A Review of Candidate Genes and Pathways in Preeclampsia–An Integrated Bioinformatical Analysis


Abstract
1. Introduction
2. Materials and Methods
2.1. Search Strategy
2.2. Inclusion Criteria
2.3. Exclusion Criteria
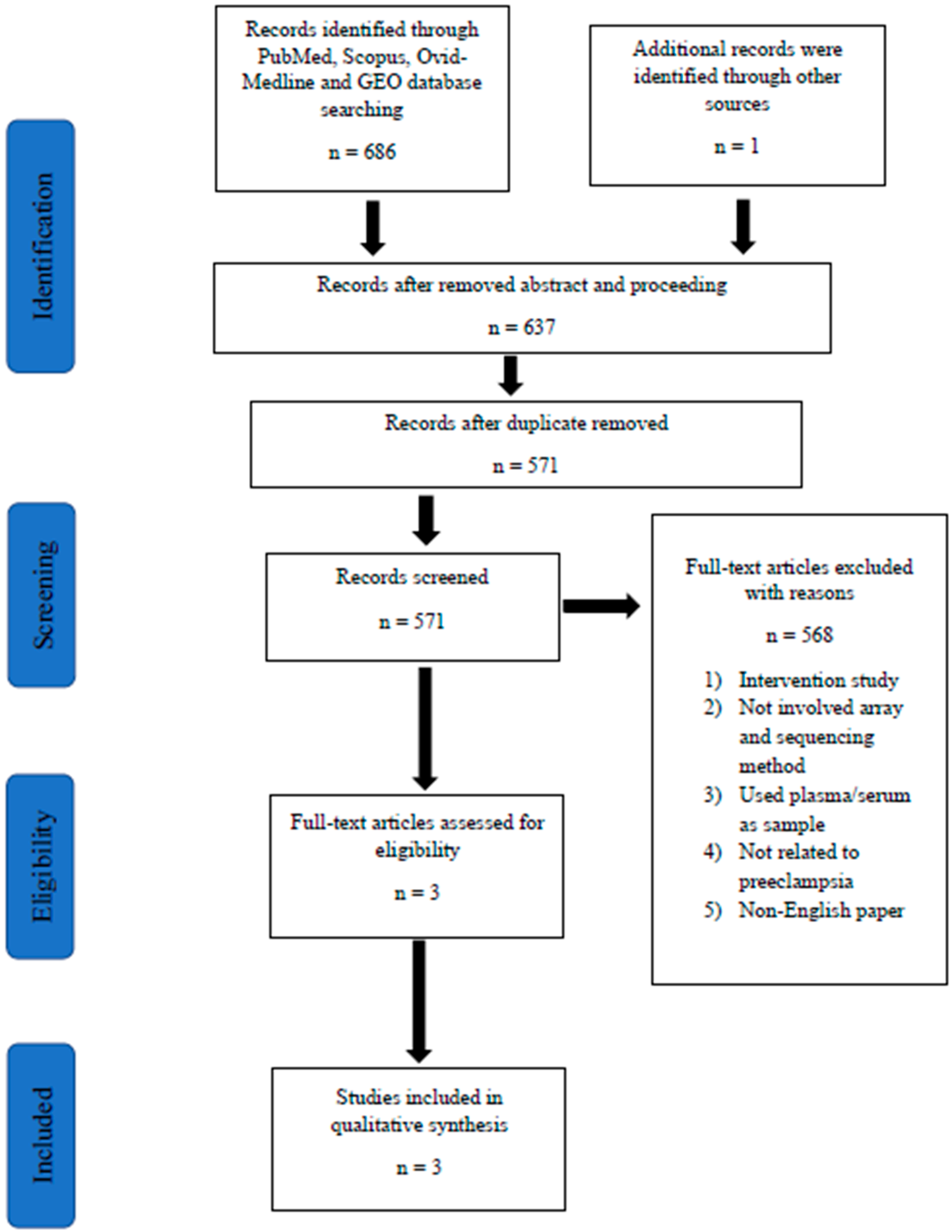
2.4. Screening of Articles for Eligibility
2.5. Data Extraction
2.6. Study Quality, Gene Ontology, and Pathway Analysis
2.7. Protein–Protein Interaction (PPI) Network
3. Results
3.1. Patient Recruitment and Categorization
3.2. Placenta Tissue Sampling
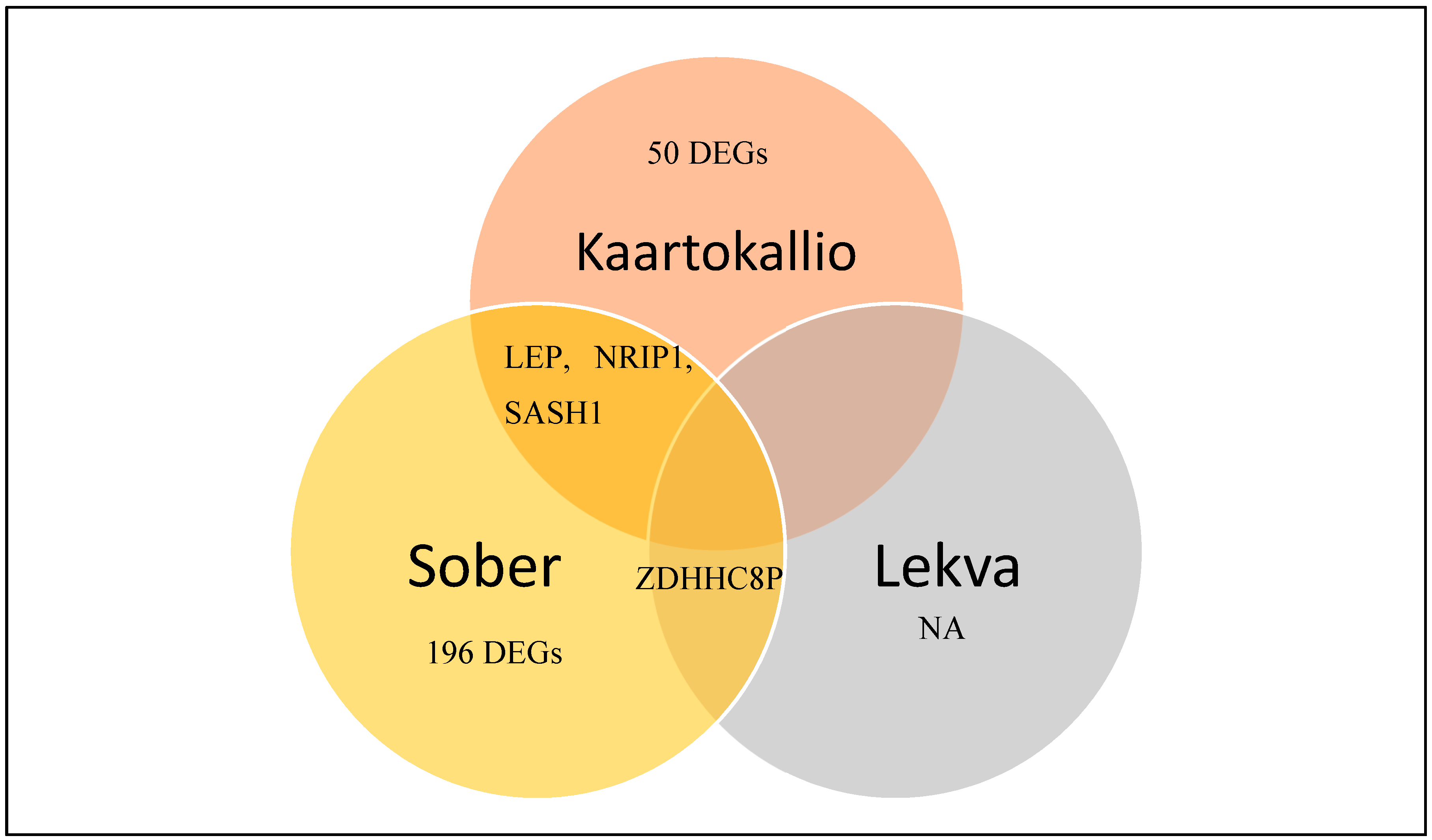
3.3. Identification of Differentially Expressed Genes in Preeclampsia
3.4. Differentially Expressed Genes’ Ontology Analysis in Preeclampsia
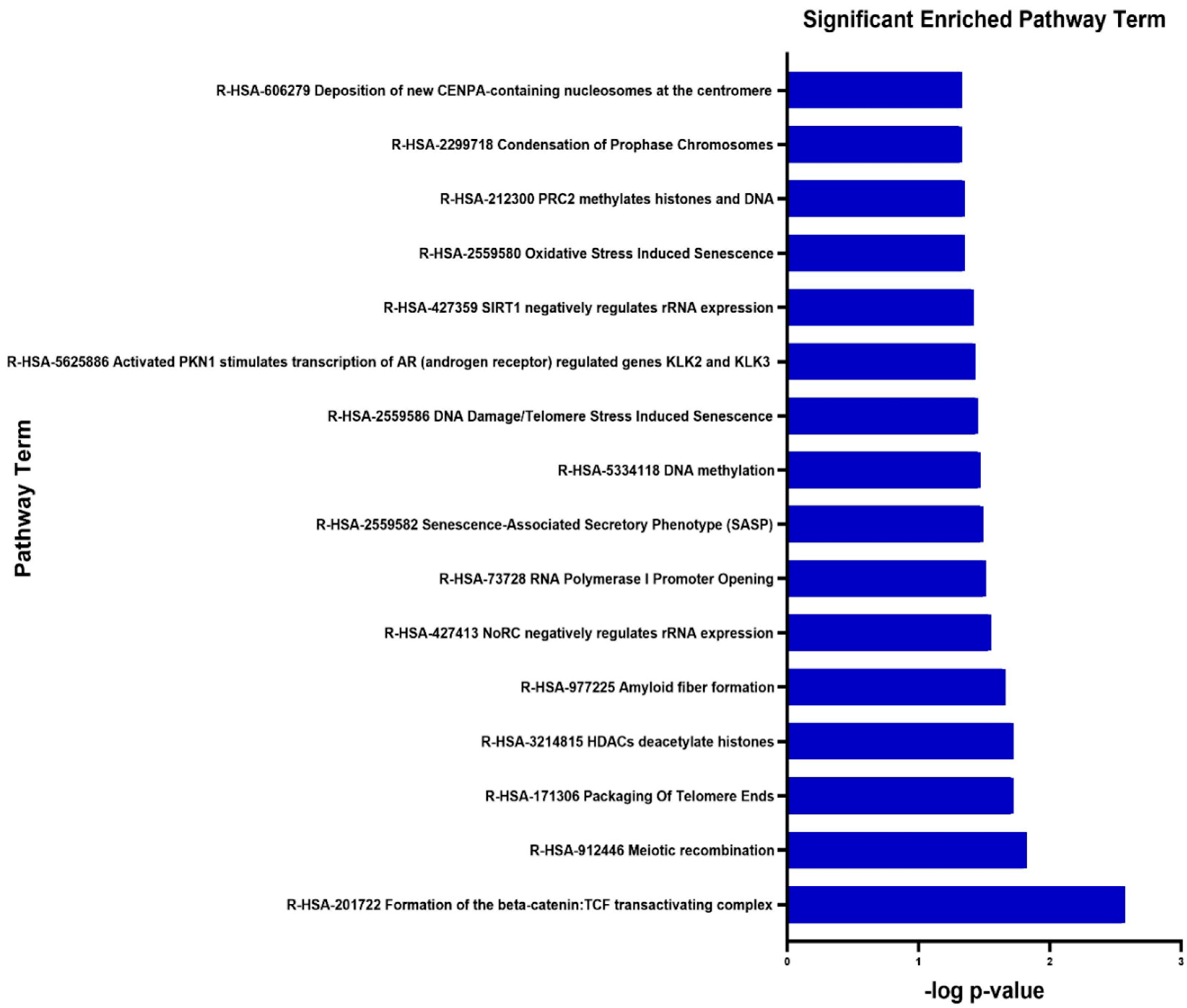
3.5. Signalling Pathway Enrichment Analysis
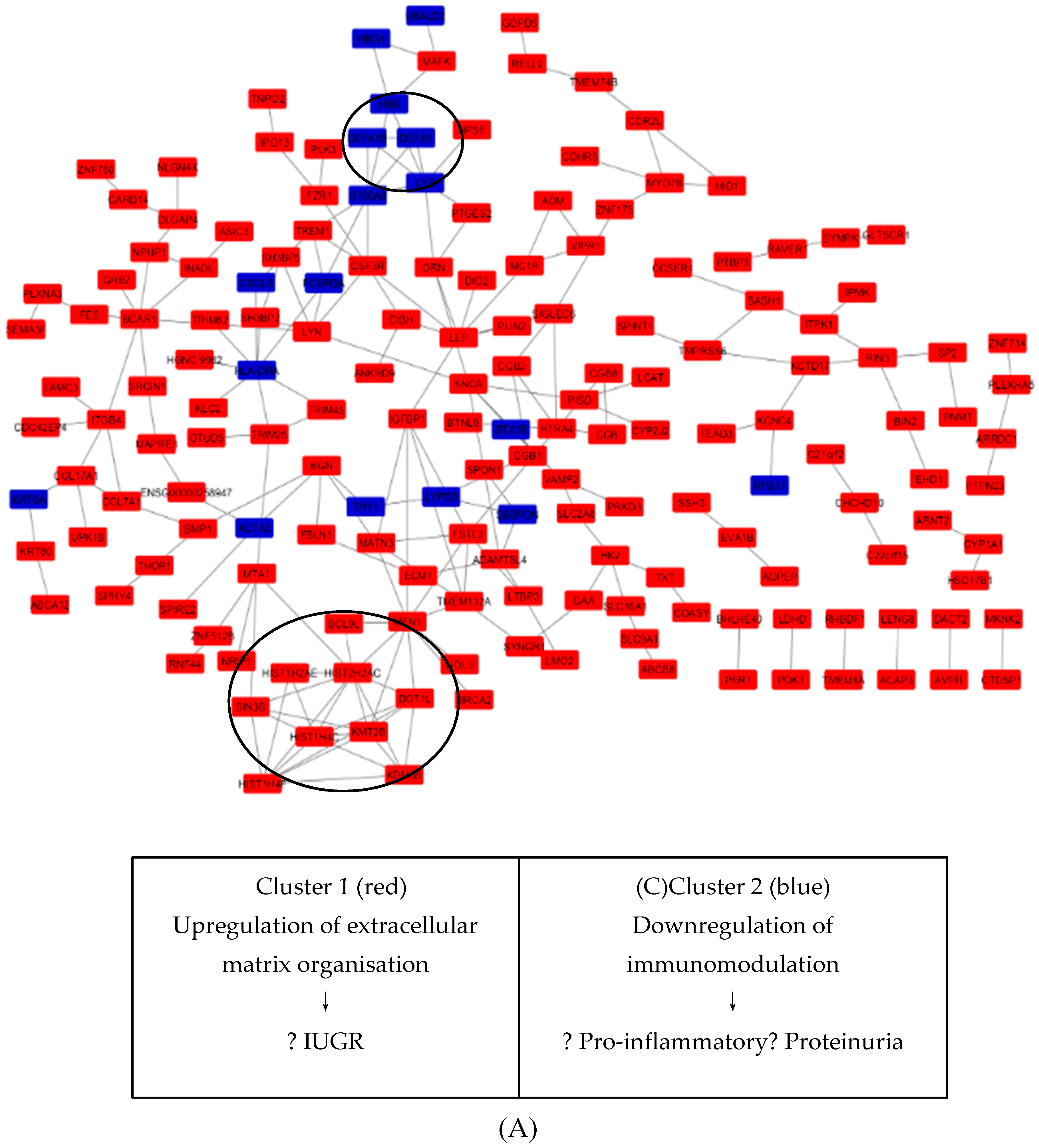
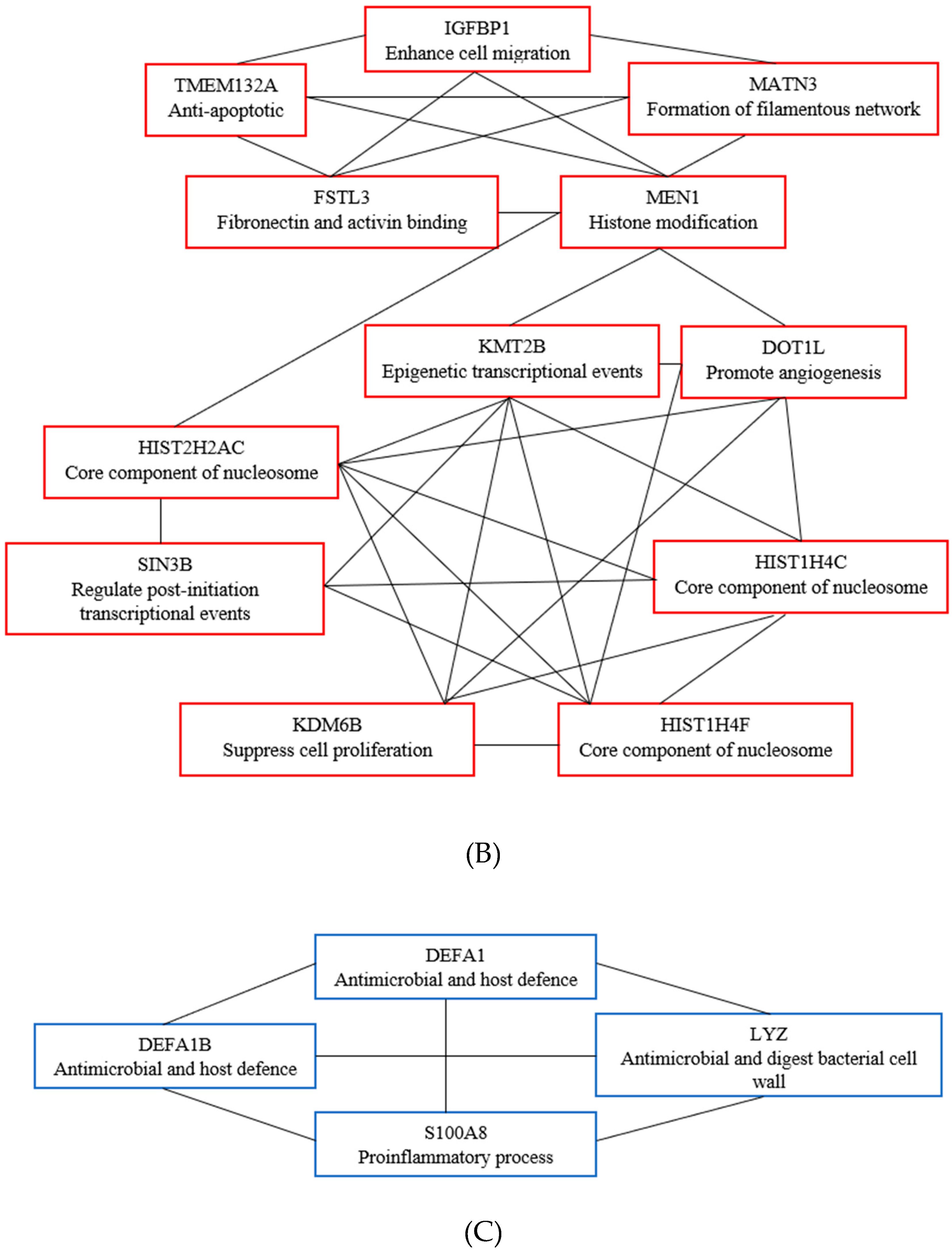
3.6. Key Candidate Genes’ and Pathways’ Identification with Differentially Expressed Genes’ Protein–Protein Interaction Network (PPI) and Modular Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tranquilli, A.L.; Dekker, G.; Magee, L.; Roberts, J.; Sibai, B.M.; Steyn, W.; Zeeman, G.G.; Brown, M.A. The classification, diagnosis and management of the hypertensive disorders of pregnancy: A revised statement from the ISSHP. Pregnancy Hypertens. 2014, 4, 97–104. [Google Scholar] [CrossRef]
- WHO. Make Every Mother and Child Count, in the World Health Report 2005; World Health Organization: Geneva, Switzerland, 2005. [Google Scholar]
- Steegers, E.A.P.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef]
- Ness, R.B.; Roberts, J.M. Heterogeneous causes constituting the single syndrome of preeclampsia: A hypothesis and its implications. Am. J. Obstet. Gynecol. 1996, 175, 1365–1370. [Google Scholar] [CrossRef]
- Dekker, G.A.; Sibai, B.M. Etiology and pathogenesis of preeclampsia: Current concepts. Am. J. Obstet. Gynecol. 1998, 179, 1359–1375. [Google Scholar] [CrossRef]
- Kaartokallio, T.; Cervera, A.; Kyllönen, A.; Laivuori, K.; Kere, J.; Laivuori, H. Gene expression profiling of pre-eclamptic placentae by RNA sequencing. Sci. Rep. 2015, 5, 14107. [Google Scholar] [CrossRef] [PubMed]
- Sõber, S.; Reiman, M.; Kikas, T.; Rull, K.; Inno, R.; Vaas, P.; Teesalu, P.; Marti, J.M.L.; Mattila, P.; Laan, M. Extensive shift in placental transcriptome profile in preeclampsia and placental origin of adverse pregnancy outcomes. Sci. Rep. 2015, 5, 13336. [Google Scholar] [CrossRef]
- Lekva, T.; Lyle, R.; Roland, M.C.P.; Friis, C.; Bianchi, D.W.; Jaffe, I.Z.; Norwitz, E.R.; Bollerslev, J.; Henriksen, T.; Ueland, T. Gene expression in term placentas is regulated more by spinal or epidural anesthesia than by late-onset preeclampsia or gestational diabetes mellitus. Sci. Rep. 2016, 6, 29715. [Google Scholar] [CrossRef]
