Abstract
Mitochondrial function declines with age, leading to a variety of age-related diseases (metabolic, central nervous system-related, cancer, etc.) and medication usage increases with age due to the increase in diseases. Drug-induced mitochondrial toxicity has been described for many different drug classes and can lead to liver, muscle, kidney and central nervous system injury and, in rare cases, to death. Many of the most prescribed medications in the geriatric population carry mitochondrial liabilities. We have demonstrated that, over the past decade, each class of drugs that demonstrated mitochondrial toxicity contained drugs with both more and less adverse effects on mitochondria. As patient treatment is often essential, we suggest using medication(s) with the best safety profile and the avoidance of concurrent usage of multiple medications that carry mitochondrial liabilities. In addition, we also recommend lifestyle changes to further improve one’s mitochondrial function, such as weight loss, exercise and nutrition.
1. Introduction
Known as the powerhouse of the cell, mitochondria are recognized for their ability to produce large amounts of a cell’s energy currency, ATP (Adenosine Triphosphate), as well as being the only other organelle that contains DNA apart from the nucleus. Virtually all eukaryotic cells employ mitochondria to produce the majority of ATP needed to perform cellular functions (e.g., replication, protein synthesis, muscle contraction, etc.) and maintain cellular viability. The electron transport chain (ETC) and ATP synthase are interdependent systems in the mitochondria that generate ATP by coupling the electrochemical potential across the inner mitochondrial membrane created by the ETC to the phosphorylation of ADP by ATP synthase. The protein complexes comprising the ETC shuttle electrons down an energy gradient to ultimately reduce molecular oxygen to water, driving oxidative phosphorylation for ATP synthesis. Compromising the function of any of these protein complexes reduces the efficiency of ATP production and endangers the cell’s ability to perform its function and can lead to cell death. Additionally, uncoupling the membrane potential from ATP synthesis through the loss of the inner mitochondrial membrane impermeability dissipates the harvested energy as heat and bypasses ATP synthesis and greatly impairs the cell’s viability. Mitochondria contain their own DNA (mtDNA) and both inherited and acquired mutations in mtDNA have been linked to well over 100 mitochondrial syndromes such as Leber’s hereditary optic neuropathy, mitochondrial myopathy, encephalomyopathy, lactic acidosis, stroke-like symptoms and myoclonic epilepsy with ragged red fibers [1]. MtDNA is susceptible to oxidative stress due to its proximity to the ETC and the fact that it does not contain histones or a repair mechanism.
As we age, mitochondrial function declines and it is described as the “mitochondrial theory of aging”, leading to a variety of age-related diseases (Figure 1).
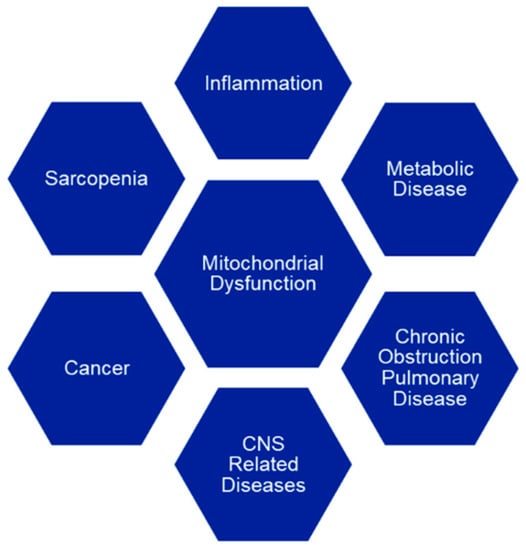
Figure 1.
Mitochondrial dysfunction is implicated in many age-related diseases such as metabolic diseases (T2DM, obesity, cardiovascular and cerebrovascular disease, Non-Alcoholic Fatty Liver Disease (NAFDL)) [2,3,4,5,6,7], CNS-related diseases (Parkinson’s, Alzheimer’s and Huntington’s disease, hearing loss, cataracts) [8,9,10,11], inflammation (osteoarthritis) [12], cancer [13,14], sarcopenia [15] and chronic obstructive pulmonary disease (COPD) [16].
These age-related diseases are often treated with multiple medications, some of which are known to cause drug-induced mitochondrial toxicity. In this review, we postulate that drug-induced mitochondrial dysfunction can increase in frequency and severity in the geriatric population due to two independent factors associated with aging, compromised mitochondrial function and polypharmacy.
2. The Mitochondrial Theory of Aging
The “Mitochondrial Theory of Aging” has gained considerable prominence in discussions of longevity, inherent vitality and the biology of mortality. First proposed in his book published in 1928, Raymond Pearl provided a lengthy account of the evidence leading him to posit that longevity is inversely proportional to “the rate of living”; by that, he is referring to the expenditure of energy. His theory is that one is born with a finite quantum of energy, the expenditure of which defines his or her mortality [17]. Although the tenants of this association may hold well for drosophila and perhaps other poikilotherms, in mammals this theory is essentially the antithesis for vitality. There is an abundance of evidence, much of which is current and clinical, demonstrating that energy expenditure is actually vital to delaying mortality and extending one’s longevity [18,19,20]. This is all summarized in the pronouncements that exercise and diet are fundamental to a long and healthy life, perhaps delaying the natural biological regression that is associated with aging.
