From Classical to Unconventional: The Immune Receptors Facilitating Platelet Responses to Infection and Inflammation


Simple Summary
Abstract
1. Introduction
2. Adhesion Receptors in Platelets Are Crucial for Platelet Aggregation and Participate in Different Infections
2.1. αIIbβ3 Is Involved in Pathogen Recognition and Molecular Mimicry
2.2. The Role of αvβ3 Integrin in Immunity
2.3. The Role of P-Selectin in Inflammation and Infection
3. Pattern Recognition Receptors (PRR) Assist Platelets in Direct Recognition of Pathogens
3.1. Toll Like Receptors Are a Major Family of Receptors Contributing to the Role of Platelets in Infections
3.2. Several Other Families of PRRs also Contribute to the Role of Platelets in Immunity
4. The Only Fc Receptor on the Platelet Surface, FcγRIIA, Binds to Opsonized Pathogens and Bacterial Surface Proteins
5. Complement Receptors on Platelets Are Important in Potentiating Immune Responses
5.1. Different Receptors on Platelets Recognize Complement Component 1q Leading to Platelet Aggregation
5.2. G protein Coupled Receptors, C3aR and C5aR, Are Expressed on Platelets and Bind to Products of Complement Activation
5.3. Complement Receptor 2 Contributes to Response of Platelets During Viral Infection
6. The Role of Scavenger Receptors in Platelet Mediated Immunity is not Well Understood
7. Platelets Express Several Other Receptors Involved in Infection and Inflammation
8. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Drelich, D.A.; Bray, P.F. The Traditional Role of Platelets in Hemostasis. In The Non-Thrombotic Role of Platelets in Health and Disease; InTech: Rijeka, Croatia, 2015. [Google Scholar]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Whiteheart, S.W. The nuts and bolts of the platelet release reaction. Platelets 2017, 28, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Cerenius, L.; Söderhäll, K. Coagulation in invertebrates. J. Innate Immun. 2011, 3, 3–8. [Google Scholar] [CrossRef]
- Kawabata, S.; Koshiba, T.; Shibata, T. The Lipopolysaccharide-Activated Innate Immune Response Network of the Horseshoe Crab. Invertebr. Surviv. J. 2009, 6, 59–77. [Google Scholar]
- Arneth, B. Coevolution of the coagulation and immune systems. Inflamm. Res. 2019, 68, 117–123. [Google Scholar] [CrossRef]
- Antoniak, S. The coagulation system in host defense. Res. Pract. Thromb. Haemost. 2018, 2, 549–557. [Google Scholar] [CrossRef]
- Lee, K. Shape transformation and cytoskeletal reorganization in activated non-mammalian thrombocytes. Cell Biol. Int. 2004, 28, 299–310. [Google Scholar] [CrossRef]
- Stosik, M.; Tokarz-Deptuła, B.; Deptuła, W. Characterisation of thrombocytes in Osteichthyes. J. Vet. Res. 2019, 63, 123–131. [Google Scholar] [CrossRef]
- Kim, S.; Radhakrishnan, U.P.; Rajpurohit, S.K.; Kulkarni, V.; Jagadeeswaran, P. Vivo-Morpholino knockdown of αIIb: A novel approach to inhibit thrombocyte function in adult zebrafish. Blood Cells Mol. Dis. 2010, 44, 169–174. [Google Scholar] [CrossRef]
- Belamarich, F.A.; Fusari, M.H.; Shepro, D.; Kien, M. In vitro studies of aggregation of non-mammalian thrombocytes. Nature 1966, 212, 1579–1580. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T.; Nakayasu, C.; Rieger, A.M.; Barreda, D.R.; Somamoto, T.; Nakao, M. Phagocytosis by Thrombocytes is a Conserved Innate Immune Mechanism in Lower Vertebrates. Front. Immunol. 2014, 5, 445. [Google Scholar] [CrossRef]
- Ferdous, F.; Scott, T.R. A comparative examination of thrombocyte/platelet immunity. Immunol. Lett. 2015, 163, 32–39. [Google Scholar] [CrossRef]
- Bennett, J.S.; Berger, B.W.; Billings, P.C. The structure and function of platelet integrins. J. Thromb. Haemost. 2009, 7, 200–205. [Google Scholar] [CrossRef]
- Bennett, J.S. Regulation of integrins in platelets. Biopolymers 2015, 104, 323–333. [Google Scholar] [CrossRef]
- Shen, B.; Delaney, M.K.; Du, X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr. Opin. Cell Biol. 2012, 24, 600–606. [Google Scholar] [CrossRef]
- Noisakran, S.; Onlamoon, N.; Pattanapanyasat, K.; Hsiao, H.-M.; Songprakhon, P.; Angkasekwinai, N.; Chokephaibulkit, K.; Villinger, F.; Ansari, A.A.; Perng, G.C. Role of CD61+ cells in thrombocytopenia of dengue patients. Int. J. Hematol. 2012, 96, 600–610. [Google Scholar] [CrossRef]
- Bettaieb, A.; Oksenhendler, E.; Duedari, N.; Bierling, P. Cross-reactive antibodies between HIV-gp120 and platelet gpIIIa (CD61) in HIV-related immune thrombocytopenic purpura. Clin. Exp. Immunol. 1996, 103, 19–23. [Google Scholar] [CrossRef]
- Lapidot, T.; Petit, I. Current understanding of stem cell mobilization: The roles of chemokines, proteolytic enzymes, adhesion molecules, cytokines, and stromal cells. Exp. Hematol. 2002, 30, 973–981. [Google Scholar] [CrossRef]
- Clark, K.B.; Noisakran, S.; Onlamoon, N.; Hsiao, H.M.; Roback, J.; Villinger, F.; Ansari, A.A.; Perng, G.C. Multiploid CD61+ Cells Are the Pre-Dominant Cell Lineage Infected during Acute Dengue Virus Infection in Bone Marrow. PLoS ONE 2012, 7, e52902. [Google Scholar] [CrossRef]
- Gavrilovskaya, I.N.; Gorbunova, E.E.; Mackow, E.R. Pathogenic Hantaviruses Direct the Adherence of Quiescent Platelets to Infected Endothelial Cells. J. Virol. 2010, 84, 4832–4839. [Google Scholar] [CrossRef]
- Hamzeh-Cognasse, H.; Damien, P.; Chabert, A.; Pozzetto, B.; Cognasse, F.; Garraud, O. Platelets and infections—Complex interactions with bacteria. Front. Immunol. 2015, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Liesenborghs, L.; Verhamme, P.; Vanassche, T. Staphylococcus aureus, master manipulator of the human hemostatic system. J. Thromb. Haemost. 2018, 16, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Petersen, H.J.; Keane, C.; Jenkinson, H.F.; Vickerman, M.M.; Jesionowski, A.; Waterhouse, J.C.; Cox, D.; Kerrigan, S.W. Human platelets recognize a novel surface protein, PadA, on Streptococcus gordonii through a unique interaction involving fibrinogen receptor GPIIbIIIa. Infect. Immun. 2010, 78, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Palankar, R.; Kohler, T.P.; Krauel, K.; Wesche, J.; Hammerschmidt, S.; Greinacher, A. Platelets kill bacteria by bridging innate and adaptive immunity via platelet factor 4 and Fcγ RIIA. J. Thromb. Haemost. 2018, 16, 1187–1197. [Google Scholar] [CrossRef]