- Von Dadelszen, P.; Magee, L.A.; Roberts, J.M. Subclassification of preeclampsia. Hypertens. Pregnancy 2003, 22, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Robertson, W.B.; Brosens, I.; Dixon, H.G. The pathological response of the vessels of the placental bed to hypertensive pregnancy. J. Pathol. Bacteriol. 1967, 93, 581–592. [Google Scholar] [CrossRef]
- Roberts, J.M.; Redman, C.W.G. Pre-eclampsia: More than pregnancy-induced hypertension. Lancet 1993, 341, 1447–1451. [Google Scholar] [CrossRef]
- Lyall, F.; Greer, I.A. Pre-eclampsia: A multifaceted vascular disorder of pregnancy. J. Hypertens. 1994, 12, 1339–1346. [Google Scholar] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Biron-Shental, T.; Schaiff, W.T.; Rimon, E.; Shim, T.L.; Nelson, D.M.; Sadovsky, Y. Hypoxia enhances the expression of follistatin-like 3 in term human trophoblasts. Placenta 2008, 29, 51–57. [Google Scholar] [CrossRef]
- Tanaka, K.; Sakai, K.; Matsushima, M.; Matsuzawa, Y.; Izawa, T.; Nagashima, T.; Furukawa, S.; Kobayashi, Y.; Iwashita, M. Branched-chain amino acids regulate insulin-like growth factor-binding protein 1 (IGFBP1) production by decidua and influence trophoblast migration through IGFBP1. Mol. Hum. Reprod. 2016, 22, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Singal, S.S.; Nygard, K.; Gratton, R.; Jansson, T.; Gupta, M.B. Increased insulin-like growth factor binding protein-1 phosphorylation in decidualized stromal mesenchymal cells in human intrauterine growth restriction placentas. J. Histochem. Cytochem. 2018, 66, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ye, Y.; Zhang, J.; Ruan, C.-C.; Gao, P.-J. Immune imbalance is associated with the development of preeclampsia. Medicine 2019, 98, e15080. [Google Scholar] [CrossRef]
- Kleinrouweler, C.E.; van Uitert, M.; Moerland, P.D.; Ris-Stalpers, C.; van der Post, J.A.M.; Afink, G.B. Differentially Expressed Genes in the Pre-Eclamptic Placenta: A Systematic Review and Meta-Analysis. PLoS ONE 2013, 8, e68991. [Google Scholar] [CrossRef]
- Brew, O.; Sullivan, M.H.F.; Woodman, A. Comparison of Normal and Pre-Eclamptic Placental Gene Expression: A Systematic Review with Meta-Analysis. PLoS ONE 2016, 11, e0161504. [Google Scholar]
- Song, Y.; Gao, J.; Qu, Y.; Wang, S.; Wang, X.; Liu, J. Serum levels of leptin, adiponectin and resistin in relation to clinical characteristics in normal pregnancy and preeclampsia. Clin. Chim. Acta 2016, 458, 133–137. [Google Scholar] [CrossRef]
