Autophagy in Cancer Cell Death



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Programmed Cell Death
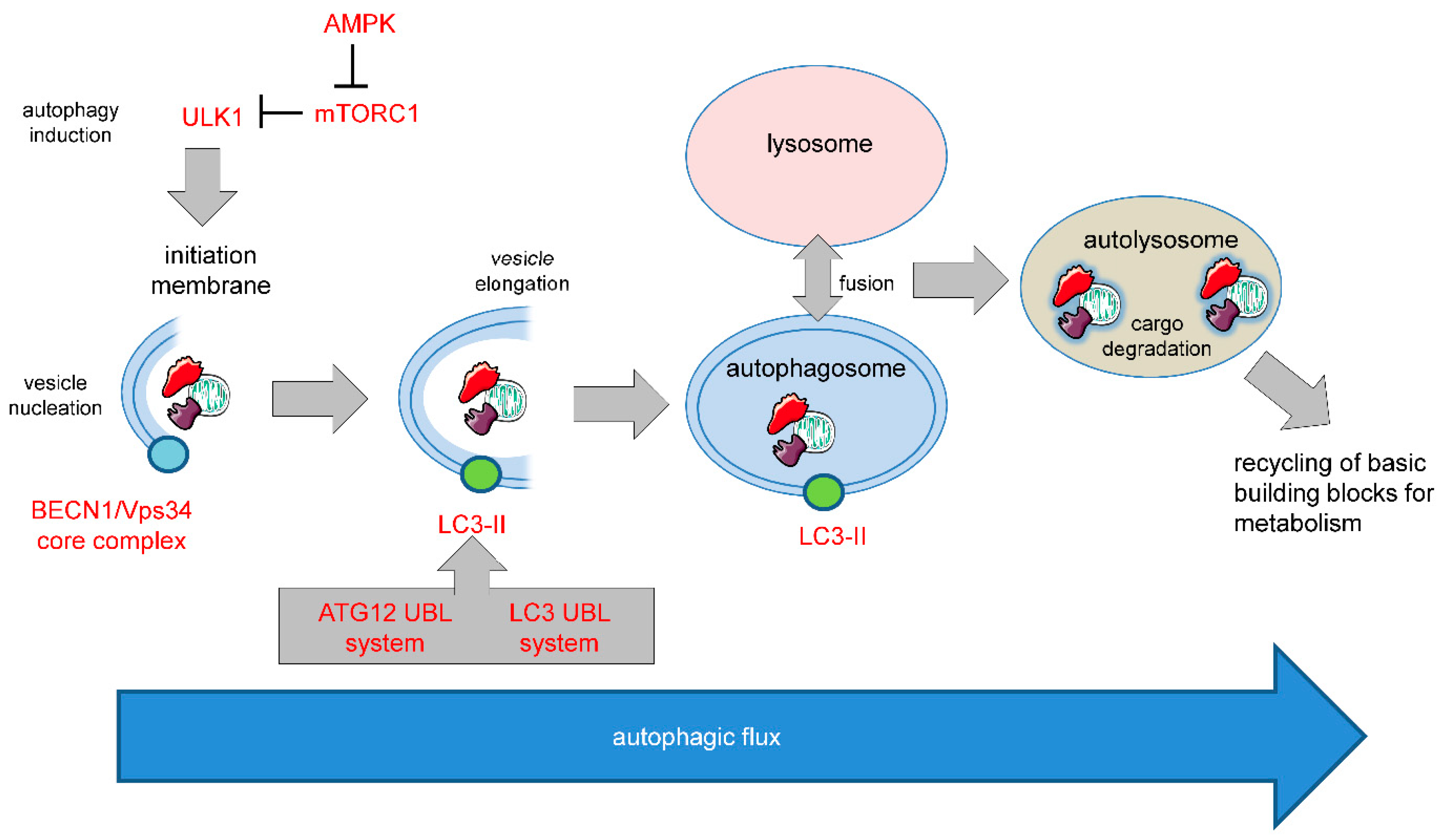
1.2. Autophagy in Cell Survival and Cell Death
2. Autophagy in Tumorigenesis and Tumor Progression
3. Autophagy in Therapy Response
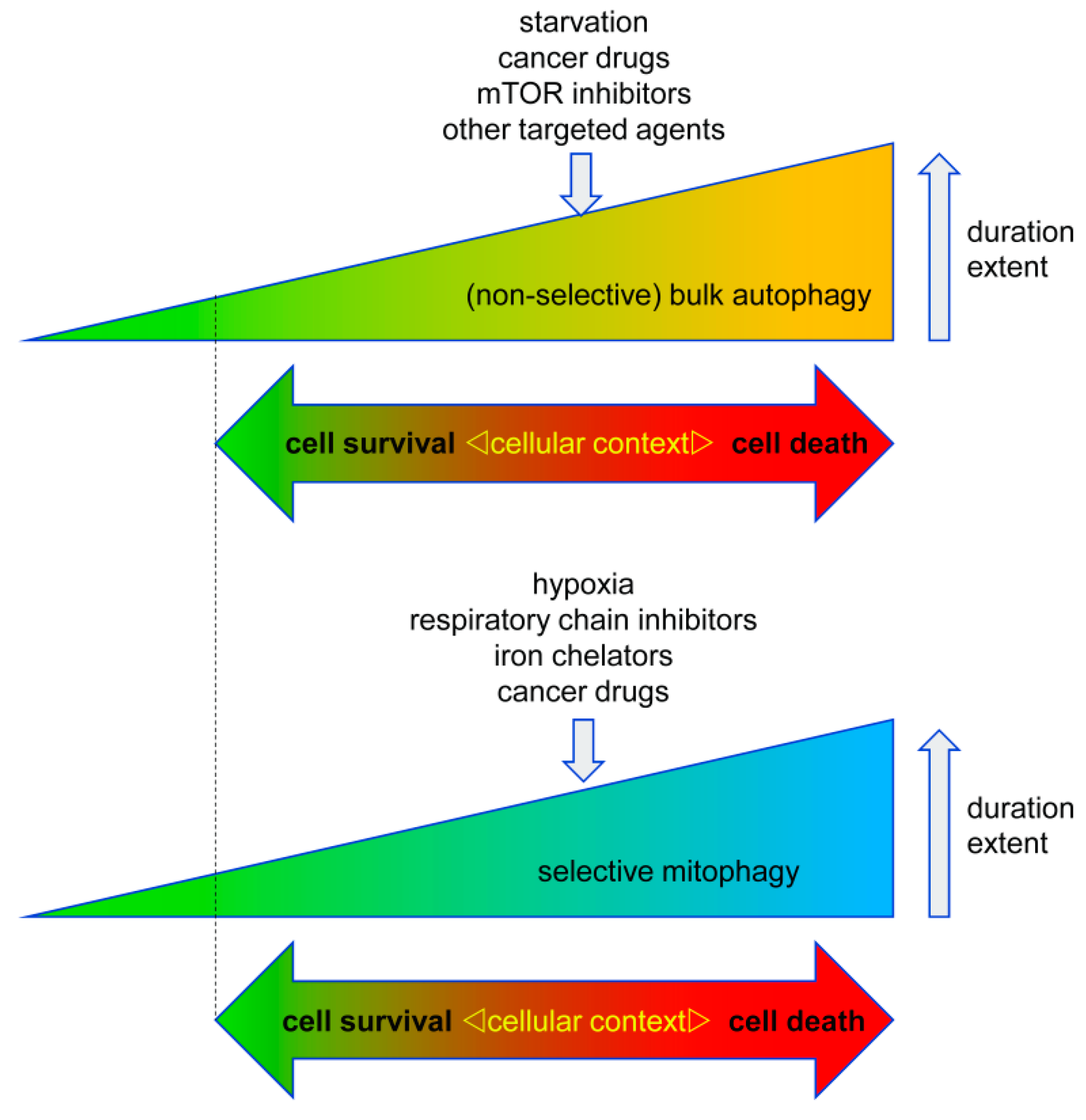
3.1. Pro-Survival Autophagy
3.2. Pro-Death Autophagy
3.2.1. Pro-Death Selective Autophagy
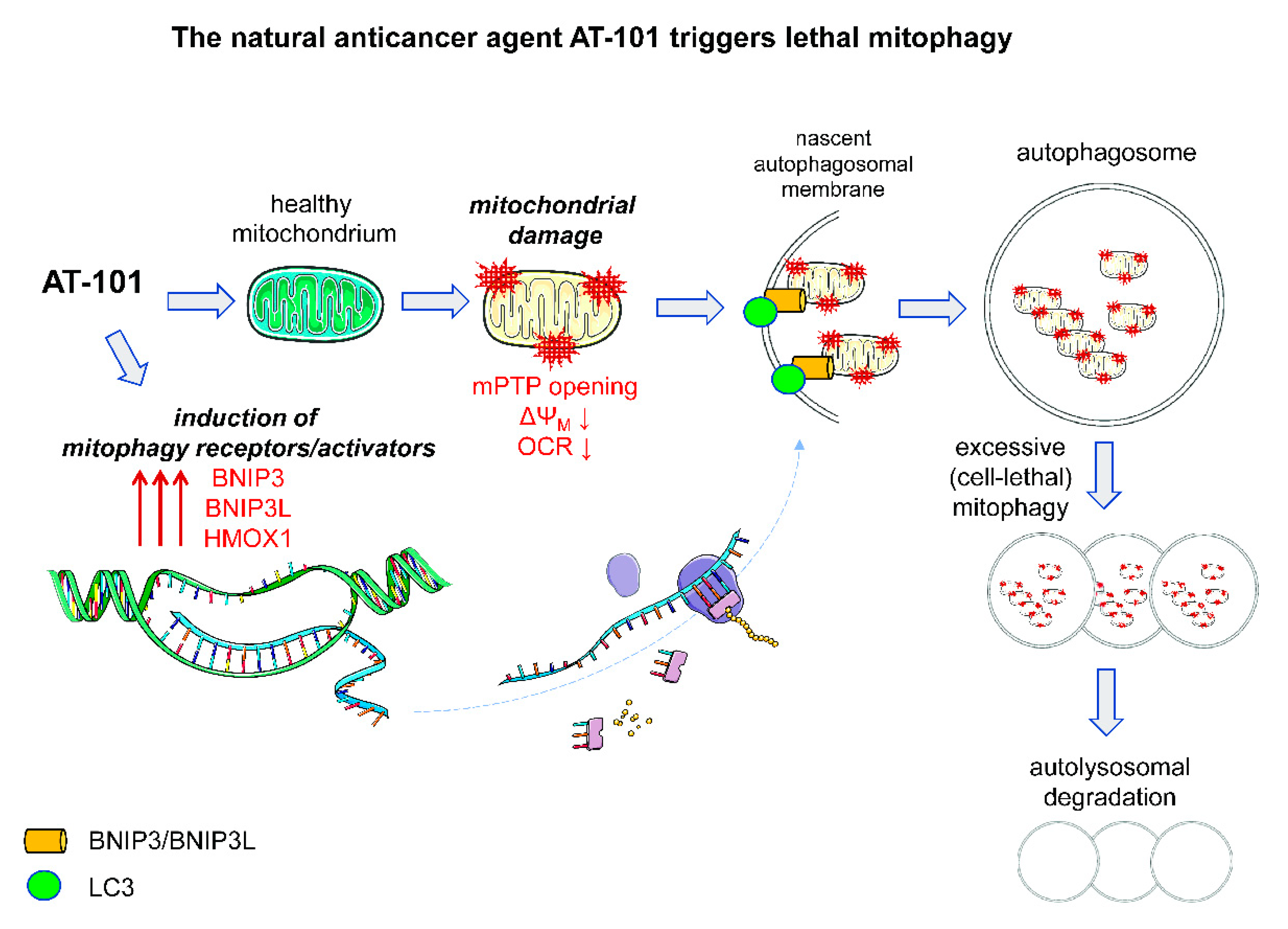
3.2.2. Pro-Death Mitophagy Triggered by Gossypol/AT-101
4. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Martin, S.J. Caspase-independent cell death. Nat. Med. 2005, 11, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell. Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Pinedo, C.; Martin, S.J. Autosis: A new addition to the cell death Tower of Babel. Cell Death Dis. 2014, 5, e1319. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1509–1518. [Google Scholar] [CrossRef]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef]
- Shen, H.M.; Codogno, P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy 2011, 7, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.L.; Baehrecke, E.H. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007, 131, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Garrido, J.; Carilla-Latorre, S.; Kubohara, Y.; Santos-Rodrigo, N.; Mesquita, A.; Soldati, T.; Golstein, P.; Escalante, R. Autophagy in Dictyostelium: Genes and pathways, cell death and infection. Autophagy 2010, 6, 686–701. [Google Scholar] [CrossRef] [PubMed]
- Samara, C.; Syntichaki, P.; Tavernarakis, N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008, 15, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, T.; Zhang, L.; Xiao, L.; Germano, I.M.; Kondo, Y.; Kondo, S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 2005, 24, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.X.; Zhang, Y.; Wu, M.L.; Liu, Y.N.; Zhang, P.; Chen, X.Y.; Kong, Q.Y.; Liu, J.; Li, H. Resveratrol and STAT inhibitor enhance autophagy in ovarian cancer cells. Cell Death Discov. 2016, 2, 15071. [Google Scholar] [CrossRef]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729. [Google Scholar] [CrossRef]
- Inbal, B.; Bialik, S.; Sabanay, I.; Shani, G.; Kimchi, A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 2002, 157, 455–468. [Google Scholar] [CrossRef]