- Guo, L.; Rondina, M.T. The Era of Thromboinflammation: Platelets Are Dynamic Sensors and Effector Cells During Infectious Diseases. Front. Immunol. 2019, 10, 2204. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, F.; Chen, X. Integrin αvβ3-targeted cancer therapy. Drug Dev. Res. 2008, 69, 329–339. [Google Scholar] [CrossRef]
- Wilder, R.L. Integrin alpha V beta 3 as a target for treatment of rheumatoid arthritis and related rheumatic diseases. Ann. Rheum. Dis. 2002, 61, ii96–ii99. [Google Scholar] [CrossRef]
- Revelle, B.M.; Scott, D.; Kogan, T.P.; Zheng, J.; Beck, P.J. Structure-function analysis of P-selectin-sialyl LewisX binding interactions: Mutagenic alteration of ligand binding specificity. J. Biol. Chem. 1996. [Google Scholar] [CrossRef]
- Johansson, D.; Shannon, O.; Rasmussen, M. Platelet and Neutrophil Responses to Gram Positive Pathogens in Patients with Bacteremic Infection. PLoS ONE 2011, 6, e26928. [Google Scholar] [CrossRef]
- Peters, M.J.; Dixon, G.; Kotowicz, K.T.; Hatch, D.J.; Heyderman, R.S.; Klein, N.J. Circulating platelet-neutrophil complexes represent a subpopulation of activated neutrophils primed for adhesion, phagocytosis and intracellular killing. Br. J. Haematol. 1999, 106, 391–399. [Google Scholar] [CrossRef]
- Ludwig, R.J.; Bergmann, P.; Garbaraviciene, J.; von Stebut, E.; Radeke, H.H.; Gille, J.; Diehl, S.; Hardt, K.; Henschler, R.; Kaufmann, R.; et al. Platelet, not endothelial, P-selectin expression contributes to generation of immunity in cutaneous contact hypersensitivity. Am. J. Pathol. 2010, 176, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Crúz, M.A.; Zhang, H.; López, J.A.; Afshar-Kharghan, V. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 2005, 201, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, C.; Wetterö, J.; Tengvall, P.; Bengtsson, T. C1q induces a rapid up-regulation of P-selectin and modulates collagen- and collagen-related peptide-triggered activation in human platelets. Immunobiology 2010, 215, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.J.; Tsai, S.; Wu, D.C.; Wu, J.Y.; Liu, T.C.; Chen, A. P-selectin-dependent platelet aggregation and apoptosis may explain the decrease in platelet count during Helicobacter pylori infection. Blood 2010, 115, 4247–4253. [Google Scholar] [CrossRef]
- Scanlon, V.M.; Teixeira, A.M.; Tyagi, T.; Zou, S.; Zhang, P.X.; Booth, C.J.; Kowalska, M.A.; Bao, J.; Hwa, J.; Hayes, V.; et al. Epithelial (E)-Cadherin is a Novel Mediator of Platelet Aggregation and Clot Stability. Thromb. Haemost. 2019, 119, 744–757. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011. [Google Scholar] [CrossRef]
- Hottz, E.D.; Bozza, F.A.; Bozza, P.T. Platelets in immune response to virus and immunopathology of viral infections. Front. Med. 2018, 5, 121. [Google Scholar] [CrossRef]
- Cognasse, F.; Hamzeh, H.; Chavarin, P.; Acquart, S.; Genin, C.; Garraud, O. Evidence of Toll-like receptor molecules on human platelets. Immunol. Cell Biol. 2005, 83, 196–198. [Google Scholar] [CrossRef]
- Uchiyama, S.; Sun, J.; Fukahori, K.; Ando, N.; Wu, M.; Schwarz, F.; Siddiqui, S.S.; Varki, A.; Marth, J.D.; Nizet, V. Dual actions of group B Streptococcus capsular sialic acid provide resistance to platelet-mediated antimicrobial killing. Proc. Natl. Acad. Sci. USA 2019, 116, 7465–7470. [Google Scholar] [CrossRef]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.J.; Patel, K.D.; Kubes, P. Platelets express functional Toll-like receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.A.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-Like Receptor 2 in Human Platelets Induces a Thromboinflammatory Response Through Activation of Phosphoinositide 3-Kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef]
- Cognasse, F.; Nguyen, K.A.; Damien, P.; McNicol, A.; Pozzetto, B.; Hamzeh-Cognasse, H.; Garraud, O. The inflammatory role of platelets via their TLRs and Siglec receptors. Front. Immunol. 2015, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.W.A.; Nestel, F.P.; Ni, H.; Lazarus, A.H.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-α production in vivo. Blood 2006. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Role of platelet TLR4 expression in pathogensis of septic thrombocytopenia. World J. Emerg. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Svensson, M.; Mörgelin, M.; Svanborg, C.; Tarr, P.I.; Mooney, J.C.; Watkins, S.L.; Johnson, R.; Karpman, D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood 2006. [Google Scholar] [CrossRef] [PubMed]
- Lopes Pires, M.E.; Clarke, S.R.; Marcondes, S.; Gibbins, J.M. Lipopolysaccharide potentiates platelet responses via toll-like receptor 4-stimulated Akt-Erk-PLA2 signalling. PLoS ONE 2017, 12, e0186981. [Google Scholar] [CrossRef] [PubMed]
- Matus, V.; Valenzuela, J.G.; Hidalgo, P.; Pozo, L.M.; Panes, O.; Wozniak, A.; Mezzano, D.; Pereira, J.; Sáez, C.G. Human platelet interaction with E. coli O111 promotes tissue-factor-dependent procoagulant activity, involving Toll like receptor 4. PLoS ONE 2017, 12, e0185431. [Google Scholar] [CrossRef]
- Claushuis, T.A.M.; Van Der Veen, A.I.P.; Horn, J.; Schultz, M.J.; Houtkooper, R.H.; Van’T Veer, C.; Van Der Poll, T. Platelet Toll-like receptor expression and activation induced by lipopolysaccharide and sepsis. Platelets 2019, 30, 296–304. [Google Scholar] [CrossRef]
