The Role of Autophagy in Pancreatic Cancer—Recent Advances

{kind=link}
Abstract
1. Introduction
2. Autophagy in Cancer
3. Conflicting Roles of Autophagy in PDAC
4. Recent Advances in the Understanding of the Control of Autophagy in PDAC
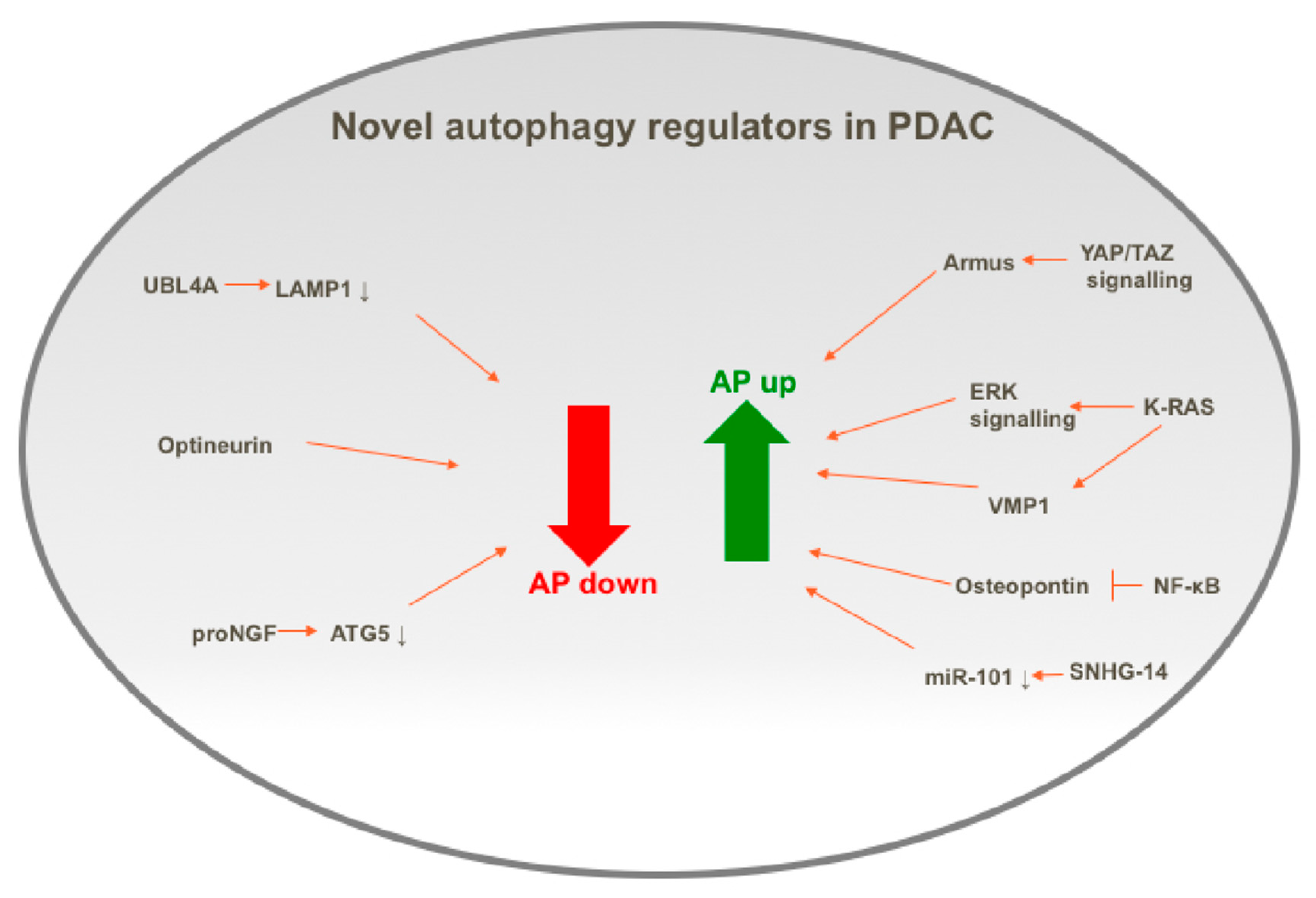
4.1. Positive Regulators of Autophagy in PDAC
4.1.1. KRAS
4.1.2. Osteopontin
4.1.3. SNHG14
4.2. Negative Regulators of Autophagy in PDAC
4.2.1. UBL4A
4.2.2. Optineurin
4.2.3. proNGF
5. Autophagy as a Therapeutic Target for Pancreatic Cancer
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef] [PubMed]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2019, 39, 517–560. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Molino, D.; Zemirli, N.; Codogno, P.; Morel, E. The Journey of the Autophagosome through Mammalian Cell Organelles and Membranes. J. Mol. Biol. 2017, 429, 497–514. [Google Scholar] [CrossRef]
- Judith, D.; Jefferies, H.B.; Boeing, S.; Frith, D.; Snijders, A.P.; Tooze, S.A. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIbeta. J. Cell Biol. 2019, 218, 1634–1652. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, P.; Wang, C.; Xin, B. SNHG14 enhances gemcitabine resistance by sponging miR-101 to stimulate cell autophagy in pancreatic cancer. Biochem. Biophys. Res. Commun. 2019, 510, 508–514. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Eng, C.H.; Wang, Z.; Tkach, D.; Toral-Barza, L.; Ugwonali, S.; Liu, S.; Fitzgerald, S.L.; George, E.; Frias, E.; Cochran, N.; et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl. Acad Sci. USA 2016, 113, 182–187. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, A.J.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients with Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, A.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 706. [Google Scholar] [CrossRef]
- Lazova, R.; Camp, R.L.; Klump, V.; Siddiqui, S.F.; Amaravadi, R.K.; Pawelek, J.M. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin. Cancer Res. 2012, 18, 370–379. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Liu, C. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef]
- Yu, Z.; Yu, Z.; Chen, Z.; Yang, L.; Ma, M.; Lu, S.; Wang, C.S.; Teng, C.B.; Nie, Y.Z. Zinc chelator TPEN induces pancreatic cancer cell death through causing oxidative stress and inhibiting cell autophagy. J. Cell. Physiol. 2019, 234, 20648–20661. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Van Den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Tittarelli, A.; Mendoza-Naranjo, A.; Farías, M.; Guerrero, I.; Ihara, F.; Wennerberg, E.; Riquelme, S.; Gleisner, A.; Kalergis, A.; Lundqvist, A.; et al. Gap junction intercellular communications regulate NK cell activation and modulate NK cytotoxic capacity. J. Immunol. 2014, 192, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Görgülü, K.; Diakopoulos, K.N.; Ai, J.; Schoeps, B.; Kabacaoglu, D.; Karpathaki, A.-F.; Kaya-Aksoy, E.; Ruess, D.A.; Berninger, A.; Kowalska, M.; et al. Levels of the Autophagy-Related 5 Protein Affect Progression and Metastasis of Pancreatic Tumors in Mice. Gastroenterology 2019, 156, 203–217.e20. [Google Scholar] [CrossRef]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef]