- Segala, G.; David, M.; de Medina, P.; Poirot, M.C.; Serhan, N.; Vergez, F.; Mougel, A.; Saland, E.; Carayon, K.; Leignadier, J.; et al. Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nat. Commun. 2017, 8, 1903. [Google Scholar] [CrossRef]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef]
- Vanzo, R.; Bartkova, J.; Merchut-Maya, J.M.; Hall, A.; Bouchal, J.; Dyrskjot, L.; Frankel, L.B.; Gorgoulis, V.; Maya-Mendoza, A.; Jaattela, M.; et al. Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nassour, J.; Radford, R.; Correia, A.; Fuste, J.M.; Schoell, B.; Jauch, A.; Shaw, R.J.; Karlseder, J. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 2019, 565, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Levine, B. The autophagy effector Beclin 1: A novel BH3-only protein. Oncogene 2008, 27 (Suppl. 1), S137–S148. [Google Scholar] [CrossRef] [PubMed]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 706. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mule, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787. [Google Scholar] [CrossRef]
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhasz, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Monkkonen, T.; Debnath, J. Inflammatory signaling cascades and autophagy in cancer. Autophagy 2018, 14, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.P.; Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, C.K.; Mishra, R.; Patil, S.; Bhutia, S.K. Autophagy regulates cisplatin-induced stemness and chemoresistance via the upregulation of CD44, ABCB1 and ADAM17 in oral squamous cell carcinoma. Cell Prolif. 2018, 51, e12411. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.; Zhou, P.; Zhang, K. Autophagy plays an important role in stemness mediation and the novel dual function of EIG121 in both autophagy and stemness regulation of endometrial carcinoma JEC cells. Int. J. Oncol. 2017, 51, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Das, C.K.; Mandal, M.; Kogel, D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. 2018, 37, 749–766. [Google Scholar] [CrossRef]
- Notte, A.; Ninane, N.; Arnould, T.; Michiels, C. Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast cancer cells: Role of autophagy and JNK activation. Cell Death Dis. 2013, 4, e638. [Google Scholar] [CrossRef]