- Moriarty, R.D.; Cox, A.; McCall, M.; Smith, S.G.J.; Cox, D. Escherichia coli induces platelet aggregation in an FcγRIIa-dependent manner. J. Thromb. Haemost. 2016. [Google Scholar] [CrossRef]
- Watson, C.N.; Kerrigan, S.W.; Cox, D.; Henderson, I.R.; Watson, S.P.; Arman, M. Human platelet activation by Escherichia coli: Roles for FcγRIIA and integrin αIIbβ3. Platelets 2016. [Google Scholar] [CrossRef]
- Berthet, J.; Damien, P.; Hamzeh-Cognasse, H.; Arthaud, C.-A.; Eyraud, M.-A.; Zéni, F.; Pozzetto, B.; McNicol, A.; Garraud, O.; Cognasse, F. Human platelets can discriminate between various bacterial LPS isoforms via TLR4 signaling and differential cytokine secretion. Clin. Immunol. 2012, 145, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Han, J.; Welch, E.J.; Ye, R.D.; Voyno-Yasenetskaya, T.A.; Malik, A.B.; Du, X.; Li, Z. Lipopolysaccharide Stimulates Platelet Secretion and Potentiates Platelet Aggregation via TLR4/MyD88 and the cGMP-Dependent Protein Kinase Pathway. J. Immunol. 2009, 182, 7997–8004. [Google Scholar] [CrossRef]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen Stimulates Macrophage Chemokine Secretion Through Toll-Like Receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015, 7, 304ra142. [Google Scholar] [CrossRef] [PubMed]
- Romani, L. Immunity to fungal infections. Nat. Rev. Immunol. 2011, 11, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Sugawara, S.; Monodane, T.; Nishijima, M.; Adachi, Y.; Akashi, S.; Miyake, K.; Hase, S.; Takada, H. Micrococcus luteus teichuronic acids activate human and murine monocytic cells in a CD14- and toll-like receptor 4-dependent manner. Infect. Immun. 2001, 69, 2025–2030. [Google Scholar] [CrossRef]
- Beaulieu, L.M.; Freedman, J.E. The Role of Inflammation in Regulating Platelet Production and Function: Toll-like Receptors in Platelets and Megakaryocytes. Thromb. Res. 2010, 125, 205–209. [Google Scholar] [CrossRef]
- Damås, J.K.; Jensenius, M.; Ueland, T.; Otterdal, K.; Yndestad, A.; Frøland, S.S.; Rolain, J.-M.; Myrvang, B.; Raoult, D.; Aukrust, P. Increased Levels of Soluble CD40L in African Tick Bite Fever: Possible Involvement of TLRs in the Pathogenic Interaction between Rickettsia africae, Endothelial Cells, and Platelets. J. Immunol. 2006, 177, 2699–2706. [Google Scholar] [CrossRef]
- Danese, S.; Katz, J.A.; Saibeni, S.; Papa, A.; Gasbarrini, A.; Vecchi, M.; Fiocchi, C. Activated platelets are the source of elevated levels of soluble CD40 ligand in the circulation of inflammatory bowel disease patients. Gut 2003, 52, 1435–1441. [Google Scholar] [CrossRef]
- Assinger, A.; Laky, M.; Badrnya, S.; Esfandeyari, A.; Volf, I. Periodontopathogens induce expression of CD40L on human platelets via TLR2 and TLR4. Thromb. Res. 2012. [Google Scholar] [CrossRef]
- Assinger, A.; Laky, M.; Schabbauer, G.; Hirschl, A.M.; Buchberger, E.; Binder, B.R.; Volf, I. Efficient phagocytosis of periodontopathogens by neutrophils requires plasma factors, platelets and TLR2. J. Thromb. Haemost. 2011, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Damien, P.; Cognasse, F.; Payrastre, B.; Spinelli, S.L.; Blumberg, N.; Arthaud, C.A.; Eyraud, M.A.; Phipps, R.P.; McNicol, A.; Pozzetto, B.; et al. NF-κB links TLR2 and PAR1 to soluble immunomodulator factor secretion in human platelets. Front. Immunol. 2017. [Google Scholar] [CrossRef]
- Assinger, A.; Kral, J.B.; Yaiw, K.C.; Schrottmaier, W.C.; Kurzejamska, E.; Wang, Y.; Mohammad, A.A.; Religa, P.; Rahbar, A.; Schabbauer, G.; et al. Human cytomegalovirus-platelet interaction triggers toll-like receptor 2-dependent proinflammatory and proangiogenic responses. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 801–809. [Google Scholar] [CrossRef]
- Hally, K.E.; Bird, G.K.; la Flamme, A.C.; Harding, S.A.; Larsen, P.D. Platelets modulate multiple markers of neutrophil function in response to in vitro Toll-like receptor stimulation. PLoS ONE 2019, 14, e0223444. [Google Scholar] [CrossRef] [PubMed]
- Anabel, A.S.; Eduardo, P.C.; Pedro Antonio, H.C.; Carlos, S.M.; Juana, N.M.; Honorio, T.A.; Nicolás, V.S.; Sergio Roberto, A.R. Human platelets express Toll-like receptor 3 and respond to poly I:C. Hum. Immunol. 2014, 75, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Vitseva, O.; MacKay, C.R.; Beaulieu, L.M.; Benjamin, E.J.; Mick, E.; Kurt-Jones, E.A.; Ravid, K.; Freedman, J.E. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 2014. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Manni, G.; Pang, C.J.; Clancy, L.; Yao, C.; Rade, J.; Levy, D.; Wang, J.P.; et al. The role of platelets in mediating a response to human influenza infection. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Thålin, C.; Hisada, Y.; Lundström, S.; Mackman, N.; Wallén, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef]
- Banerjee, M.; Huang, Y.; Joshi, S.; Popa, G.J.; Mendenhall, M.D.; Wang, Q.J.; Garvy, B.A.; Myint, T.; Whiteheart, S.W. Platelets Endocytose Viral Particles and Are Activated via TLR (Toll-Like Receptor) Signaling. Arterioscler. Thromb. Vasc. Biol. 2020. [Google Scholar] [CrossRef]
- Cervantes, J.L.; Weinerman, B.; Basole, C.; Salazar, J.C. TLR8: The forgotten relative revindicated. Cell. Mol. Immunol. 2012, 9, 434–438. [Google Scholar] [CrossRef]
- Moen, S.H.; Ehrnström, B.; Kojen, J.F.; Yurchenko, M.; Beckwith, K.S.; Afset, J.E.; Damås, J.K.; Hu, Z.; Yin, H.; Espevik, T.; et al. Human Toll-like Receptor 8 (TLR8) Is an Important Sensor of Pyogenic Bacteria, and Is Attenuated by Cell Surface TLR Signaling. Front. Immunol. 2019, 10, 1209. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Bortolus, C.; Lecointe, K.; Parny, M.; Charlet, R.; Sendid, B.; Jawhara, S. Fungal Chitin Reduces Platelet Activation Mediated via TLR8 Stimulation. Front. Cell. Infect. Microbiol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Mick, E.; Mikhalev, E.; Benjamin, E.J.; Tanriverdi, K.; Freedman, J.E. Sex differences in platelet toll-like receptors and their association with cardiovascular risk factors. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The role of TLR2 ininfection and immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef]