It is a well-established fact that mitochondrial numbers, function and bioenergetic capacity decline progressively with age [21,22]; Harman summarized this when he coined the term “Mitochondrial Clock” in 1972 [23], which is rooted in his seminal thesis dating from 1956 on the free radical theory of aging [24,25]. This theory is based on the demonstrated accumulation of oxidative damage to lipid, protein, and nucleic acids in aged tissues [26], which Harman attributed to the increased free radical generation observed with aging. The thought is that as biochemical efficiency and physiological function secede with age, the rate of free radical generation increases, leading to increased levels of oxidative damage. This line of thinking continues by prospecting that increased oxidative damage to the ETC within the mitochondrial compartment leads to a progressive loss of coupling efficiency and increased rates of free radical liberation from the electron transport chain. Accordingly, rather than being the consequence of aging, mitochondria are also implicated as the progenitor of the aging process [25]. It becomes quite apparent that the “Mitochondrial Theory of Aging” and the “Free Radical Theory of Aging” are closely intertwined, and it is subject to debate as to which is the cause and which is the consequence of aging (the chicken vs. egg conundrum).
Regardless of whether it is the cause or consequence, hallmarks of mitochondrial aging [19] include a decrease in the number and an increase in the size of mitochondria [27], a net decrease in the cellular mitochondrial membrane potential, a decreased ADP:O ratio (oxidative phosphorylation) and respiratory control index (RCI), and a loss of individual ETC enzyme activities [21,22]; the steady-state [ATP] concentration, however, tends to be unaffected.
Although each of these factors can individually contribute to the progressive loss of mitochondrial function with age, it is far more plausible that they reflect a much broader and concerted decline in homeostatic regulation directed from a molecular level [28]. Molecular hallmarks associated with mitochondrial aging include a decreased mtDNA copy number [29] accompanied by the accumulation of mutations to the mitochondrial genome [30,31], reduced abundance of both nuclear and mitochondrial encoded mRNA [22,32], decreased mitochondrial protein synthesis [33,34,35,36], and the dysregulation of the homeostatic balance between mitochondrial biogenesis, fission and mitophagy [19,28].
Ristow and Zarse [20] were first to apply the term “mitohormesis” to conceptualize the loss of the adaptive response of mitochondria by means of regulating mitochondrial homeostasis, contributing to cell and organismal senescence. This has been followed by a number of reviews suggesting that the age-related accumulation of defective mitochondria, accompanied by the loss of ability to clear these from the cell by way of mitophagy and replace them with healthy mitochondria through mitochondrial fission and biogenesis, underpins the mitochondrial theory of aging [19,28,37,38,39]. This loss of hormesis or mitochondrial homeostasis is equated to a progressive decline in the processes devoted to quality control (QCmt) to sustain mitochondrial fidelity and capacity within the aging cell [19,28,40].
Two major surveillance mechanisms exist within the cell for sensing dysfunctional mitochondria: (1) bioenergetic or nutrient signaling and (2) mitochondrial proteostasis [19,28]. Nutrient sensing is largely by way of activating AMP kinase (AMPK), SIRT1, and the sestrins for a negative nutrient balance, and mTOR for nutrient surplus (Figure 2).
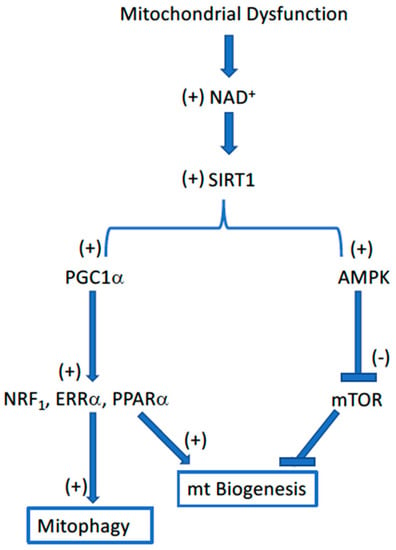
Figure 2.
Flow diagram illustrating the interrelationships governing mitochondrial homeostasis in response to the loss of mitochondrial function, such as that which occurs with aging. The Sirt1-dependent regulation of both PGC1α and AMPK provides a well-controlled integration of the disposal of dysfunctional mitochondria (mitophagy) and their replacement with new, supposedly fully functional, mitochondria (biogenesis).