- Sagawa, N.; Yura, S.; Itoh, H.; Kakui, K.; Takemura, M.; Nuamah, M.A.; Ogawa, Y.; Masuzaki, H.; Nakao, K.; Fujii, S. Possible role of placental leptin in pregnancy. Endocrine 2002, 19, 65–71. [Google Scholar] [CrossRef]
- Iwagaki, S.; Yokoyama, Y.; Tang, L.; Takahashi, Y.; Nakagawa, Y.; Tamaya, T. Augmentation of leptin and hypoxia-inducible factor 1alpha mRNAs in the pre-eclamptic placenta. Gynecol. Endocrinol. 2004, 18, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Maymó, J.L.; Pérez Pérez, A.; Gambino, Y.; Calvo, J.C.; Sánchez-Margalet, V.; Varone, C.L. Review: Leptin gene expression in the placenta—Regulation of a key hormone in trophoblast proliferation and survival. Placenta 2011, 32, S146–S153. [Google Scholar] [CrossRef] [PubMed]
- Hoggard, N.; Haggarty, P.; Thomas, L.; Lea, R.G. Leptin expression in placental and fetal tissues: Does leptin have a functional role? Biochem. Soc. Trans. 2001, 29, 57–63. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Maymó, J.; Gambino, Y.; Dueñas, J.L.; Goberna, R.; Varone, C.; Sánchez-Margalet, V. Leptin Stimulates Protein Synthesis-Activating Translation Machinery in Human Trophoblastic Cells1. Biol. Reprod. 2009, 81, 826–832. [Google Scholar]
- Magariños, M.P.; Sánchez-Margalet, V.; Kotler, M.; Calvo, J.C.; Varone, C.L. Leptin Promotes Cell Proliferation and Survival of Trophoblastic Cells1. Biol. Reprod. 2007, 76, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Van Uitert, M.; Moerland, P.D.; Enquobahrie, D.A.; Laivuori, H.; van der Post, J.A.; Ris-Stalpers, C.; Afink, G.B. Meta-analysis of placental transcriptome data Identifies a novel molecular pathway related to preeclampsia. PLoS ONE 2015, 10, e0132468. [Google Scholar]
- Molvarec, A.; Szarka, A.; Walentin, S.; Bekő, G.; Karádi, I.; Prohászka, Z.; Rigó, J. Serum leptin levels in relation to circulating cytokines, chemokines, adhesion molecules and angiogenic factors in normal pregnancy and preeclampsia. Reprod. Biol. Endocrinol. 2011, 9, 124. [Google Scholar] [CrossRef]
- Ahmed, A.B.; Ahmed, M.R.; Mohamed, H.S. Maternal serum leptin as a marker of preeclampsia. Arch. Gynecol. Obstet. 2013, 288, 1317–1322. [Google Scholar]
- Salimi, S.; Farajian-Mashhadi, F.; Naghavi, A.; Mokhtari, M.; Shahrakipour, M.; Saravani, M.; Yaghmaei, M. Different profile of serum Leptin between early onset and late onset preeclampsia. Dis. Markers 2014, 2014, 628476. [Google Scholar] [CrossRef]
- Doster, Y.; Cetinkaya Demir, B.; Atalay, M.A.; Durusoy, E.E.; Kucukkomurcu, S. The possible role of serum leptin in preeclampsia. Clin. Exp. Obstet. Gynecol. 2016, 43, 98–102. [Google Scholar] [PubMed]