- Thon, J.N.; Peters, C.G.; Machlus, K.R.; Aslam, R.; Rowley, J.; Macleod, H.; Devine, M.T.; Fuchs, T.A.; Weyrich, A.S.; Semple, J.W.; et al. T granules in human platelets function in TLR9 organization and signaling. J. Cell Biol. 2012. [Google Scholar] [CrossRef]
- Panigrahi, S.; Ma, Y.; Hong, L.; Gao, D.; West, X.Z.; Salomon, R.G.; Byzova, T.V.; Podrez, E.A. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circ. Res. 2013. [Google Scholar] [CrossRef]
- Hally, K.E.; La Flamme, A.C.; Larsen, P.D.; Harding, S.A. Toll-like receptor 9 expression and activation in acute coronary syndrome patients on dual anti-platelet therapy. Thromb. Res. 2016. [Google Scholar] [CrossRef]
- Chiffoleau, E. C-type lectin-like receptors as emerging orchestrators of sterile inflammation represent potential therapeutic targets. Front. Immunol. 2018, 9, 227. [Google Scholar] [CrossRef]
- Suzuki-Inoue, K.; Fuller, G.L.J.; García, Á.; Eble, J.A.; Pöhlmann, S.; Inoue, O.; Gartner, T.K.; Hughan, S.C.; Pearce, A.C.; Laing, G.D.; et al. Anovel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood 2006, 107, 542–554. [Google Scholar] [CrossRef]
- Boukour, S.; Massé, J.M.; Bénit, L.; Dubart-Kupperschmitt, A.; Cramer, E.M. Lentivirus degradation and DC-SIGN expression by human platelets and megakaryocytes. J. Thromb. Haemost. 2006. [Google Scholar] [CrossRef]
- Chaipan, C.; Soilleux, E.J.; Simpson, P.; Hofmann, H.; Gramberg, T.; Marzi, A.; Geier, M.; Stewart, E.A.; Eisemann, J.; Steinkasserer, A.; et al. DC-SIGN and CLEC-2 Mediate Human Immunodeficiency Virus Type 1 Capture by Platelets. J. Virol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Beck, Z.; Jagodzinski, L.L.; Eller, M.A.; Thelian, D.; Matyas, G.R.; Kunz, A.N.; Alving, C.R. Platelets and erythrocyte-bound platelets bind infectious HIV-1 in plasma of chronically infected patients. PLoS ONE 2013, 8, e81002. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.Y.; Sutherland, M.R.; Pryzdial, E.L.G. Dengue virus binding and replication by platelets. Blood 2015, 126, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.H.; Wu, W.C.; Lai, Y.C.; Tsai, P.J.; Perng, G.C.; Lin, Y.S.; Yeh, T.M. Dengue virus nonstructural protein 1 activates platelets via Toll-like receptor 4, leading to thrombocytopenia and hemorrhage. PLoS Pathog. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tomo, S.; Mohan, S.; Ramachandrappa, V.S.; Samadanam, D.M.; Suresh, S.; Pillai, A.B.; Tamilarasu, K.; Ramachandran, R.; Rajendiran, S. Dynamic modulation of DC-SIGN and FcYR2A receptors expression on platelets in dengue. PLoS ONE 2018, 13, e0206346. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.S.; Huang, T.F.; Hsieh, S.L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat. Commun. 2019, 10, 2402. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, J.R.; Cook, C.N.; Bobat, S.; Ross, E.A.; Flores-Langarica, A.; Lowe, K.L.; Khan, M.; Coral Dominguez-Medina, C.; Lax, S.; Carvalho-Gaspar, M.; et al. Inflammation drives thrombosis after Salmonella infection via CLEC-2 on platelets. J. Clin. Investig. 2015, 125, 4429–4446. [Google Scholar] [CrossRef]
- Kim, Y.K.; Shin, J.S.; Nahm, M.H. NOD-like receptors in infection, immunity, and diseases. Yonsei Med. J. 2016, 57, 5–14. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, S.; Hu, L.; Zhai, L.; Xue, R.; Ye, J.; Chen, L.; Cheng, G.; Mruk, J.; Kunapuli, S.P.; et al. Nucleotide-binding oligomerization domain 2 receptor is expressed in platelets and enhances platelet activation and thrombosis. Circulation 2015, 131, 1160–1170. [Google Scholar] [CrossRef]
- Ló pez, J.A. The platelet Fc receptor: A new role for an old actor. Blood 2013, 121, 1674–1675. [Google Scholar] [CrossRef]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Arman, M.; Krauel, K. Human platelet IgG Fc receptor FcγRIIA in immunity and thrombosis. J. Thromb. Haemost. 2015, 13, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Cabral, C.; Kushner, L.; Salzman, E.W. Membrane glycoproteins and platelet cytoskeleton in immune complex-induced platelet activation. Blood 1993, 81, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Antczak, A.J.; Singh, N.; Gay, S.R.; Worth, R.G. IgG-complex stimulated platelets: A source of sCD40L and RANTES in initiation of inflammatory cascade. Cell. Immunol. 2010. [Google Scholar] [CrossRef]
- Berlacher, M.D.; Vieth, J.A.; Heflin, B.C.; Gay, S.R.; Antczak, A.J.; Tasma, B.E.; Boardman, H.J.; Singh, N.; Montel, A.H.; Kahaleh, M.B.; et al. FcgammaRIIa Ligation Induces Platelet Hypersensitivity to Thrombotic Stimuli. Am. J. Pathol. 2013, 182, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Bernard, N.J. Preventing immune-complex-mediated disease. Nat. Rev. Rheumatol. 2019, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.J.C.; Snowden, N.; Carthy, D.; Papasteriades, C.; Hajeer, A.; Ollier, W.E.R. FcγRIIa polymorphism in systemic lupus erythematosus. Ann. Rheum. Dis. 1997, 56, 744–746. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.G.C.; Clatworthy, M.R. FcγRIIB in autoimmunity and infection: Evolutionary and therapeutic implications. Nat. Rev. Immunol. 2010, 10, 328–343. [Google Scholar] [CrossRef]
- Yu, X.; Lazarus, A.H. Targeting FcγRs to treat antibody-dependent autoimmunity. Autoimmun. Rev. 2016, 15, 510–512. [Google Scholar] [CrossRef]