- Min, H.; Xu, M.; Chen, Z.R.; Zhou, J.D.; Huang, M.; Zheng, K.; Zou, X.P. Bortezomib induces protective autophagy through AMP-activated protein kinase activation in cultured pancreatic and colorectal cancer cells. Cancer Chemother. Pharmacol. 2014, 74, 167–176. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Towers, C.G.; Sullivan, K.D.; Andrysik, Z.; Hoh, M.; Ludwig, M.; O’Prey, J.; Ryan, K.M.; Espinosa, J.M.; Morgan, M.J.; et al. Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev. Cell 2018, 44, 555–565. [Google Scholar] [CrossRef]
- Gump, J.M.; Staskiewicz, L.; Morgan, M.J.; Bamberg, A.; Riches, D.W.; Thorburn, A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat. Cell Biol. 2014, 16, 47–54. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Sun, L.; Jin, Y.; An, Q.; Weng, C.; Zheng, J. Silencing of BAG3 promotes the sensitivity of ovarian cancer cells to cisplatin via inhibition of autophagy. Oncol. Rep 2017, 38, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVorkin, L.; Hattersley, M.; Kim, P.; Ries, J.; Spowart, J.; Anglesio, M.S.; Levi, S.M.; Huntsman, D.G.; Amaravadi, R.K.; Winkler, J.D.; et al. Autophagy Inhibition Enhances Sunitinib Efficacy in Clear Cell Ovarian Carcinoma. Mol. Cancer Res. 2017, 15, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Pelloski, C.E.; Ballman, K.V.; Furth, A.F.; Zhang, L.; Lin, E.; Sulman, E.P.; Bhat, K.; McDonald, J.M.; Yung, W.K.; Colman, H.; et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J. Clin. Oncol. 2007, 25, 2288–2294. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Jutten, B.; Keulers, T.G.; Peeters, H.J.M.; Schaaf, M.B.E.; Savelkouls, K.G.M.; Compter, I.; Clarijs, R.; Schijns, O.; Ackermans, L.; Teernstra, O.P.M.; et al. EGFRvIII expression triggers a metabolic dependency and therapeutic vulnerability sensitive to autophagy inhibition. Autophagy 2018, 14, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife 2017, 6, e19671. [Google Scholar] [CrossRef]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Sukumaran, P.; Singh, B.B. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol. Carcinog. 2016, 55, 818–831. [Google Scholar] [CrossRef]
- Fukuda, T.; Oda, K.; Wada-Hiraike, O.; Sone, K.; Inaba, K.; Ikeda, Y.; Makii, C.; Miyasaka, A.; Kashiyama, T.; Tanikawa, M.; et al. Autophagy inhibition augments resveratrol-induced apoptosis in Ishikawa endometrial cancer cells. Oncol. Lett. 2016, 12, 2560–2566. [Google Scholar] [CrossRef] [Green Version]