- Nautiyal, J.; Christian, M.; Parker, M.G. Distinct functions for RIP140 in development, inflammation, and metabolism. Trends Endocrinol. Metab. 2013, 24, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Ashar-Patel, A.; Kaymaz, Y.; Rajakumar, A.; Bailey, J.A.; Karumanchi, S.A.; Moore, M.J. FLT1 and transcriptome-wide polyadenylation site (PAS) analysis in preeclampsia. Sci. Rep. 2017, 7, 12139. [Google Scholar] [CrossRef]
- White, R.; Morganstein, D.; Christian, M.; Seth, A.; Herzog, B.; Parker, M.G. Role of RIP140 in metabolic tissues: Connections to disease. FEBS Lett. 2008, 582, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Zhao, H.; Shang, G.; Zou, R.; Dai, Z.; Zhou, D.; Huang, Q.; Xu, Y. RIP140 is Associated with Subclinical Inflammation in Type 2 Diabetic Patients. Exp. Clin. Endocrinol. Diabetes 2013, 121, 37–42. [Google Scholar] [CrossRef]
- Zhou, N.; Liu, C.; Wang, X.; Mao, Q.; Jin, Q.; Li, P. Downregulated SASH1 expression indicates poor clinical prognosis in gastric cancer. Hum. Pathol. 2018, 74, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Zhang, W.; Zhang, J.; Lv, Q.Y.; Luan, Y.F. Downregulation of SASH1 correlates with poor prognosis in cervical cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3781–3786. [Google Scholar]
- Dauphinee, S.M.; Clayton, A.; Hussainkhel, A.; Yang, C.; Park, Y.J.; Fuller, M.E.; Blonder, J.; Veenstra, T.D.; Karsan, A. SASH1 Is a Scaffold Molecule in Endothelial TLR4 Signaling. J. Immunol. 2013, 191, 892–901. [Google Scholar] [CrossRef]
- Zeller, C.; Hinzmann, B.; Seitz, S.; Prokoph, H.; Burkhard-Goettges, E.; Fischer, J.; Jandrig, B.; Schwarz, L.-P.; Rosenthal, A.; Scherneck, S. SASH1: A candidate tumor suppressor gene on chromosome 6q24.3 is downregulated in breast cancer. Oncogene 2003, 22, 2972–2983. [Google Scholar] [CrossRef]
- He, P.; Zhang, H.-X.; Sun, C.-Y.; Chen, C.-Y.; Jiang, H.-Q. Overexpression of SASH1 Inhibits the Proliferation, Invasion, and EMT in Hepatocarcinoma Cells. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2016, 24, 25–32. [Google Scholar] [CrossRef]
- Zong, W.; Yu, C.; Wang, P.; Dong, L. Overexpression of SASH1 Inhibits TGF-β1-Induced EMT in Gastric Cancer Cells. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2016, 24, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, T.; Zhang, Y.; Li, Q.; Jin, L.K. LncRNA-ZDHHC8P1 promotes the progression and metastasis of colorectal cancer by targeting miR-34a. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1476–1486. [Google Scholar] [PubMed]