- Baroletti, S.A.; Goldhaber, S.Z. Heparin-induced thrombocytopenia. Circulation 2006, 110, e454–e458. [Google Scholar] [CrossRef]
- Karnes, J.H. Pharmacogenetics to prevent heparin-induced thrombocytopenia: What do we know? Pharmacogenomics 2018, 19, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Arman, M.; Krauel, K.; Tilley, D.O.; Weber, C.; Cox, D.; Greinacher, A.; Kerrigan, S.W.; Watson, S.P. Amplification of bacteria-induced platelet activation is triggered by FcγRIIA, integrin αIIbβ3, and platelet factor 4. Blood 2014, 123, 3166–3174. [Google Scholar] [CrossRef] [PubMed]
- Loughman, A.; Fitzgerald, J.R.; Brennan, M.P.; Higgins, J.; Downer, R.; Cox, D.; Foster, T.J. Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol. Microbiol. 2005. [Google Scholar] [CrossRef]
- Tilley, D.O.; Arman, M.; Smolenski, A.; Cox, D.; O’Donnell, J.S.; Douglas, C.W.I.; Watson, S.P.; Kerrigan, S.W. Glycoprotein Ibα and FcγRIIa play key roles in platelet activation by the colonizing bacterium, Streptococcus oralis. J. Thromb. Haemost. 2013. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E.; Paré, G.; Rousseau, M.; Cloutier, N.; Dubuc, I.; Lévesque, T.; Borgeat, P.; Flamand, L. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood 2014. [Google Scholar] [CrossRef] [PubMed]
- Kelton, J.G.; Smith, J.W.; Warkentin, T.E.; Hayward, C.P.M.; Denomme, G.A.; Horsewood, P. Immunoglobulin G from patients with heparin-induced thrombocytopenia binds to a complex of heparin and platelet factor 4. Blood 1994. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V. Complement and clot. Blood 2017, 129, 2214–2215. [Google Scholar] [CrossRef]
- Nepomuceno, R.R.; Tenner, A.J. C1qRP, the C1q receptor that enhances phagocytosis, is detected specifically in human cells of myeloid lineage, endothelial cells, and platelets. J. Immunol. 1998, 160, 1929–1935. [Google Scholar]
- Arbesu, I.; Bucsaiova, M.; Fischer, M.B.; Mannhalter, C. Platelet-borne complement proteins and their role in platelet–bacteria interactions. J. Thromb. Haemost. 2016. [Google Scholar] [CrossRef]
- Kishore, U.; Reid, K.B.M. C1q: Structure, function, and receptors. Immunopharmacology 2000, 49, 159–170. [Google Scholar] [CrossRef]
- Peerschke, E.I.; Ghebrehiwet, B. Human blood platelets possess specific binding sites for C1q. J. Immunol. 1987, 138, 1537–1541. [Google Scholar]
- Pednekar, L.; Pathan, A.A.; Paudyal, B.; Tsolaki, A.G.; Kaur, A.; Abozaid, S.M.; Kouser, L.; Khan, H.A.; Peerschke, E.I.; Shamji, M.H.; et al. Analysis of the Interaction between Globular Head Modules of Human C1q and Its Candidate Receptor gC1qR. Front. Immunol. 2016, 7, 567. [Google Scholar] [CrossRef]
- Peerschke, E.I.B.; Yin, W.; Grigg, S.E.; Ghebrehiwet, B. Blood platelets activate the classical pathway of human complement. J. Thromb. Haemost. 2006, 4, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Peerschke, E.I.B.; Ghebrehiwet, B. Platelet receptors for the complement component C1q: Implications for hemostasis and thrombosis. Immunobiology 1998, 199, 239–249. [Google Scholar] [CrossRef]
- Peerschke, E.I.B.; Murphy, T.K.; Ghebrehiwet, B. Activation-dependent surface expression of gC1qR/p33 on human blood platelets. Thromb. Haemost. 2003, 89, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Donat, C.; Kölm, R.; Csorba, K.; Tuncer, E.; Tsakiris, D.A.; Trendelenburg, M. Complement C1q Enhances Primary Hemostasis. Front. Immunol. 2020, 11, 1522. [Google Scholar] [CrossRef]
- Ghebrehiwet, B.; Lim, B.L.; Peerschke, E.I.B.; Willis, A.C.; Reid, K.B.M. Isolation, cdna clonlng, and overexpression of a 33-kd cell surface glycoprotein that binds to the globular “heads” of clq. J. Exp. Med. 1994. [Google Scholar] [CrossRef]
- Nguyen, T.; Ghebrehiwet, B.; Peerschke, E.I.B. Staphylococcus aureus protein A recognizes platelet gC1qR/p33: A novel mechanism for staphylococcal interactions with platelets. Infect. Immun. 2000. [Google Scholar] [CrossRef]
- Ford, I.; Douglas, C.W.I.; Cox, D.; Rees, D.G.C.; Heath, J.; Preston, F.E. The role of immunoglobulin G and fibrinogen in platelet aggregation by Streptococcus sanguis. Br. J. Haematol. 1997, 97, 737–7446. [Google Scholar] [CrossRef]
- Palm, F.; Sjöholm, K.; Malmström, J.; Shannon, O. Complement Activation Occurs at the Surface of Platelets Activated by Streptococcal M1 Protein and This Results in Phagocytosis of Platelets. J. Immunol. 2019. [Google Scholar] [CrossRef]
- Panessa-Warren, B.; Wong, S.S.; Ghebrehiwet, B.; Tortora, G.T.; Warren, J.B. Carbon Nanotube Membrane Probes:Immuno-Labeling by LM, AFM, Tem, & FESEM. Microsc. Microanal. 2002. [Google Scholar] [CrossRef]
- Ghebrehiwet, B.; Lim, B.L.; Kumar, R.; Feng, X.; Peerschke, E.I.B. gC1q-R/p33, a member of a new class of multifunctional and multicompartmental cellular proteins, is involved in inflammation and infection. Immunol. Rev. 2001, 180, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Tantral, L.; Panessa-Warren, B.; Tortora, G.; Wong, S.; Warren, J.; Ghebrehiwet, B. The process of B. cereus spore attachment induces upregulation of cell surface gC1q-R/p33. Mol. Immunol. 2004. [Google Scholar] [CrossRef]
- Peerschke, E.I.; Ghebrehiwet, B. C1q augments platelet activation in response to aggregated Ig. J. Immunol. 1997, 159, 5594–5598. [Google Scholar] [PubMed]
- Coulthard, L.G.; Woodruff, T.M. Is the Complement Activation Product C3a a Proinflammatory Molecule? Re-evaluating the Evidence and the Myth. J. Immunol. 2015. [Google Scholar] [CrossRef]
- Sauter, R.J.; Sauter, M. Functional relevance of the Anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation 2018. [Google Scholar] [CrossRef]
- Guidetti, G.F.; Torti, M. The Small GTPase Rap1b: A Bidirectional Regulator of Platelet Adhesion Receptors. J. Signal Transduct. 2012. [Google Scholar] [CrossRef]
