. WERAM: A database of writers, erasers and readers of histone acetylation and methylation in eukaryotes. Nucleic Acids Res. 2016, 45, D264–D270. [Google Scholar] [CrossRef]
- Gendler, K.; Paulsen, T.; Napoli, C. ChromDB: The Chromatin Database. Nucleic Acids Res. 2007, 36, D298–D302. [Google Scholar] [CrossRef]
- Choi, J.; Kim, K.-T.; Huh, A.; Kwon, S.; Hong, C.; Asiegbu, F.O.; Jeon, J.; Lee, Y.-H. dbHiMo: A web-based epigenomics platform for histone-modifying enzymes. Database 2015, 2015, bav052. [Google Scholar] [CrossRef]
- Draizen, E.J.; Shaytan, A.; Mariño-Ramírez, L.; Talbert, P.B.; Landsman, D.; Panchenko, A.R. HistoneDB 2.0: A histone database with variants—An integrated resource to explore histones and their variants. Database 2016, 2016. [Google Scholar] [CrossRef]
- El Kennani, S.; Adrait, A.; Shaytan, A.; Khochbin, S.; Bruley, C.; Panchenko, A.R.; Landsman, D.; Pflieger, D.; Govin, J. MS_HistoneDB, a manually curated resource for proteomic analysis of human and mouse histones. Epigenetics Chromatin 2017, 10, 2. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, C.J.A.; Cerutti, L.; De Castro, E.; Langendijk-Genevaux, P.; Bulliard, V.; Bairoch, A.; Hulo, N. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 2009, 38, D161–D166. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method Name | Type of Data Collected | Information Obtained | Difficulty | Popularity | Limitations |
|---|---|---|---|---|---|
| Stochastic Optical Reconstruction Microscopy (STORM) | Super-resolution images | Higher-order chromatin structures | High | Medium | Number of fluorescent probes, tissue autofluorescence |
| Förster Resonance Energy Transfer (FRET) | Fluorophore distance, images, and intensity | Nucleosome organization, effector binding, chromatin state | Medium | Very High | Number of fluorescent probes, specific dimension of data |
| Optical tweezers | Force | Chromatin assembly, histone displacement, enzyme force | High | High | Very specific dimension of data, live tissue damage |
| Methyl-DNA Immunoprecipitation Sequencing (MeDIP-seq) | Regions of enriched methylation | Methylated DNA regions | Easy | Medium | Cost, broad information, non-specific binding can occur |
| Assay for Transposase-Accessible Chromatin Sequencing (ATAC-seq) | Regions of accessible chromatin | Exposed DNA regions | Easy | High | Cost, broad information, cryopreserved tissue may not work |
| Chromatin Immunoprecipitation Sequencing (ChIP-seq) | DNA associated with specific proteins | Protein–DNA interactions, histone organization | Easy | High | Cost, non-specific binding can occur |
| Micrococcal Nuclease Sequencing (MNase-seq) | Nucleosome concentration | Condensed DNA regions | Easy | Medium | Cost, broad information |
| DNase I Hypersensitive Sites Sequencing (DNase-seq) | Regions of accessible DNA | Exposed DNA regions | Easy | Medium | Cost, broad information, more time-intensive than ATAC-seq |
| Histone surface accessibility | Regions of accessible histones | Chromatin structure and dynamics | Medium | Low | Requires cysteine-lacking histones |
| Hi-C | Crosslinked regions of DNA | Chromatin arrangement, organization, and long-range interactions | High | High | Broad information, possible lack of long-range contacts |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stransky, S.; Aguilan, J.; Lachowicz, J.; Madrid-Aliste, C.; Nieves, E.; Sidoli, S. Mass Spectrometry to Study Chromatin Compaction. Biology 2020, 9, 140. https://doi.org/10.3390/biology9060140
Stransky S, Aguilan J, Lachowicz J, Madrid-Aliste C, Nieves E, Sidoli S. Mass Spectrometry to Study Chromatin Compaction. Biology. 2020; 9(6):140. https://doi.org/10.3390/biology9060140
Chicago/Turabian StyleStransky, Stephanie, Jennifer Aguilan, Jake Lachowicz, Carlos Madrid-Aliste, Edward Nieves, and Simone Sidoli. 2020. "Mass Spectrometry to Study Chromatin Compaction" Biology 9, no. 6: 140. https://doi.org/10.3390/biology9060140
APA StyleStransky, S., Aguilan, J., Lachowicz, J., Madrid-Aliste, C., Nieves, E., & Sidoli, S. (2020). Mass Spectrometry to Study Chromatin Compaction. Biology, 9(6), 140. https://doi.org/10.3390/biology9060140