- Maynard, S.E.; Karumanchi, S.A. Angiogenic factors and preeclampsia. In Seminars in Nephrology; WB Saunders: Philadelphia, PA, USA, 2011; Volume 31, pp. 33–46. [Google Scholar]
- Nevo, O.; Soleymanlou, N.; Wu, Y.; Xu, J.; Kingdom, J.; Many, A.; Zamudio, S.; Caniggia, I. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1085–R1093. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title and Reference | Year | Study Design | Study Population | Findings | |
|---|---|---|---|---|---|
| Gene Expression Analysis Parameters | Genes Upregulated and Down Regulated | ||||
| Gene expression profiling of pre-eclamptic placentae by RNA sequencing (Kaartokallio, Cervera et al. 2015) | 2015 | Case-control study | RNASeq (n = 18, 3 groups) qPCR (n = 39, 3 groups) |
| Upregulated 32 genes Downregulated 21 genes |
| Extensive shift in placental transcriptome profile in preeclampsia and placental origin of adverse pregnancy outcomes (Sõber, Reiman et al. 2015) | 2015 | Case-control study | RNASeq (n = 40, 5 groups), qPCR (n = 120, 5 groups) |
| Upregulated 215 genes Downregulated 10 genes |
| Gene expression in term placentas is regulated more by spinal or epidural anaesthesia than by late-onset preeclampsia or gestational diabetes mellitus (Lekva, Lyle et al. 2016) | 2016 | Case-control study | RNASeq (n = 30, 3 groups), qPCR (n = 475) |
| Upregulated 1 gene Downregulated 4 genes |
| Differentially Expressed Gene | Gene Name |
|---|---|
| Upregulated | ABCA12, ABCB8, ACAP3, ADAMTSL4, ADM, AHDC1, ANKRD33, ANKRD9, APBA3, LVRN, ARMS2, ARNT2, ARRDC1, ASIC3, AVPI1, BCAR1, BCL9L, BGN, BHLHE40, BIN2, BMP1, BRCA2, BTNL9, ADIRF, C10orf54, C12orf75, C15orf39, NATD1, C1QTNF1, C21orf2, C22orf15, C7orf43, CARD14, CCSAP, CCSER1, CD320, CDC42EP4, CDHR5, CDR2L, CGB1, CGB2, CGB8, CHCHD10, CISH, CLPTM1, CNTNAP3B, COASY, COL17A1, COL7A1, CORO2A, CPZ, CRLF1, CRTC2, CSF3R, DDX43P1, CTDSP1, CYP2J2, CYP51A1P3, DACT2, DDX60L, DIO2, DLGAP4, DLX3, DLX4, DNAH11, DOT1L, DPPA4, ECM1, EHD1, EMC10, EVA1B, FAM109A, FAM160B2, FBLN1, FER1L4, FES, FOXO6, FSTL3, FZR1, GAA, GDPD5, GLTSCR1, GRAMD1A, GRB7, GRINA, GRN, GUSBP3, HID1, HIST1H2AE, HIST1H4C, HIST1H4F, HIST2H2AC, HK2, HPSE, HSD11B2, HSD17B1, HSD17B1P1, HTRA4, IGFBP1, IGHA1, IGHA2, IPMK, IPO13, ITGB4, ITPK1, ITPK1-AS1, KCNC4, KCTD17, KDM6B, KIAA1211, KIAA1522, KLC2, KRT80, LAMC3, LCAT, LDHD, LENG8, LEP, LMO2, LRCH4, LTBP3, LTBR, LYN, MAFK, MAGEA11, MAP1S, MAPRE3, MATN3 MC1R, MEN1, MIR205HG, MKNK2, MTA1, MYO7B, NDRG1, NLGN4X, NOL3, NPHP1, NRIP1, OTUD5, P2RY6, PDK3, PER1, PIGT, PIM3, GSAP, PISD, PLEKHA5, PLEKHA7, PLIN2, PLK3, PLXNA3, PNMT, PRKD3, PTBP3, PTGES2, PTPN23, RAB4B-EGLN2, RAVER1, RELL2, RFX1, RHBDF1, RIN3, RNF44, RTEL1-TNFRSF6B, SASH1, SCAF1, SEMA3F, SERPINB9, SH3BP2, SH3BP5, SH3GLB2, SIGLEC6, SIN3B, SLC16A1, SLC25A35, SLC26A11, SLC2A8, SLC9A1, SLCO3A1 