- Ward, P.A. Functions of C5a receptors. J. Mol. Med. 2009, 87, 375–378. [Google Scholar] [CrossRef]
- Sarma, J.V.; Ward, P.A. New developments in C5a receptor signaling. Cell Health Cytoskelet. 2012, 4, 73–82. [Google Scholar]
- Nording, H.; Giesser, A.; Patzelt, J.; Sauter, R.; Emschermann, F.; Stellos, K.; Gawaz, M.; Langer, H.F. Platelet bound oxLDL shows an inverse correlation with plasma anaphylatoxin C5a in patients with coronary artery disease. Platelets 2016. [Google Scholar] [CrossRef]
- Calame, D.G.; Mueller-Ortiz, S.L.; Morales, J.E.; Wetsel, R.A. The C5a Anaphylatoxin Receptor (C5aR1) Protects against Listeria monocytogenes Infection by Inhibiting Type 1 IFN Expression. J. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Galbusera, M.; Gastoldi, S.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Pagani, C.; Noris, M.; Gobbi, M.; Stravalaci, M.; et al. Alternative Pathway Activation of Complement by Shiga Toxin Promotes Exuberant C3a Formation That Triggers Microvascular Thrombosis. J. Immunol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Muenstermann, M.; Strobel, L.; Klos, A.; Wetsel, R.A.; Woodruff, T.M.; Köhl, J.; Johswich, K.O. Distinct roles of the anaphylatoxin receptors C3aR, C5aR1 and C5aR2 in experimental meningococcal infections. Virulence 2019. [Google Scholar] [CrossRef] [PubMed]
- Hannan, J.; Young, K.; Szakonyi, G.; Overduin, M.J.; Perkins, S.J.; Chen, X.; Holers, V.M. Structure of complement receptor (CR) 2 and CR2-C3d complexes. Biochem. Soc. Trans. 2002, 30 (Pt 6), 983–989. [Google Scholar] [CrossRef]
- Asokan, R.; Banda, N.K.; Szakonyi, G.; Chen, X.S.; Holers, V.M. Human complement receptor 2 (CR2/CD21) as a receptor for DNA: Implications for its roles in the immune response and the pathogenesis of systemic lupus erythematosus (SLE). Mol. Immunol. 2013. [Google Scholar] [CrossRef]
- Ahmad, A.; Menezes, J. Binding of the Epstein-Barr Virus to Human Platelets Causes the Release of Transforming Growth Factor-β. J. Immunol. 1997, 159, 3984–3988. [Google Scholar]
- Meyer, A.; Wang, W.; Qu, J.; Croft, L.; Degen, J.L.; Coller, B.S.; Ahamed, J. Platelet TGF-β1 contributions to plasma TGF-β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood 2012. [Google Scholar] [CrossRef]
- PrabhuDas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.E.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J. Immunol. 2017. [Google Scholar] [CrossRef]
- Zani, I.; Stephen, S.; Mughal, N.; Russell, D.; Homer-Vanniasinkam, S.; Wheatcroft, S.; Ponnambalam, S. Scavenger Receptor Structure and Function in Health and Disease. Cells 2015, 4, 178–201. [Google Scholar] [CrossRef]
- Valiyaveettil, M.; Podrez, E.A. Platelet hyperreactivity, scavenger receptors and atherothrombosis. J. Thromb. Haemost. 2009, 7 (Suppl. S1), 218–221. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of cell signaling by the scavenger receptor CD36: Implications in atherosclerosis and thrombosis. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 206–220. [Google Scholar] [PubMed]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Li, W.; Febbraio, M.; Espinola, R.G.; McCrae, K.R.; Cockrell, E.; Silverstein, R.L. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J. Clin. Investig. 2008. [Google Scholar] [CrossRef] [PubMed]
- Stuart, L.M.; Deng, J.; Silver, J.M.; Takahashi, K.; Tseng, A.A.; Hennessy, E.J.; Ezekowitz, R.A.B.; Moore, K.J. Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J. Cell Biol. 2005, 170, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Sharif, O.; Matt, U.; Saluzzo, S.; Lakovits, K.; Haslinger, I.; Furtner, T.; Doninger, B.; Knapp, S. The Scavenger Receptor CD36 Downmodulates the Early Inflammatory Response while Enhancing Bacterial Phagocytosis during Pneumococcal Pneumonia. J. Immunol. 2013, 190, 5640–5648. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.; Cockburn, I.A.; Swaim, A.M.; Thompson, L.E.; Tripathi, A.; Fletcher, C.A.; Shirk, E.M.; Sun, H.; Kowalska, M.A.; Fox-Talbot, K.; et al. Platelet Factor 4 Mediates Inflammation in Experimental Cerebral Malaria. Cell Host Microbe 2008, 4, 179–187. [Google Scholar] [CrossRef]
- Brill, A.; Yesilaltay, A.; De Meyer, S.F.; Kisucka, J.; Fuchs, T.A.; Kocher, O.; Krieger, M.; Wagner, D.D. Extrahepatic high-density lipoprotein receptor SR-BI and ApoA-I protect against deep vein thrombosis in mice. Arterioscler. Thromb. Vasc. Biol. 2012. [Google Scholar] [CrossRef]
- Trigatti, B.L.; Fuller, M. HDL signaling and protection against coronary artery atherosclerosis in mice. J. Biomed. Res. 2016, 30, 94–100. [Google Scholar]
- Areschoug, T.; Gordon, S. Scavenger receptors: Role in innate immunity and microbial pathogenesis. Cell. Microbiol. 2009, 11, 1160–1169. [Google Scholar] [CrossRef]
- Schäfer, G.; Guler, R.; Murray, G.; Brombacher, F.; Brown, G.D. The role of scavenger receptor B1 in infection with mycobacterium tuberculosis in a murine model. PLoS ONE 2009, 4, e8448. [Google Scholar] [CrossRef]
- Dempsey, L.A. Scavenger receptor B1 in lung immunity. Nat. Immunol. 2015, 16, 64. [Google Scholar] [CrossRef]
- Chen, M.; Kakutani, M.; Masaki, T.; Sawamura, T.; Chen, M.; Narumiya, S.; Naruko, T.; Ueda, M.; Sawamura, T. Activation-dependent surface expression of LOX-1 in human platelets. Biochem. Biophys. Res. Commun. 2001. [Google Scholar] [CrossRef] [PubMed]
- Marwali, M.R.; Hu, C.P.; Mohandas, B.; Dandapat, A.; Deonikar, P.; Chen, J.; Cawich, I.; Sawamura, T.; Kavdia, M.; Mehta, J.L. Modulation of ADP-induced platelet activation by aspirin and pravastatin: Role of lectin-like oxidized low-density lipoprotein receptor-1, nitric oxide, oxidative stress, and inside-out integrin signaling. J. Pharmacol. Exp. Ther. 2007, 322, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, T.; Kume, N.; Minami, M.; Hayashida, K.; Sawamura, T.; Kita, T.; Yonehara, S. LOX-1 Supports Adhesion of Gram-Positive and Gram-Negative Bacteria. J. Immunol. 2001, 166, 5108–5114. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhao, G.; Che, C.; Lin, J.; Li, N.; Hu, L.; Jiang, N.; Liu, Y. The role of LOX-1 in innate immunity to Aspergillus fumigatus in corneal epithelial cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3593–3603. [Google Scholar] [CrossRef]