- Rausch, V.; Liu, L.; Apel, A.; Rettig, T.; Gladkich, J.; Labsch, S.; Kallifatidis, G.; Kaczorowski, A.; Groth, A.; Gross, W.; et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J. Pathol. 2012, 227, 325–335. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, D.; Zhang, L.; Xie, X.; Wu, Y.; Liu, Y.; Shao, G.; Su, Z. Upregulation of autophagy by hypoxia-inducible factor-1alpha promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol Rep. 2014, 32, 935–942. [Google Scholar] [CrossRef]
- Yang, M.C.; Wang, H.C.; Hou, Y.C.; Tung, H.L.; Chiu, T.J.; Shan, Y.S. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol. Cancer 2015, 14, 179. [Google Scholar] [CrossRef]
- Totaro, A.; Zhuang, Q.; Panciera, T.; Battilana, G.; Azzolin, L.; Brumana, G.; Gandin, A.; Brusatin, G.; Cordenonsi, M.; Piccolo, S. Cell phenotypic plasticity requires autophagic flux driven by YAP/TAZ mechanotransduction. Proc. Natl. Acad. Sci. USA 2019, 116, 17848–17857. [Google Scholar] [CrossRef]
- New, M.; Van Acker, T.; Sakamaki, J.I.; Jiang, M.; Saunders, R.E.; Long, J.; Wang, V.M.-Y.; Behrens, A.; Cerveira, J.; Sudhakar, P.; et al. MDH1 and MPP7 Regulate Autophagy in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 1884–1898. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019, 33, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, L.; Fagman, J.B.; Kim, J.Y.; Todoric, J.; Gukovsky, I.; Mackey, M.; Ellisman, M.H.; Karin, M. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6166–E6174. [Google Scholar] [CrossRef]
- Hausmann, S.; Kong, B.; Michalski, C.; Erkan, M.; Friess, H. The role of inflammation in pancreatic cancer. Adv. Exp. Med. Biol. 2014, 816, 129–151. [Google Scholar] [PubMed]
- Huang, C.; Wang, H.Y.; Wang, M.E.; Hsu, M.C.; Wu, Y.H.S.; Jiang, Y.F.; Wu, L.-S.; Jong, D.-S.; Chiu, C.-H. Kisspeptin-Activated Autophagy Independently Suppresses Non-Glucose-Stimulated Insulin Secretion from Pancreatic beta-Cells. Sci. Rep. 2019, 9, 17451. [Google Scholar] [CrossRef] [PubMed]
- New, M.; Van Acker, T.; Long, J.S.; Sakamaki, J.I.; Ryan, K.M.; Tooze, S.A. Molecular Pathways Controlling Autophagy in Pancreatic Cancer. Front Oncol. 2017, 7, 28. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, D.S.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Lee, C.S.; Lee, L.C.; Yuan, T.L.; Chakka, S.; Fellmann, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4508–4517. [Google Scholar] [CrossRef]
- Ré, A.E.L.; Fernández-Barrena, M.G.; Almada, L.L.; Mills, L.D.; Elsawa, S.F.; Lund, G.; Ropolo, A.; Molejon, M.I.; Vaccaro, M.I.; Fernandez-Zapico, M.E. Novel AKT1-GLI3-VMP1 pathway mediates KRAS oncogene-induced autophagy in cancer cells. J. Biol. Chem. 2012, 287, 25325–25334. [Google Scholar]
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell. Sci. 2014, 127 (Pt 18), 4089–4102. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.H.; Tian, C.; Meng, Y.; Qin, Y.W.; Du, Y.H.; Du, J.; Li, H.H. Osteopontin stimulates autophagy via integrin/CD44 and p38 MAPK signaling pathways in vascular smooth muscle cells. J. Cell Physiol. 2012, 227, 127–135. [Google Scholar] [CrossRef]
