Metabolic Cancer-Macrophage Crosstalk in the Tumor Microenvironment



{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
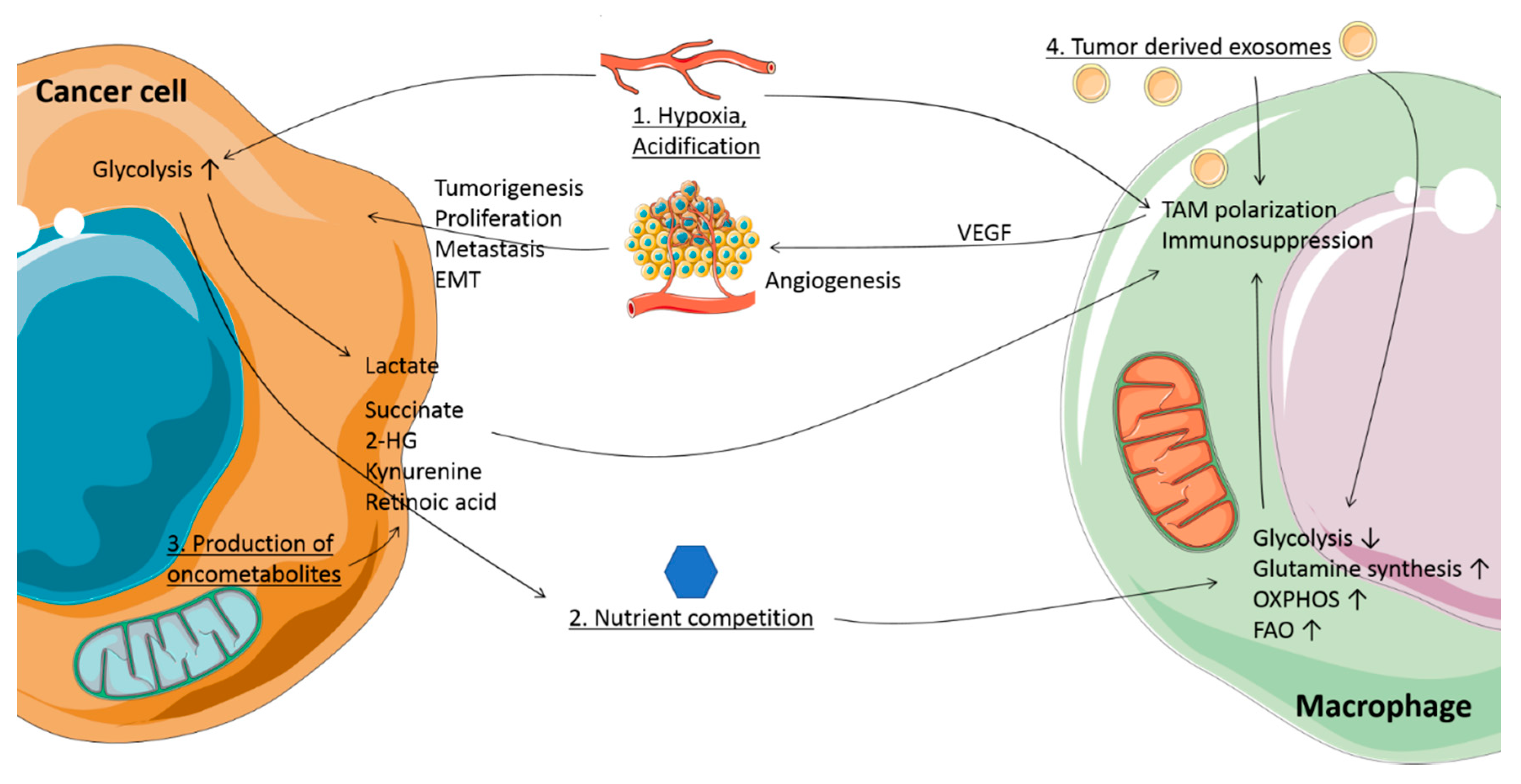
2. Metabolic Conditions Affect the TME
3. Oncometabolites Accumulate due to Mutations in TCA Cycle Enzyme Genes
4. Oncometabolites Regulate Macrophage Function and Heterogeneity
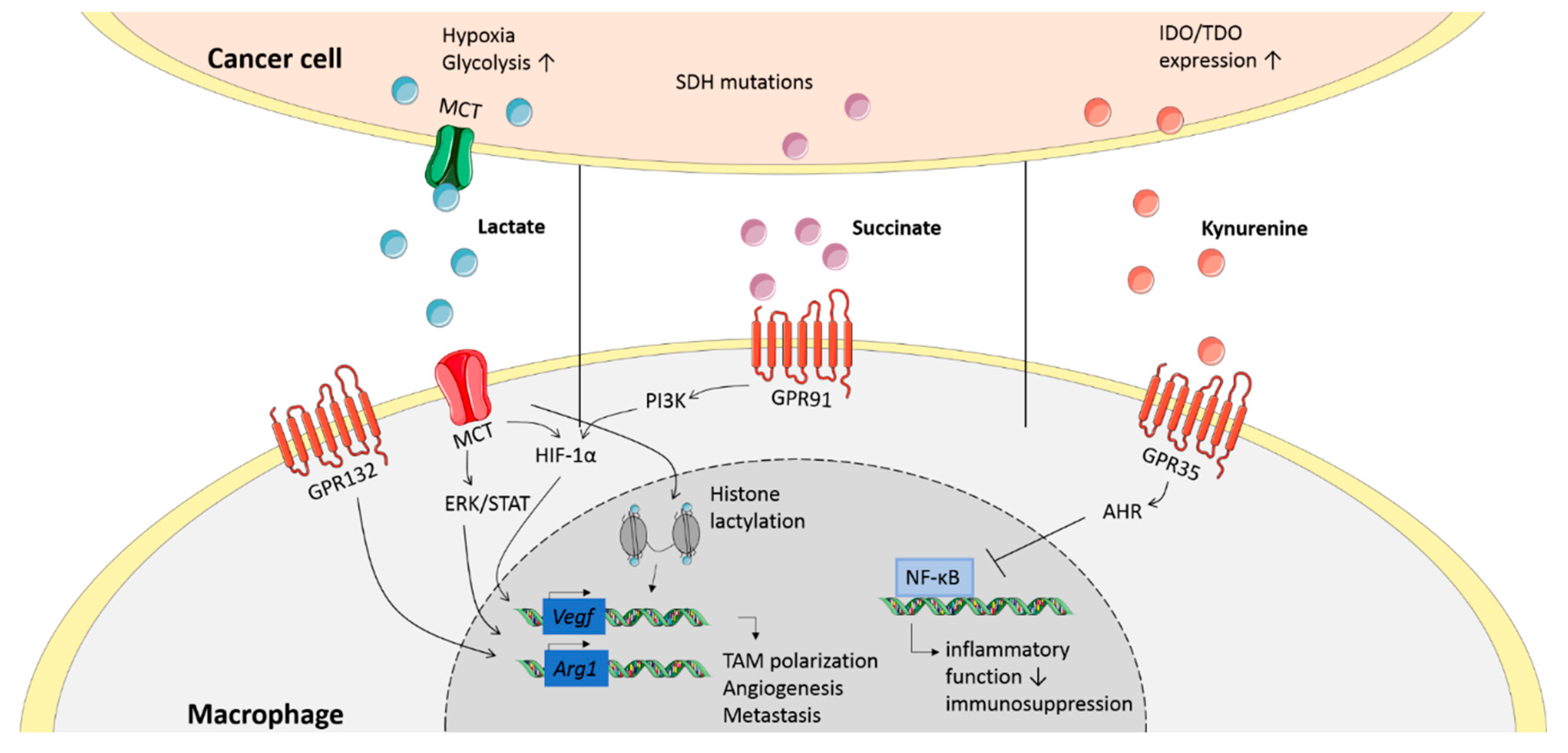
4.1. Cancer-Derived Lactate Induces TAMs that Promote Angiogenesis and Tumor Metastasis
4.2. Succinate Induces Tumor-Promoting Cytokine Production in Macrophages
4.3. D-2HG Suppresses the Immune Response and Reduces Immune Cell Infiltration
4.4. Kynurenine and Other Tryptophan Metabolites Inhibit the Anti-Tumor Response in TAMs
4.5. Production of Retinoic Acid by Tumor Cells Induces TAM Differentiation
5. Tumor-Derived Exosomes as Transporters of Oncometabolites
6. Effect of TAM Metabolism on Tumor Progression
7. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kelly, P.M.; Davison, R.S.; Bliss, E.; McGee, J.O. Macrophages in human breast disease: A quantitative immunohistochemical study. Br. J. Cancer 1988, 57, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-W.; Liu, L.; Gong, C.-Y.; Shi, H.-S.; Zeng, Y.-H.; Wang, X.-Z.; Zhao, Y.-W.; Wei, Y.-Q. Prognostic Significance of Tumor-Associated Macrophages in Solid Tumor: A Meta-Analysis of the Literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Bolli, E.; Movahedi, K.; Laoui, D.; Van Ginderachter, J.A. Novel insights in the regulation and function of macrophages in the tumor microenvironment. Curr. Opin. Oncol. 2017, 29, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Fontaine, C.; Deforet, M.; Akkari, L.; Thompson, C.B.; Joyce, J.A.; Xavier, J.B. Metabolic origins of spatial organization in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2017, 114, 2934–2939. [Google Scholar] [CrossRef]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Honkanen, T.J.; Tikkanen, A.; Karihtala, P.; Makinen, M.; Vayrynen, J.P.; Koivunen, J.P. Prognostic and predictive role of tumour-associated macrophages in HER2 positive breast cancer. Sci. Rep. 2019, 9, 10961. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Sica, A.; Saccani, A.; Bottazzi, B.; Polentarutti, N.; Vecchi, A.; van Damme, J.; Mantovani, A. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J. Immunol. 2000, 164, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Liao, J.; Liu, J.; Huang, D.; He, C.; Chen, F.; Yang, L.; Wu, W.; Chen, J.; Lin, L.; et al. Blocking the recruitment of naive CD4(+) T cells reverses immunosuppression in breast cancer. Cell Res. 2017, 27, 461–482. [Google Scholar] [CrossRef]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e413. [Google Scholar] [CrossRef]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef]