- Dasari, S.K.; Bialik, S.; Levin-Zaidman, S.; Levin-Salomon, V.; Merrill, A.H., Jr.; Futerman, A.H.; Kimchi, A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017, 24, 1288–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F.; et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 2012, 120, 3519–3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginet, V.; Puyal, J.; Rummel, C.; Aubry, D.; Breton, C.; Cloux, A.J.; Majjigapu, S.R.; Sordat, B.; Vogel, P.; Bruzzone, S.; et al. A critical role of autophagy in antileukemia/lymphoma effects of APO866, an inhibitor of NAD biosynthesis. Autophagy 2014, 10, 603–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, S.; Takabe, K.; Newton, J.; Saurabh, K.; Young, M.M.; Leopoldino, A.M.; Hait, N.C.; Roberts, J.L.; Wang, H.G.; Dent, P.; et al. TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy 2018, 14, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Linder, B.; Wehle, A.; Hehlgans, S.; Bonn, F.; Dikic, I.; Rodel, F.; Seifert, V.; Kogel, D. Arsenic Trioxide and (-)-Gossypol Synergistically Target Glioma Stem-Like Cells via Inhibition of Hedgehog and Notch Signaling. Cancers 2019, 11, 350. [Google Scholar] [CrossRef]
- Li, C.L.; Wei, H.L.; Chen, J.; Wang, B.; Xie, B.; Fan, L.L.; Li, L.J. Arsenic trioxide induces autophagy and antitumor effects in Burkitt’s lymphoma Raji cells. Oncol. Rep. 2014, 32, 1557–1563. [Google Scholar] [CrossRef]
- Goussetis, D.J.; Altman, J.K.; Glaser, H.; McNeer, J.L.; Tallman, M.S.; Platanias, L.C. Autophagy is a critical mechanism for the induction of the antileukemic effects of arsenic trioxide. J. Biol. Chem. 2010, 285, 29989–29997. [Google Scholar] [CrossRef]
- Wang, H.; Cao, F.; Li, J.; Li, L.; Li, Y.; Shi, C.; Lan, W.; Li, D.; Zhao, H.; Zhang, Y.; et al. Arsenic trioxide and mannitol for the treatment of acute promyelocytic leukemia relapse in the central nervous system. Blood 2014, 124, 1998–2000. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef]