- Sun, T. Long noncoding RNAs act as regulators of autophagy in cancer. Pharmacol. Res. 2018, 129, 151–155. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Zhang, W.; Li, J.; Liu, W.; Lu, W. Long non-coding RNA SNHG14 exerts oncogenic functions in non-small cell lung cancer through acting as an miR-340 sponge. Biosci. Rep. 2019, 39, BSR20180941. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Wang, Y.; Ning, H.; Ren, F.; Zhang, Y.; Rong, Y.; Wang, Y.; Su, F.; Cai, F.; Jin, Z.; Li, Z.; et al. GdX/UBL4A specifically stabilizes the TC45/STAT3 association and promotes dephosphorylation of STAT3 to repress tumorigenesis. Mol. Cell. 2014, 53, 752–765. [Google Scholar] [CrossRef]
- Mariappan, M.; Li, X.; Stefanovic, S.; Sharma, A.; Mateja, A.; Keenan, R.J.; Hegde, R.S. A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature 2010, 466, 1120–1124. [Google Scholar] [CrossRef]
- Chen, H.; Li, L.; Hu, J.; Zhao, Z.; Ji, L.; Cheng, C.; Zhang, G.; Zhang, T.; Li, Y.; Chen, L.; et al. UBL4A inhibits autophagy-mediated proliferation and metastasis of pancreatic ductal adenocarcinoma via targeting LAMP1. J. Exp. Clin. Cancer Res. 2019, 38, 297. [Google Scholar] [CrossRef]
- Ali, D.M.; Ansari, S.S.; Zepp, M.; Knapp-Mohammady, M.; Berger, M.R. Optineurin downregulation induces endoplasmic reticulum stress, chaperone-mediated autophagy, and apoptosis in pancreatic cancer cells. Cell Death Discov. 2019, 5, 128. [Google Scholar] [CrossRef]
- Xu, J.; Song, J.; Yang, X.; Guo, J.; Wang, T.; Zhuo, W. ProNGF siRNA inhibits cell proliferation and invasion of pancreatic cancer cells and promotes anoikis. Biomed. Pharmacother. 2019, 111, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Elliott, I.A.; Dann, A.M.; Xu, S.; Kim, S.S.; Abt, E.R.; Kim, W.; Poddar, S.; Moore, A.; Zhou, L.; Williams, J.L.; et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 6842–6847. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; McKenna, W.G.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Kowalski, S.; Wyrzykowski, D.; Hac, S.; Rychlowski, M.; Radomski, M.W.; Inkielewicz-Stepniak, I. New Oxidovanadium(IV) Coordination Complex Containing 2-Methylnitrilotriacetate Ligands Induces Cell Cycle Arrest and Autophagy in Human Pancreatic Ductal Adenocarcinoma Cell Lines. Int. J. Mol. Sci. 2019, 20, 261. [Google Scholar] [CrossRef]
- Xue, N.; Lai, F.; Du, T.; Ji, M.; Liu, D.; Yan, C.; Zhang, S.; Yu, X.; Jin, J.; Chen, X.; et al. Chaperone-mediated autophagy degradation of IGF-1Rbeta induced by NVP-AUY922 in pancreatic cancer. Cell. Mol. Life Sci. 2019, 76, 3433–3447. [Google Scholar] [CrossRef]
- Brancaccio, M.; Russo, M.; Masullo, M.; Palumbo, A.; Russo, G.L.; Castellano, I. Sulfur-containing histidine compounds inhibit gamma-glutamyl transpeptidase activity in human cancer cells. J. Biol. Chem. 2019, 294, 14603–14614. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Ng, S.-C.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
New, M.; Tooze, S. The Role of Autophagy in Pancreatic Cancer—Recent Advances. Biology 2020, 9, 7. https://doi.org/10.3390/biology9010007
New M, Tooze S. The Role of Autophagy in Pancreatic Cancer—Recent Advances. Biology. 2020; 9(1):7. https://doi.org/10.3390/biology9010007
Chicago/Turabian StyleNew, Maria, and Sharon Tooze. 2020. "The Role of Autophagy in Pancreatic Cancer—Recent Advances" Biology 9, no. 1: 7. https://doi.org/10.3390/biology9010007
APA StyleNew, M., & Tooze, S. (2020). The Role of Autophagy in Pancreatic Cancer—Recent Advances. Biology, 9(1), 7. https://doi.org/10.3390/biology9010007