- Huang, S.C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Wu, J.Y.; Huang, T.W.; Hsieh, Y.T.; Wang, Y.F.; Yen, C.C.; Lee, G.L.; Yeh, C.C.; Peng, Y.J.; Kuo, Y.Y.; Wen, H.T.; et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol. Cell 2020, 77, 213–227.e215. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Gaustad, J.V.; Egeland, T.A.; Mathiesen, B.; Galappathi, K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int. J. Cancer 2010, 127, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.Y.; Hughes, R.; Murdoch, C.; Coffelt, S.B.; Biswas, S.K.; Harris, A.L.; Johnson, R.S.; Imityaz, H.Z.; Simon, M.C.; Fredlund, E.; et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 2009, 114, 844–859. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 2008, 118, 3930–3942. [Google Scholar] [CrossRef]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef]
- Tripathi, C.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bhatt, M.L.; Bhadauria, S. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014, 5, 5350–5368. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chang, C.; Lu, N.; Wang, X.; Lu, Q.; Ren, X.; Ren, P.; Zhao, D.; Wang, L.; Zhu, Y.; et al. Comprehensive Proteomics Analysis Reveals Metabolic Reprogramming of Tumor-Associated Macrophages Stimulated by the Tumor Microenvironment. J. Proteome Res. 2017, 16, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See Chi Ee, P.; San Luis, B.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. OncoImmunology 2016, 5, e1191731. [Google Scholar] [CrossRef]
- Arts, R.J.; Plantinga, T.S.; Tuit, S.; Ulas, T.; Heinhuis, B.; Tesselaar, M.; Sloot, Y.; Adema, G.J.; Joosten, L.A.; Smit, J.W.; et al. Transcriptional and metabolic reprogramming induce an inflammatory phenotype in non-medullary thyroid carcinoma-induced macrophages. OncoImmunology 2016, 5, e1229725. [Google Scholar] [CrossRef]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martin-Perez, R.; Serneels, J.; Prenen, H.; Ghesquiere, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef]
- Zhihua, Y.; Yulin, T.; Yibo, W.; Wei, D.; Yin, C.; Jiahao, X.; Runqiu, J.; Xuezhong, X. Hypoxia decreases macrophage glycolysis and M1 percentage by targeting microRNA-30c and mTOR in human gastric cancer. Cancer Sci. 2019, 110, 2368–2377. [Google Scholar] [CrossRef]
- Zhou, R.; Pantel, A.R.; Li, S.; Lieberman, B.P.; Ploessl, K.; Choi, H.; Blankemeyer, E.; Lee, H.; Kung, H.F.; Mach, R.H.; et al. [(18)F](2S,4R)4-Fluoroglutamine PET Detects Glutamine Pool Size Changes in Triple-Negative Breast Cancer in Response to Glutaminase Inhibition. Cancer Res. 2017, 77, 1476–1484. [Google Scholar] [CrossRef]
- Choi, J.; Stradmann-Bellinghausen, B.; Yakubov, E.; Savaskan, N.E.; Regnier-Vigouroux, A. Glioblastoma cells induce differential glutamatergic gene expressions in human tumor-associated microglia/macrophages and monocyte-derived macrophages. Cancer Biol. Ther. 2015, 16, 1205–1213. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Menga, A.; Martin-Perez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquiere, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef]