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 2015, 1853(Pt. B), 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Cordani, M.; Somoza, A. Targeting autophagy using metallic nanoparticles: A promising strategy for cancer treatment. Cell. Mol. Life Sci. 2019, 76, 1215–1242. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Wang, Y.; Chen, H.Z.; Xing, Y.Z.; Li, F.W.; Zhang, Q.; Zhou, B.; Zhang, H.K.; Zhang, J.; Bian, X.L.; et al. Orphan nuclear receptor TR3 acts in autophagic cell death via mitochondrial signaling pathway. Nat. Chem. Biol. 2014, 10, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Dany, M.; Gencer, S.; Nganga, R.; Thomas, R.J.; Oleinik, N.; Baron, K.D.; Szulc, Z.M.; Ruvolo, P.; Kornblau, S.; Andreeff, M.; et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 2016, 128, 1944–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.H.; Choi, K.S. A critical role of superoxide anion in selenite-induced mitophagic cell death. Autophagy 2008, 4, 76–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.H.; Sohn, S.; Kwon, H.J.; Kim, S.U.; Kim, M.J.; Lee, S.J.; Choi, K.S. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007, 67, 6314–6324. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, E.M. Gossypol: A contraceptive for men. Contraception 2002, 65, 259–263. [Google Scholar] [CrossRef]
- Ren, T.; Shan, J.; Li, M.; Qing, Y.; Qian, C.; Wang, G.; Li, Q.; Lu, G.; Li, C.; Peng, Y.; et al. Small-molecule BH3 mimetic and pan-Bcl-2 inhibitor AT-101 enhances the antitumor efficacy of cisplatin through inhibition of APE1 repair and redox activity in non-small-cell lung cancer. Drug Des. Dev. Ther. 2015, 9, 2887–2910. [Google Scholar]
- Kaza, N.; Kohli, L.; Graham, C.D.; Klocke, B.J.; Carroll, S.L.; Roth, K.A. BNIP3 regulates AT101 [(-)-gossypol] induced death in malignant peripheral nerve sheath tumor cells. PLoS ONE 2014, 9, e96733. [Google Scholar] [CrossRef]
- Antonietti, P.; Gessler, F.; Dussmann, H.; Reimertz, C.; Mittelbronn, M.; Prehn, J.H.; Kogel, D. AT-101 simultaneously triggers apoptosis and a cytoprotective type of autophagy irrespective of expression levels and the subcellular localization of Bcl-xL and Bcl-2 in MCF7 cells. Biochim. Biophys. Acta 2016, 1863, 499–509. [Google Scholar] [CrossRef]
- Lan, L.; Appelman, C.; Smith, A.R.; Yu, J.; Larsen, S.; Marquez, R.T.; Liu, H.; Wu, X.; Gao, P.; Roy, A.; et al. Natural product (-)-gossypol inhibits colon cancer cell growth by targeting RNA-binding protein Musashi-1. Mol. Oncol. 2015, 9, 1406–1420. [Google Scholar] [CrossRef]