SMOC1, SNCA, SNX8, SP2, SPINT1, SPIRE2, SPON1, SPRY4, SRCIN1, SSH3, ST8SIA4, STX1B, SYMPK, SYNGR1, TCTEX1D4, TEAD3, TET3, THOP1, TKT, TMEM132A, TMEM74B, TMEM8A, TMPRSS6, TNPO2 TPBG, TREM1, TRIM25, TRIM45, TRIM62, TUBB3, UBALD1, UBALD2, ULK1, UPK1B, VAMP2, VIPR1, KMT2B, ZDHHC8P1, ZNF124, ZNF175, ZNF512B, ZNF714, ZNF750, ZNF76, ZNF90, CGB3, PATJ, NECTIN4 |
| Downregulated | ACTA2, CXCL9, CYP1A1, DEFA1, DEFA1B, FCGR3A, HBB, HBG1, HLA-DRA, KRT6A, LGALS14, LOC100129345, LYPD5, LYZ, MAGEA4, NCMAP, PSG10P, RPS17, S100A8, SBSPON, THY1, ZDHHC1 |
| Term | Description | Count | p-Value | -Log10 p Value |
|---|---|---|---|---|
| Up-regulated | ||||
| GO:0030198 BP | extracellular matrix organization | 10 | 3.80 × 10−4 | 3.42 |
| GO:0005578 CC | proteinaceous extracellular matrix | 11 | 7.80 × 10−4 | 3.10 |
| GO:0005604 CC | basement membrane | 6 | 1.80 × 10−4 | 2.74 |
| GO:0031012 CC | extracellular matrix | 9 | 1.70 × 10−4 | 1.77 |
| Downregulated | ||||
| GO:0006955 BP | immune response | 6 | 6.30 × 10−5 | 4.20 |
| GO:0050832 BP | defense response to fungus | 3 | 3.70 × 10−4 | 3.43 |
| GO:0050830 BP | defense response to Gram-positive bacterium | 3 | 3.70 × 10−3 | 2.43 |
| GO:0006935 BP | chemotaxis | 3 | 7.40 × 10−3 | 2.13 |
| GO:0005615 CC | extracellular space | 6 | 8.30 × 10−3 | 2.08 |
| GO:0051607 BP | defense response to virus | 3 | 1.30 × 10−2 | 1.88 |
| GO:0005576 CC | extracellular region | 6 | 1.70 × 10−2 | 1.77 |
| Pathway | Name | Genes | p-Value | -Log10 p Value |
|---|---|---|---|---|
| R-HSA-201722 | Formation of the beta-catenin: TCF transactivating complex | BCL9L, HIST1H2AE, HIST1H4C, HIST1H4F, HIST2H2AC, MEN1 | 2.70 × 10−3 | 2.57 |
| R-HSA-912446 | Meiotic recombination | BRCA2, HIST1H2AE, HIST1H4C, HIST1H4F, HIST2H2AC | 1.50 × 10−2 | 1.82 |
| R-HSA-171306 | Packaging Of Telomere Ends | HIST1H2AE, HIST1H4C, HIST1H4F, HIST2H2AC | 1.90 × 10−2 | 1.72 |
| R-HSA-3214815 | HDACs deacetylate histones | HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC MTA1 | 1.90 × 10−2 | 1.72 |
| R-HSA-977225 | Amyloid fibre formation | HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC SNCA | 2.20 × 10−2 | 1.66 |
| R-HSA-427413 | NoRC negatively regulates rRNA expression | SIN3B HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC | 2.80 × 10−2 | 1.55 |
| R-HSA-73728 | RNA Polymerase I Promoter Opening | HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC | 3.10 × 10−2 | 1.51 |