- Kostourou, V.; Cartwright, J.E.; Johnstone, A.P.; Boult, J.K.; Cullis, E.R.; Whitley, G.; Robinson, S.P. The role of tumour-derived iNOS in tumour progression and angiogenesis. Br. J. Cancer 2011, 104, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Theate, I.; van Baren, N.; Pilotte, L.; Moulin, P.; Larrieu, P.; Renauld, J.C.; Herve, C.; Gutierrez-Roelens, I.; Marbaix, E.; Sempoux, C.; et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol. Res. 2015, 3, 161–172. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Zea, A.H.; DeSalvo, J.; Culotta, K.S.; Zabaleta, J.; Quiceno, D.G.; Ochoa, J.B.; Ochoa, A.C. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J. Immunol. 2003, 171, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003, 1001–1015. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- Burnichon, N.; Briere, J.J.; Libe, R.; Vescovo, L.; Riviere, J.; Tissier, F.; Jouanno, E.; Jeunemaitre, X.; Benit, P.; Tzagoloff, A.; et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Crespin, M.; Nau, V.; Khau Van Kien, P.; Corvol, P.; Plouin, P.F.; Jeunemaitre, X.; et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003, 63, 5615–5621. [Google Scholar] [PubMed]
- Bourdeau, I.; Grunenwald, S.; Burnichon, N.; Khalifa, E.; Dumas, N.; Binet, M.C.; Nolet, S.; Gimenez-Roqueplo, A.P. A SDHC Founder Mutation Causes Paragangliomas (PGLs) in the French Canadians: New Insights on the SDHC-Related PGL. J. Clin. Endocrinol. Metab. 2016, 101, 4710–4718. [Google Scholar] [CrossRef]
- Habano, W.; Sugai, T.; Nakamura, S.; Uesugi, N.; Higuchi, T.; Terashima, M.; Horiuchi, S. Reduced expression and loss of heterozygosity of the SDHD gene in colorectal and gastric cancer. Oncol. Rep. 2003, 10, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Looper, R.E.; Koivunen, P.; Lee, S.; Schneider, R.K.; McMahon, C.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G., Jr. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625. [Google Scholar] [CrossRef]
- Letouze, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Lameirinhas, A.; Macedo-Silva, C.; Lobo, J.; C Dias, P.; Ferreira, V.; Henrique, R.; Jeronimo, C. Lactate Increases Renal Cell Carcinoma Aggressiveness through Sirtuin 1-Dependent Epithelial Mesenchymal Transition Axis Regulation. Cells 2020, 9, 1053. [Google Scholar] [CrossRef]
- Aspuria, P.P.; Lunt, S.Y.; Varemo, L.; Vergnes, L.; Gozo, M.; Beach, J.A.; Salumbides, B.; Reue, K.; Wiedemeyer, W.R.; Nielsen, J.; et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014, 2, 21. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Goncalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite D-2-Hydroxyglurate Directly Induces Epithelial-Mesenchymal Transition and is Associated with Distant Metastasis in Colorectal Cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef]