- Voss, V.; Senft, C.; Lang, V.; Ronellenfitsch, M.W.; Steinbach, J.P.; Seifert, V.; Kogel, D. The pan-Bcl-2 inhibitor (-)-gossypol triggers autophagic cell death in malignant glioma. Mol. Cancer Res. 2010, 8, 1002–1016. [Google Scholar] [CrossRef]
- Wang, J.; Peng, Y.; Liu, Y.; Yang, J.; Huang, M.; Tan, W. AT-101 inhibits hedgehog pathway activity and cancer growth. Cancer Chemother. Pharmacol. 2015, 76, 461–469. [Google Scholar] [CrossRef]
- Meng, Y.; Tang, W.; Dai, Y.; Wu, X.; Liu, M.; Ji, Q.; Ji, M.; Pienta, K.; Lawrence, T.; Xu, L. Natural BH3 mimetic (-)-gossypol chemosensitizes human prostate cancer via Bcl-xL inhibition accompanied by increase of Puma and Noxa. Mol. Cancer Ther. 2008, 7, 2192–2202. [Google Scholar] [CrossRef] [Green Version]
- Paoluzzi, L.; Gonen, M.; Gardner, J.R.; Mastrella, J.; Yang, D.; Holmlund, J.; Sorensen, M.; Leopold, L.; Manova, K.; Marcucci, G.; et al. Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood 2008, 111, 5350–5358. [Google Scholar] [CrossRef]
- Wolter, K.G.; Wang, S.J.; Henson, B.S.; Wang, S.; Griffith, K.A.; Kumar, B.; Chen, J.; Carey, T.E.; Bradford, C.R.; D’Silva, N.J. (-)-gossypol inhibits growth and promotes apoptosis of human head and neck squamous cell carcinoma in vivo. Neoplasia 2006, 8, 163–172. [Google Scholar] [CrossRef]
- Balakrishnan, K.; Wierda, W.G.; Keating, M.J.; Gandhi, V. Gossypol, a BH3 mimetic, induces apoptosis in chronic lymphocytic leukemia cells. Blood 2008, 112, 1971–1980. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.D.; Li, L.Y.; Li, P.H.; You, T.; Wang, F.H.; Sun, W.J.; Zheng, Z.Q. Gossypol induces cell death by activating apoptosis and autophagy in HT-29 cells. Mol. Med. Rep. 2017, 16, 2128–2132. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Wu, X.; He, F.; Karnak, D.; Tang, W.; Meng, Y.; Xiang, D.; Ji, M.; Lawrence, T.S.; Xu, L. A natural BH3 mimetic induces autophagy in apoptosis-resistant prostate cancer via modulating Bcl-2-Beclin1 interaction at endoplasmic reticulum. Cell Death Differ. 2011, 18, 60–71. [Google Scholar] [CrossRef]
- Meyer, N.; Zielke, S.; Michaelis, J.B.; Linder, B.; Warnsmann, V.; Rakel, S.; Osiewacz, H.D.; Fulda, S.; Mittelbronn, M.; Munch, C.; et al. AT 101 induces early mitochondrial dysfunction and HMOX1 (heme oxygenase 1) to trigger mitophagic cell death in glioma cells. Autophagy 2018, 14, 1693–1709. [Google Scholar] [CrossRef] [PubMed]