| R-HSA-2559582 | Senescence-Associated Secretory Phenotype (SASP) | FZR1 HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC | 3.20 × 10−2 | 1.49 |
| R-HSA-5334118 | DNA methylation | HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC | 3.40 × 10−2 | 1.47 |
| R-HSA-2559586 | DNA Damage/Telomere Stress Induced Senescence | HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC | 3.50 × 10−2 | 1.45 |
| R-HSA-5625886 | Activated PKN1 stimulates transcription of AR (androgen receptor) regulated genes KLK2 and KLK3 | HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC | 3.70 × 10−2 | 1.43 |
| R-HSA-427359 | SIRT1 negatively regulates rRNA expression | HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC | 3.80 × 10−2 | 1.42 |
| R-HSA-2559580 | Oxidative Stress Induced Senescence | KDM6B HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC | 4.50 × 10−2 | 1.35 |
| R-HSA-212300 | PRC2 methylates histones and DNA | HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC | 4.50 × 10−2 | 1.35 |
| R-HSA-2299718 | Condensation of Prophase Chromosomes | HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC | 4.70 × 10−2 | 1.33 |
| R-HSA-606279 | Deposition of new CENPA-containing nucleosomes at the centromere | HIST1H2AE HIST1H4C HIST1H4F HIST2H2AC | 4.70 × 10−2 | 1.33 |
| Term | Description | Number of Genes | p-Value |
|---|---|---|---|
| R-HSA-3214841 | PKMTs methylate histone lysines | 4 | 3.88 × 10−5 |
| R-HSA-201722 | Formation of the beta-catenin:TCF transactivating complex | 4 | 1.01 × 10−4 |
| GO:0032200 | Telomere organization | 3 | 1.11 × 10−4 |
| R-HSA-427413 | NoRC negatively regulates rRNA expression | 4 | 7.51 × 10−5 |
| R-HSA-2559580 | Oxidative Stress Induced Senescence | 4 | 2.72 × 10−4 |
| GO:1904837 | Beta-catenin-TCF complex assembly | 3 | 2.85 × 10−4 |
| GO:0000786 | Nucleosome | 3 | 1.4 × 10−3 |
| R-HSA-171306 | Packaging of Telomere Ends | 3 | 1.4 × 10−3 |
| GO:0043234 | Protein complex | 4 | 1.65 × 10−3 |
| R-HSA-73728 | RNA Polymerase I Promoter Opening | 3 | 2.06 × 10−3 |
| R-HSA-5334118 | DNA methylation | 3 | 2.19 × 10−3 |
| R-HSA-2559586 | DNA Damage/Telomere Stress Induced Senescence | 3 | 2.26 × 10−3 |
| R-HSA-5625886 | Activated PKN1 stimulates transcription of AR (androgen receptor) regulated genes KLK2 and KLK3 | 3 | 2.32 × 10−3 |
| R-HSA-427359 | SIRT1 negatively regulates rRNA expression | 3 | 2.39 × 10−3 |
| GO:0000784 | Nuclear chromosome, telomeric region | 3 | 2.66 × 10−3 |
| R-HSA-212300 | PRC2 methylates histones and DNA | 3 | 2.75 × 10−3 |
| R-HSA-2299718 | Condensation of Prophase Chromosomes | 3 | 2.83 × 10−3 |
| R-HSA-606279 | Deposition of new CENPA-containing nucleosomes at the centromere | 3 | 2.83 × 10−3 |