- Delahousse, J.; Verlingue, L.; Broutin, S.; Legoupil, C.; Touat, M.; Doucet, L.; Ammari, S.; Lacroix, L.; Ducreux, M.; Scoazec, J.Y.; et al. Circulating oncometabolite D-2-hydroxyglutarate enantiomer is a surrogate marker of isocitrate dehydrogenase-mutated intrahepatic cholangiocarcinomas. Eur. J. Cancer 2018, 90, 83–91. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Singh, A.R.; Zulcic, M.; Durden, D.L. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1alpha and HIF2alpha stability and tumor growth, angiogenesis, and metastasis. Mol. Cancer Res. 2014, 12, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Harber, K.J.; de Goede, K.E.; Verberk, S.G.S.; Meinster, E.; de Vries, H.E.; van Weeghel, M.; de Winther, M.P.J.; Van den Bossche, J. Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages. Metabolites 2020, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.L.; Phan, A.T.; Velica, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef]
- Ugele, I.; Cardenas-Conejo, Z.E.; Hammon, K.; Wehrstein, M.; Bruss, C.; Peter, K.; Singer, K.; Gottfried, E.; Boesch, J.; Oefner, P.; et al. D-2-Hydroxyglutarate and L-2-Hydroxyglutarate Inhibit IL-12 Secretion by Human Monocyte-Derived Dendritic Cells. Int. J. Mol. Sci. 2019, 20, 742. [Google Scholar] [CrossRef]
- Han, C.J.; Zheng, J.Y.; Sun, L.; Yang, H.C.; Cao, Z.Q.; Zhang, X.H.; Zheng, L.T.; Zhen, X.C. The oncometabolite 2-hydroxyglutarate inhibits microglial activation via the AMPK/mTOR/NF-kappaB pathway. Acta Pharmacol. Sin. 2019, 40, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- SongTao, Q.; Lei, Y.; Si, G.; YanQing, D.; HuiXia, H.; XueLin, Z.; LanXiao, W.; Fei, Y. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; He, L.; Lugano, R.; Roodakker, K.; Bergqvist, M.; Smits, A.; Dimberg, A. IDH mutation status is associated with distinct vascular gene expression signatures in lower-grade gliomas. Neuro Oncol. 2018, 20, 1505–1516. [Google Scholar] [CrossRef]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef]
- Venkateswaran, N.; Lafita-Navarro, M.C.; Hao, Y.H.; Kilgore, J.A.; Perez-Castro, L.; Braverman, J.; Borenstein-Auerbach, N.; Kim, M.; Lesner, N.P.; Mishra, P.; et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 2019, 33, 1236–1251. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutierrez-Vazquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. NeuroSci. 2019, 22, 729–740. [Google Scholar] [CrossRef]
- Yu, C.P.; Song, Y.L.; Zhu, Z.M.; Huang, B.; Xiao, Y.Q.; Luo, D.Y. Targeting TDO in cancer immunotherapy. Med. Oncol. 2017, 34, 73. [Google Scholar] [CrossRef] [PubMed]
- Sekkai, D.; Guittet, O.; Lemaire, G.; Tenu, J.P.; Lepoivre, M. Inhibition of nitric oxide synthase expression and activity in macrophages by 3-hydroxyanthranilic acid, a tryptophan metabolite. Arch. Biochem. Biophys. 1997, 340, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Su, Y.; Mei, Y.; Leng, Q.; Leng, B.; Liu, Z.; Stass, S.A.; Jiang, F. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Invest. 2010, 90, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Khoury, T.; Ademuyiwa, F.O.; Chandrasekhar, R.; Jabbour, M.; Deleo, A.; Ferrone, S.; Wang, Y.; Wang, X. Aldehyde dehydrogenase 1A1 expression in breast cancer is associated with stage, triple negativity, and outcome to neoadjuvant chemotherapy. Mod. Pathol. 2012, 25, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Mangiarotti, R.; Danova, M.; Alberici, R.; Pellicciari, C. All-trans retinoic acid (ATRA)-induced apoptosis is preceded by G1 arrest in human MCF-7 breast cancer cells. Br. J. Cancer 1998, 77, 186–191. [Google Scholar] [CrossRef]