- Warnsmann, V.; Meyer, N.; Hamann, A.; Kogel, D.; Osiewacz, H.D. A novel role of the mitochondrial permeability transition pore in (-)-gossypol-induced mitochondrial dysfunction. Mech. Ageing Dev. 2018, 170, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Constantin, M.; Choi, A.J.; Cloonan, S.M.; Ryter, S.W. Therapeutic potential of heme oxygenase-1/carbon monoxide in lung disease. Int. J. Hypertens. 2012, 2012, 859235. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [Green Version]
- Hull, T.D.; Boddu, R.; Guo, L.; Tisher, C.C.; Traylor, A.M.; Patel, B.; Joseph, R.; Prabhu, S.D.; Suliman, H.B.; Piantadosi, C.A.; et al. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 2016, 1, e85817. [Google Scholar] [CrossRef]
- Carchman, E.H.; Rao, J.; Loughran, P.A.; Rosengart, M.R.; Zuckerbraun, B.S. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology 2011, 53, 2053–2062. [Google Scholar] [CrossRef]
- Suliman, H.B.; Keenan, J.E.; Piantadosi, C.A. Mitochondrial quality-control dysregulation in conditional HO-1(-/-) mice. JCI Insight 2017, 2, e89676. [Google Scholar] [CrossRef]
- Zukor, H.; Song, W.; Liberman, A.; Mui, J.; Vali, H.; Fillebeen, C.; Pantopoulos, K.; Wu, T.D.; Guerquin-Kern, J.L.; Schipper, H.M. HO-1-mediated macroautophagy: A mechanism for unregulated iron deposition in aging and degenerating neural tissues. J. Neurochem. 2009, 109, 776–791. [Google Scholar] [CrossRef]
- Bansal, S.; Biswas, G.; Avadhani, N.G. Mitochondria-targeted heme oxygenase-1 induces oxidative stress and mitochondrial dysfunction in macrophages, kidney fibroblasts and in chronic alcohol hepatotoxicity. Redox Biol. 2014, 2, 273–283. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linder, B.; Kögel, D. Autophagy in Cancer Cell Death. Biology 2019, 8, 82. https://doi.org/10.3390/biology8040082
Linder B, Kögel D. Autophagy in Cancer Cell Death. Biology. 2019; 8(4):82. https://doi.org/10.3390/biology8040082
Chicago/Turabian StyleLinder, Benedikt, and Donat Kögel. 2019. "Autophagy in Cancer Cell Death" Biology 8, no. 4: 82. https://doi.org/10.3390/biology8040082
APA StyleLinder, B., & Kögel, D. (2019). Autophagy in Cancer Cell Death. Biology, 8(4), 82. https://doi.org/10.3390/biology8040082