| R-HSA-3214858 | RMTs methylate histone arginines | 3 | 3.06 × 10−3 |
| R-HSA-1221632 | Meiotic synapsis | 3 | 3.22 × 10−3 |
| hsa05322 | Systemic lupus erythematosus | 3 | 3.62 × 10−3 |
| R-HSA-912446 | Meiotic recombination | 3 | 3.89 × 10−3 |
| R-HSA-73777 | - | 3 | 4.16 × 10−3 |
| R-HSA-5250924 | B-WICH complex positively regulates rRNA expression | 3 | 4.25 × 10−3 |
| R-HSA-3214815 | HDACs deacetylate histones | 3 | 4.53 × 10−3 |
| R-HSA-977225 | Amyloid fiber formation | 3 | 4.91 × 10−3 |
| R-HSA-5578749 | Transcriptional regulation by small RNAs | 3 | 5.72 × 10−3 |
| R-HSA-2559582 | Senescence-Associated Secretory Phenotype (SASP) | 3 | 6.15 × 10−3 |
| hsa05034 | Alcoholism | 3 | 6.26 × 10−3 |
| R-HSA-5617472 | Activation of anterior HOX genes in hindbrain development during early embryogenesis | 3 | 7.52 × 10−3 |
| R-HSA-3214847 | HATs acetylate histones | 3 | 1.01 × 10−2 |
| GO:0003677 | DNA binding | 5 | 1.24 × 10−2 |
| GO:0046982 | protein heterodimerization activity | 3 | 2.94 × 10−2 |
| Term | Description | Number of Genes | p-Value |
|---|---|---|---|
| GO:0050832 | Defense response to fungus | 3 | 7.46 × 10−6 |
| GO:0050830 | Defense response to Gram-positive bacterium | 3 | 7.57 × 10−5 |
| GO:0005615 | Extracellular space | 4 | 4.03 × 10−4 |
| GO:0005576 | Extracellular region | 4 | 6.88 × 10−4 |
| GO:0045087 | Innate immune response | 3 | 1.92 × 10−3 |
| GO:0070062 | Extracellular exosome | 4 | 3.67 × 10−3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamad, M.A.; Mohd Manzor, N.F.; Zulkifli, N.F.; Zainal, N.; Hayati, A.R.; Ahmad Asnawi, A.W. A Review of Candidate Genes and Pathways in Preeclampsia–An Integrated Bioinformatical Analysis. Biology 2020, 9, 62. https://doi.org/10.3390/biology9040062
Mohamad MA, Mohd Manzor NF, Zulkifli NF, Zainal N, Hayati AR, Ahmad Asnawi AW. A Review of Candidate Genes and Pathways in Preeclampsia–An Integrated Bioinformatical Analysis. Biology. 2020; 9(4):62. https://doi.org/10.3390/biology9040062
Chicago/Turabian StyleMohamad, Muhammad Aliff, Nur Fariha Mohd Manzor, Noor Fadzilah Zulkifli, Nurzaireena Zainal, Abd Rahman Hayati, and Asral Wirda Ahmad Asnawi. 2020. "A Review of Candidate Genes and Pathways in Preeclampsia–An Integrated Bioinformatical Analysis" Biology 9, no. 4: 62. https://doi.org/10.3390/biology9040062
APA StyleMohamad, M. A., Mohd Manzor, N. F., Zulkifli, N. F., Zainal, N., Hayati, A. R., & Ahmad Asnawi, A. W. (2020). A Review of Candidate Genes and Pathways in Preeclampsia–An Integrated Bioinformatical Analysis. Biology, 9(4), 62. https://doi.org/10.3390/biology9040062