- Devalaraja, S.; To, T.K.J.; Folkert, I.W.; Natesan, R.; Alam, M.Z.; Li, M.; Tada, Y.; Budagyan, K.; Dang, M.T.; Zhai, L.; et al. Tumor-Derived Retinoic Acid Regulates Intratumoral Monocyte Differentiation to Promote Immune Suppression. Cell 2020, 180, 1098–1114.e1016. [Google Scholar] [CrossRef]
- Liss, C.; Fekete, M.J.; Hasina, R.; Lingen, M.W. Retinoic acid modulates the ability of macrophages to participate in the induction of the angiogenic phenotype in head and neck squamous cell carcinoma. Int. J. Cancer 2002, 100, 283–289. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Cianciaruso, C.; Beltraminelli, T.; Duval, F.; Nassiri, S.; Hamelin, R.; Mozes, A.; Gallart-Ayala, H.; Ceada Torres, G.; Torchia, B.; Ries, C.H.; et al. Molecular Profiling and Functional Analysis of Macrophage-Derived Tumor Extracellular Vesicles. Cell Rep. 2019, 27, 3062–3080.e3011. [Google Scholar] [CrossRef]
- Casadei, L.; Calore, F.; Creighton, C.J.; Guescini, M.; Batte, K.; Iwenofu, O.H.; Zewdu, A.; Braggio, D.A.; Bill, K.L.; Fadda, P.; et al. Exosome-Derived miR-25-3p and miR-92a-3p Stimulate Liposarcoma Progression. Cancer Res. 2017, 77, 3846–3856. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [PubMed]
- Boussadia, Z.; Lamberti, J.; Mattei, F.; Pizzi, E.; Puglisi, R.; Zanetti, C.; Pasquini, L.; Fratini, F.; Fantozzi, L.; Felicetti, F.; et al. Acidic microenvironment plays a key role in human melanoma progression through a sustained exosome mediated transfer of clinically relevant metastatic molecules. J. Exp. Clin. Cancer Res. 2018, 37, 245. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Zhao, W.L.; Ye, Y.Y.; Bai, X.C.; Liu, R.Q.; Chang, L.F.; Zhou, Q.; Sui, S.F. Cellular internalization of exosomes occurs through phagocytosis. Traffic 2010, 11, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Wu, Q.; Wu, X.; Zhu, Q.; Wang, X.; Jiang, L.; Chen, X.; Wang, X. Epithelial ovarian cancer-secreted exosomal miR-222-3p induces polarization of tumor-associated macrophages. Oncotarget 2016, 7, 43076–43087. [Google Scholar] [CrossRef] [PubMed]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A.; et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Song, T.N.; Wang, F.R.; Yin, C.; Li, Z.; Lin, J.P.; Meng, Y.Q.; Feng, H.M.; Jing, T. Tumor-derived exosomal HMGB1 promotes esophageal squamous cell carcinoma progression through inducing PD1(+) TAM expansion. OncoGenesis 2019, 8, 17. [Google Scholar] [CrossRef]