- Ruggeri, Z.M. Platelet GPIb: Sensing force and responding. Blood 2015, 125, 423–424. [Google Scholar] [CrossRef]
- Clemetson, K.J.; Clemetson, J.M. Platelet GPIb-V-IX complex. Structure, function, physiology, and pathology. Semin. Thromb. Hemost. 1995, 21, 130–136. [Google Scholar] [CrossRef]
- Plummer, C.; Wu, H.; Kerrigan, S.W.; Meade, G.; Cox, D.; Douglas, C.W.I. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br. J. Haematol. 2005, 129, 101–109. [Google Scholar] [CrossRef]
- Bensing, B.A.; López, J.A.; Sullam, P.M. The Streptococcus gordonii surface proteins GspB and Hsa mediate binding to sialylated carbohydrate epitopes on the platelet membrane glycoprotein Ibα. Infect. Immun. 2004, 72, 6528–6537. [Google Scholar] [CrossRef]
- Byrne, M.F.; Kerrigan, S.W.; Corcoran, P.A.; Atherton, J.C.; Murray, F.E.; Fitzgerald, D.J.; Cox, D.M. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology 2003, 124, 1846–1854. [Google Scholar] [CrossRef]
- Claes, J.; Vanassche, T.; Peetermans, M.; Liesenborghs, L.; Vandenbriele, C.; Vanhoorelbeke, K.; Missiakas, D.; Schneewind, O.; Hoylaerts, M.F.; Heying, R.; et al. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor—Binding protein. Blood 2014, 124, 1669–1676. [Google Scholar] [CrossRef]
- Erpenbeck, L.; Nieswandt, B.; Schön, M.; Pozgajova, M.; Schön, M.P. Inhibition of platelet GPIbα and promotion of melanoma metastasis. J. Investig. Dermatol. 2010, 130, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, E.E. Proteolytic processing of platelet receptors. Res. Pract. Thromb. Haemost. 2018, 2, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Claushuis, T.A.M.; de Vos, A.F.; Nieswandt, B.; Boon, L.; Roelofs, J.J.T.H.; de Boer, O.J.; van’t Veer, C.; van der Poll, T. Platelet glycoprotein VI aids in local immunity during pneumonia-derived sepsis caused by gram-negative bacteria. Blood 2018, 131, 864–876. [Google Scholar] [CrossRef]
- Zahn, A.; Jennings, N.; Ouwehand, W.H.; Allain, J.P. Hepatitis C virus interacts with human platelet glycoprotein VI. J. Gen. Virol. 2006, 87, 2243–2251. [Google Scholar] [CrossRef]
- Pierre, S.; Linke, B.; Suo, J.; Tarighi, N.; Del Turco, D.; Thomas, D.; Ferreiros, N.; Stegner, D.; Frölich, S.; Sisignano, M.; et al. GPVI and Thromboxane Receptor on Platelets Promote Proinflammatory Macrophage Phenotypes during Cutaneous Inflammation. J. Investig. Dermatol. 2017, 137, 686–695. [Google Scholar] [CrossRef]
- Maurer, S.; Kropp, K.N.; Klein, G.; Steinle, A.; Haen, S.P.; Walz, J.S.; Hinterleitner, C.; Märklin, M.; Kopp, H.G.; Salih, H.R. Platelet-mediated shedding of NKG2D ligands impairs NK cell immune-surveillance of tumor cells. Oncoimmunology 2018, 7, e1364827. [Google Scholar] [CrossRef]
- Powers, M.E.; Becker, R.E.N.; Sailer, A.; Turner, J.R.; Bubeck Wardenburg, J. Synergistic Action of Staphylococcus aureus α-Toxin on Platelets and Myeloid Lineage Cells Contributes to Lethal Sepsis. Cell Host Microbe 2015, 17, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Colciaghi, F.; Borroni, B.; Pastorino, L.; Marcello, E.; Zimmermann, M.; Cattabeni, F.; Padovani, A.; Di Luca, M. α-secretase ADAM10 as well as αAPPs is reduced in platelets and CSF of Alzheimer disease patients. Mol. Med. 2002, 8, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, B.M.; Murray, J.L.; Li, G.; Sheng, J.; Hodge, T.W.; Rubin, D.H.; O’Brien, W.A.; Ferguson, M.R. A Functional Role for ADAM10 in Human Immunodeficiency Virus Type-1 Replication. Retrovirology 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- MacAuley, M.S.; Crocker, P.R.; Paulson, J.C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Netravali, I.A.; Cariappa, A.; Mattoo, H. Siglecs and Immune Regulation. Annu. Rev. Immunol. 2012, 30, 357–392. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.A.; Hamzeh-Cognasse, H.; Palle, S.; Anselme-Bertrand, I.; Arthaud, C.-A.; Chavarin, P.; Pozzetto, B.; Garraud, O.; Cognasse, F. Role of Siglec-7 in Apoptosis in Human Platelets. PLoS ONE 2014, 9, e106239. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.Y.; Yin, W.W.; Zhang, Q.F.; Liu, Q.; Peng, M.L.; Hu, H.D.; Hu, P.; Ren, H.; Zhang, D.Z. Siglec-7 Defines a Highly Functional Natural Killer Cell Subset and Inhibits Cell-Mediated Activities. Scand. J. Immunol. 2016, 84, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ma, X.; Su, D.; Zhang, Y.; Yu, L.; Jiang, F.; Zhou, X.; Feng, Y.; Ma, F. The Roles of Siglec7 and Siglec9 on Natural Killer Cells in Virus Infection and Tumour Progression. J. Immunol. Res. 2020, 2020, 6243819. [Google Scholar] [CrossRef]
- Herr, N.; Bode, C.; Duerschmied, D. The Effects of Serotonin in Immune Cells. Front. Cardiovasc. Med. 2017, 4, 48. [Google Scholar] [CrossRef]
- Masri, M.F.B.; Mantri, C.K.; Rathore, A.P.S.; St. John, A.L. Peripheral serotonin causes dengue virus-induced thrombocytopenia through 5HT2 receptors. Blood 2019, 133, 2325–2337. [Google Scholar] [CrossRef]
- Nicolson, P.L.R.; Nock, S.H.; Hinds, J.; Garcia-Quintanilla, L.; Smith, C.W.; Campos, J.; Brill, A.; Pike, J.A.; Khan, A.O.; Poulter, N.S.; et al. Low dose Btk inhibitors selectively block platelet activation by CLEC-2. Haematologica 2020. [Google Scholar] [CrossRef]
- Chen, B.; Vousden, K.A.; Naiman, B.; Turman, S.; Sun, H.; Wang, S.; Vinall, L.M.K.; Kemp, B.P.; Kasturiangan, S.; Rees, D.G.; et al. Humanised effector-null Fcγ3RIIA antibody inhibits immune complex-mediated proinflammatory responses. Ann. Rheum. Dis. 2019, 78, 228–237. [Google Scholar] [CrossRef]