- Gerloff, D.; Lutzkendorf, J.; Moritz, R.K.C.; Wersig, T.; Mader, K.; Muller, L.P.; Sunderkotter, C. Melanoma-Derived Exosomal miR-125b-5p Educates Tumor Associated Macrophages (TAMs) by Targeting Lysosomal Acid Lipase A (LIPA). Cancers 2020, 12, 464. [Google Scholar] [CrossRef]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. OncoGene 2019, 38, 5158–5173. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, S.; Zhou, Y.; Meng, K.; Chen, Z.; Cui, Y.; Shi, Y.; Wang, T.; He, Q.Y. Motile hepatocellular carcinoma cells preferentially secret sugar metabolism regulatory proteins via exosomes. Proteomics 2017, 17. [Google Scholar] [CrossRef]
- Vallabhaneni, K.C.; Penfornis, P.; Dhule, S.; Guillonneau, F.; Adams, K.V.; Mo, Y.Y.; Xu, R.; Liu, Y.; Watabe, K.; Vemuri, M.C.; et al. Extracellular vesicles from bone marrow mesenchymal stem/stromal cells transport tumor regulatory microRNA, proteins, and metabolites. Oncotarget 2015, 6, 4953–4967. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Rabold, K.; Aschenbrenner, A.; Thiele, C.; Boahen, C.K.; Schiltmans, A.; Smit, J.W.A.; Schultze, J.L.; Netea, M.G.; Adema, G.J.; Netea-Maier, R.T. Enhanced lipid biosynthesis in human tumor-induced macrophages contributes to their protumoral characteristics. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Peres, C.M.; de Bittencourt, P.I., Jr.; da Costa, M.; Curi, R. Transference of fatty acids from macrophages to lymphocytes in culture. Biochem. Soc. Trans. 1997, 25, 344S. [Google Scholar] [CrossRef]
- Peres, C.M.; Homem de Bittencourt, P.I., Jr.; Mendonca, J.R.; Curi, R. Evidence that macrophages transfer arachidonic acid and cholesterol to tissues in vivo. Cell Biochem. Funct. 2003, 21, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.I.; Liao, J.C.; Kuo, L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res. 2001, 61, 1100–1106. [Google Scholar]
- Jeong, H.; Kim, S.; Hong, B.J.; Lee, C.J.; Kim, Y.E.; Bok, S.; Oh, J.M.; Gwak, S.H.; Yoo, M.Y.; Lee, M.S.; et al. Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res. 2019, 79, 795–806. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, G.; Chu, H.; Wang, X.; Xiong, L.; Cai, G.; Liu, R.; Gao, H.; Tao, B.; Li, W.; et al. Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol. Cell 2018, 71, 201–215.e207. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.; Dang, W.Q.; Cao, M.F.; Xiao, J.F.; Lv, S.Q.; Jiang, W.J.; Yao, X.H.; Lu, H.M.; Miao, J.Y.; et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab. Invest. 2020, 100, 619–629. [Google Scholar] [CrossRef]
- Chen, F.; Chen, J.; Yang, L.; Liu, J.; Zhang, X.; Zhang, Y.; Tu, Q.; Yin, D.; Lin, D.; Wong, P.P.; et al. Extracellular vesicle-packaged HIF-1alpha-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat. Cell Biol. 2019, 21, 498–510. [Google Scholar] [CrossRef]
- Lin, S.; Sun, L.; Lyu, X.; Ai, X.; Du, D.; Su, N.; Li, H.; Zhang, L.; Yu, J.; Yuan, S. Lactate-activated macrophages induced aerobic glycolysis and epithelial-mesenchymal transition in breast cancer by regulation of CCL5-CCR5 axis: A positive metabolic feedback loop. Oncotarget 2017, 8, 110426–110443. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e1396. [Google Scholar] [CrossRef]
- Cassetta, L.; Kitamura, T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 38. [Google Scholar] [CrossRef]
- Li, M.; Li, M.; Yang, Y.; Liu, Y.; Xie, H.; Yu, Q.; Tian, L.; Tang, X.; Ren, K.; Li, J.; et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K-gamma and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J. Control. Release 2020, 321, 23–35. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Kruspig, B.; Valter, K.; Skender, B.; Zhivotovsky, B.; Gogvadze, V. Targeting succinate:ubiquinone reductase potentiates the efficacy of anticancer therapy. Biochim. Biophys. Acta 2016, 1863, 2065–2071. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Lang, R. Targeting L-Lactate Metabolism to Overcome Resistance to Immune Therapy of Melanoma and Other Tumor Entities. J. Oncol. 2019, 2019, 2084195. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Wang, Q.; Bi, E.; Ma, X.; Liu, L.; Yang, M.; Qian, J.; Yi, Q. Enhanced Lipid Accumulation and Metabolism Are Required for the Differentiation and Activation of Tumor-Associated Macrophages. Cancer Res. 2020, 80, 1438–1450. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, X.; Zheng, L.; Zhao, H.; Yan, G.; Zhang, Q.; Zhou, Y.; Lei, J.; Zhang, J.; Wang, J.; et al. RIPK3 Orchestrates Fatty Acid Metabolism in Tumor-Associated Macrophages and Hepatocarcinogenesis. Cancer Immunol. Res. 2020, 8, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; He, Y.; Zhao, P.; Hu, Y.; Tao, J.; Chen, J.; Huang, Y. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin beta3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics 2019, 9, 265–278. [Google Scholar] [CrossRef]
- Oh, M.H.; Sun, I.H.; Zhao, L.; Leone, R.D.; Sun, I.M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H.; et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Invest. 2020, 130, 3865–3884. [Google Scholar] [CrossRef]
- Patel, C.H.; Leone, R.D.; Horton, M.R.; Powell, J.D. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discov. 2019, 18, 669–688. [Google Scholar] [CrossRef]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic Instruction of Immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Goede, K.E.; Driessen, A.J.M.; Van den Bossche, J. Metabolic Cancer-Macrophage Crosstalk in the Tumor Microenvironment. Biology 2020, 9, 380. https://doi.org/10.3390/biology9110380
de Goede KE, Driessen AJM, Van den Bossche J. Metabolic Cancer-Macrophage Crosstalk in the Tumor Microenvironment. Biology. 2020; 9(11):380. https://doi.org/10.3390/biology9110380
Chicago/Turabian Stylede Goede, Kyra E., Amber J. M. Driessen, and Jan Van den Bossche. 2020. "Metabolic Cancer-Macrophage Crosstalk in the Tumor Microenvironment" Biology 9, no. 11: 380. https://doi.org/10.3390/biology9110380
APA Stylede Goede, K. E., Driessen, A. J. M., & Van den Bossche, J. (2020). Metabolic Cancer-Macrophage Crosstalk in the Tumor Microenvironment. Biology, 9(11), 380. https://doi.org/10.3390/biology9110380