- Daly, J.; Carlsten, M.; O’Dwyer, M. Sugar free: Novel immunotherapeutic approaches targeting siglecs and sialic acids to enhance natural killer cell cytotoxicity against cancer. Front. Immunol. 2019, 10, 1047. [Google Scholar] [CrossRef]
- Chu, S.; Zhu, X.; You, N.; Zhang, W.; Zheng, F.; Cai, B.; Zhou, T.; Wang, Y.; Sun, Q.; Yang, Z.; et al. The fab fragment of a human anti-Siglec-9 monoclonal antibody suppresses LPS-induced inflammatory responses in human macrophages. Front. Immunol. 2016, 7, 649. [Google Scholar] [CrossRef] [PubMed]
- Hashemzadeh, M.; Furukawa, M.; Goldsberry, S.; Movahed, M.R. Chemical structures and mode of action of intravenous glycoprotein IIb/IIIa receptor blockers: A review. Exp. Clin. Cardiol. 2008, 13, 192–197. [Google Scholar]
- Pustylnikov, S.; Dave, R.S.; Khan, Z.K.; Porkolab, V.; Rashad, A.A.; Hutchinson, M.; Fieschi, F.; Chaiken, I.; Jain, P. Short Communication: Inhibition of DC-SIGN-Mediated HIV-1 Infection by Complementary Actions of Dendritic Cell Receptor Antagonists and Env-Targeting Virus Inactivators. AIDS Res. Hum. Retrovir. 2016, 32, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Mangold, S.L.; Prost, L.R.; Kiessling, L.L. Quinoxalinone inhibitors of the lectin DC-SIGN. Chem. Sci. 2012, 3, 772–777. [Google Scholar] [CrossRef]
- Tsukiji, N.; Osada, M.; Sasaki, T.; Shirai, T.; Satoh, K.; Inoue, O.; Umetani, N.; Mochizuki, C.; Saito, T.; Kojima, S.; et al. Cobalt hematoporphyrin inhibits CLEC-2–podoplanin interaction, tumor metastasis, and arterial/venous thrombosis in mice. Blood Adv. 2018, 2, 2214–2225. [Google Scholar] [CrossRef]
- Nemerow, G.R.; Mullen, J.J.; Dickson, P.W.; Cooper, N.R. Soluble recombinant CR2 (CD21) inhibits Epstein-Barr virus infection. J. Virol. 1990, 64, 1348–1352. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |


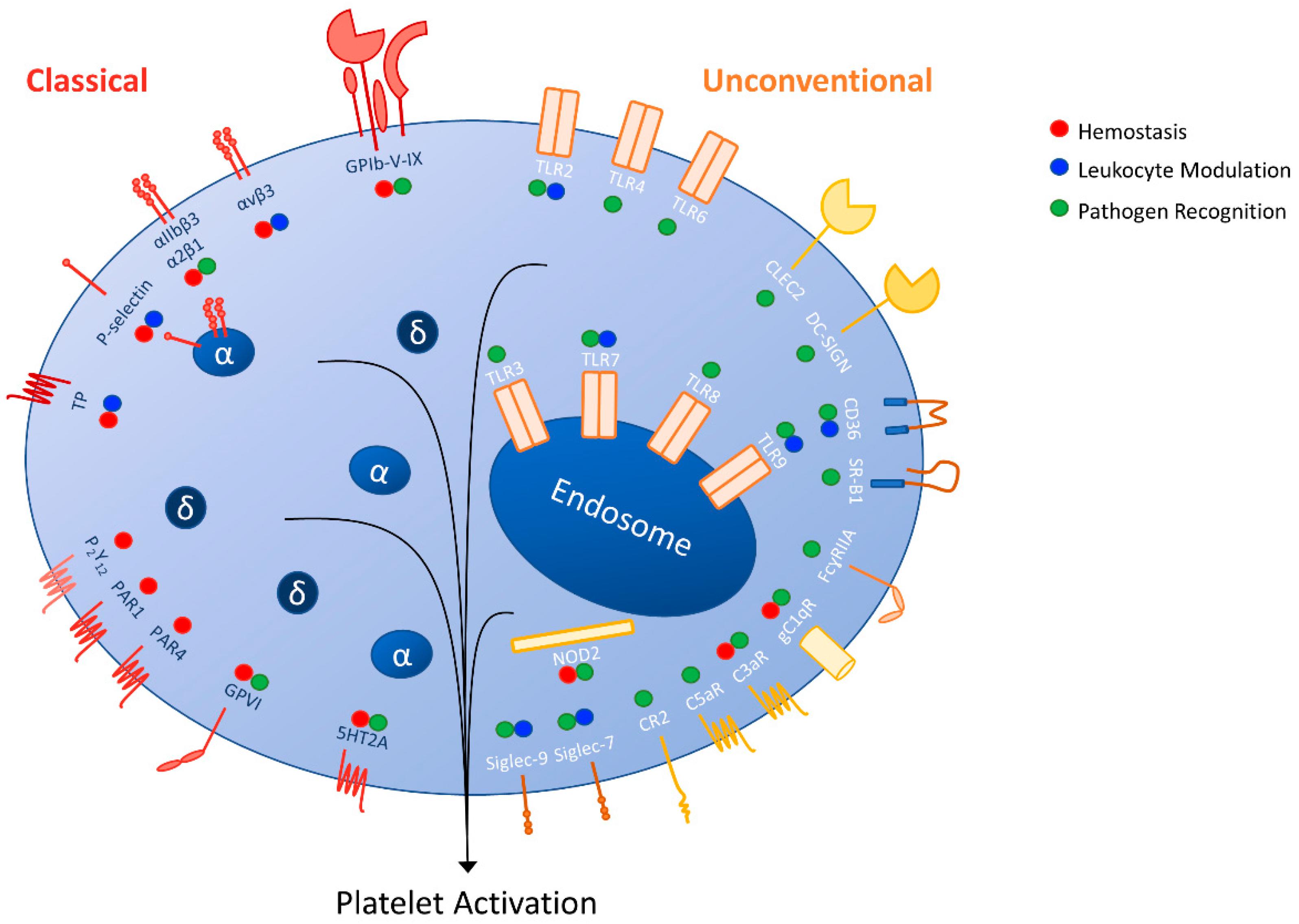{kind=link}
| Integrin Subtype | CD Marker | Ligands | Result | References |
|---|---|---|---|---|
| αIIbβ3 | CD41/61 | Collagen; Fibrinogen; DENV; Hantavirus; Fibrinogen binding proteins (FBPs) from bacteria; Streptococcus gordonii surface proteins | Platelet activation; Platelet aggregation; Virus recognition and internalization; Upregulation of thromboinflammation | [14,16,17,18,21,22,23,24,26] |
| αvβ3 | CD49e/CD61 | Vitronectin | Platelet activation Platelet aggregation Leukocyte migration Endothelial growth Angiogenesis Tumor metastasis Osteoclast bone resorption | [14,27,28] |
| TLRs | Agonists | Functions | References |
|---|---|---|---|
| TLR2 | Bacterial Lipoproteins; Pam3CSK4; HCMV; P. gingivalis | sCD40L upregulation and release; Platelet aggregation; Platelet activation; Platelet-neutrophil aggregates; Increased phagocytosis by neutrophils | [58,59,61,62,63,64,65,75] |
| TLR3 | Double stranded RNA; Poly I:C | P-selectin expression; Increased calcium concentration | [66] |
| TLR4 | Gram-negative bacterial LPS | Platelet activation; Platelet accumulation; sCD40L release; Increased oxidative phosphorylation capacity; ROS production; Infection-induced thrombocytopenia | [41,43,44,45,46,47,48,49] |
| TLR7 | Single stranded RNA | Increased association with granulocytes; Upregulation of NET formation | [67,69] |
| TLR9 | Synthetic unmethylated Type C CpG oligonucleotides; CAP adducts | Platelet activation and P-selectin expression; Platelet aggregation Platelet leukocyte aggregates | [70,76,77,78] |
| Platelet Receptors | Pathogen Recognized | Adverse Consequences Facilitated by Receptor Activation | Potential Receptor Targeting Therapeutics | References |
|---|---|---|---|---|
| αIIbβ3 | HIV DENV Hantavirus S. aureus E. coli | ITP; Virus induced thrombocytopenia; Altered endothelial cell properties; Platelet aggregation; Thromboinflammation | Abiciximab Eptifibatibe Tirofiban | [21,23,38,183] |
| DC-SIGN | HIV DENV | Viral entry and replication; NET formation | Dextran Isomaltooligosaccharides Quinoxalinones | [184,185] |
| CLEC-2 | DENV HIV Salmonella typhimurium | Viral entry and replication; Thrombosis | Cobalt hematoporphyrin Ibrutinib | [179,186] |
| FcγRIIA | S. aureus E. coli DENV | Sepsis associated thrombocytopenia HIT SLE APS Kawasaki disease | Anti-FcγRIIA antibodies (IV.3 & VIB9600) | [180] |
| CR2 | EBV | TGF-b release | Soluble recombinant CR2 proteins | [187] |
| Siglec 9 | Group B Streptococci | Resistance against platelet mediated killing | Anti-Siglec 9 antibodies | [182,183] |
| 5HT2A | DENV | Thrombocytopenia | Ketanserin | [178] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gautam, I.; Storad, Z.; Filipiak, L.; Huss, C.; Meikle, C.K.; Worth, R.G.; Wuescher, L.M. From Classical to Unconventional: The Immune Receptors Facilitating Platelet Responses to Infection and Inflammation. Biology 2020, 9, 343. https://doi.org/10.3390/biology9100343
Gautam I, Storad Z, Filipiak L, Huss C, Meikle CK, Worth RG, Wuescher LM. From Classical to Unconventional: The Immune Receptors Facilitating Platelet Responses to Infection and Inflammation. Biology. 2020; 9(10):343. https://doi.org/10.3390/biology9100343
Chicago/Turabian StyleGautam, Iluja, Zachary Storad, Louis Filipiak, Chadwick Huss, Claire K. Meikle, Randall G. Worth, and Leah M. Wuescher. 2020. "From Classical to Unconventional: The Immune Receptors Facilitating Platelet Responses to Infection and Inflammation" Biology 9, no. 10: 343. https://doi.org/10.3390/biology9100343
APA StyleGautam, I., Storad, Z., Filipiak, L., Huss, C., Meikle, C. K., Worth, R. G., & Wuescher, L. M. (2020). From Classical to Unconventional: The Immune Receptors Facilitating Platelet Responses to Infection and Inflammation. Biology, 9(10), 343. https://doi.org/10.3390/biology9100343