Pyridine nucleotide redox status (NAD+/NADH) is a key factor linking the nutritional status of the cell to both free radical generation and mitochondrial homeostasis. Nutritional deficiency (i.e., mitochondrial dysfunction) is associated with a high NAD+/NADH ratio and low rates of free radical generation. Under these same conditions, the accumulation of high [NAD+] is also associated with high rates of mitochondrial proliferation (biogenesis) and disposal of defective mitochondria (mitophagy), both of which occur by way of the NAD+-dependent activation of SIRT1. The activation of SIRT1 by NAD+ stimulates mitochondrial biogenesis, both by stimulating the PGC1α-dependent activation of mitochondrial biogenesis combined with the AMPK-dependent inhibition of the negative regulator mTOR. The second surveillance pathway, mitochondrial proteostasis, actuates the unfolded protein response (UPRmt) indigenous to mitochondria, where AMPK and mTOR are competing regulators. Collectively, these nutrient sensor pathways regulate both the disposal of defective mitochondria and their replenishment with supposedly fully functional mitochondria.
There is growing evidence that these various mitochondrial signaling pathways are compromised with aging. It may be as simple as the age-associated decrease of [NAD+] [41,42] that is responsible for the lower rates of both mitochondrial biogenesis and mitophagy in aged tissues. Likewise, AMPK and the sirtuins are down-regulated with aging [43,44,45] as are the concentrations of PGC1α and the rate of mitophagic elimination of dysfunctional mitochondria [46]. These changes suggest loss of molecular mechanisms regulating quality control as being responsible for the ineffective nutrient sensing of mitochondria in aging tissues.
Regardless of the cause, the bioenergetic phenotype of mitochondria from most tissues of aged individuals is very different from that of young adults, which may be a significant factor accounting for the higher rates of drug-induced adverse events in geriatric populations. There is growing appreciation of the possibility that the decline in molecular regulation of mitochondrial hormesis is the primary factor that impedes the ability of aging mitochondria to withstand or adapt to external inputs [26,28,47]. It may be this loss of homeostatic regulation that underlies the sometimes-greater susceptibility of elderly patients to drug-induced adverse events.
3. Drug-Induced Mitochondrial Toxicity
Drug-induced mitochondrial toxicity has been studied in academic settings for well over 50 years. Indeed, many such studies relied on xenobiotic inhibitors, such as rotenone, antimycin and oligomycin, to deduce the function of the ETC, and uncouplers such as 2,4 dinitrophenol, to understand how mitochondria generate ATP. As a result, we should not be surprised that pharmaceutical xenobiotics intended to be therapeutics could also have deleterious ‘side effects’ on mitochondrial function. Drugs can inhibit mitochondrial function in many different ways such as through the inhibition of ETC protein complexes, inhibition of ATP synthase, inhibition of enzymes of the citric acid cycle, inhibition of various mitochondrial transporters, inhibition of the mitochondrial transcription and translational machinery, as well as through the uncoupling of the ETC from ATP synthase. However, most of these side effects were not detected in preclinical animal studies. This is due to the fact that in vivo toxicity studies are usually done in drug-naïve, young adult animals that have robust mitochondrial reserves; lack of sufficient genetic diversity to allow for idiosyncratic responses; absence of environmental factors; co-medication; insensitivity of histopathology for revealing mitochondrial failure. Much progress has been made in the past decade to develop a variety of high-throughput applicable organelle based and in vitro cell models preclinically [48]. Whereas, traditionally, cells were cultured in high glucose and were unresponsive to mitochondrial toxicity due to shifting to glycolysis for energy production, recently developed cell models grown in galactose can correctly identify potential drug-induced mitochondrial toxicity with greater sensitivity [49,50]. Drugs can also now be tested for potentially causing mitochondrial toxicity in 96- and 384-well formats using solid and soluble oxygen sensors [51,52].
Drug-induced mitochondrial toxicity has been recognized to cause organ toxicity to the liver, skeletal muscle, kidney, heart and the central nervous system. Drug classes identified to cause mitochondrial toxicity are anti-diabetic drugs (thiazolidinediones, fibrates, biguanides), cholesterol lowering drugs (statins), anti-depressants (SARIs), pain medications (NSAIDs), certain antibiotics (fluroquinolones, macrolide), and anti-cancer drugs (kinase inhibitors and anthracyclins) [48]. Most of these observations have been made through studies in isolated mitochondria and cell lines [48].
Over the past decade, our laboratories examined the effects of many different drug classes and found that in each drug class, some members of the class displayed greater potency for adverse effects in vitro than others and the rank order of toxicity observed mimicked the safety profile reported in patients. For example, the anti-diabetic drug Resulin (triglitazone), which was discontinued from the market for liver toxicity, caused greater mitochondrial toxicity when tested in isolated mitochondria than Actos (pioglitazone), which is on the market with a much better safety profile [53]. The same is true for the anti-depressant Zerzone (nefazadone), which belongs to the class of serotonin antagonist reuptake inhibitors. Whereas Zerzone was attrited due to liver toxicity caused at least in part by mitochondrial toxicity, Buspar (buspirone) is still on the market and is well tolerated [54]. Another class of anti-diabetic drugs is the biguanides. Phenformin was discontinued because it caused death by lactic acidosis which is considered a hallmark of mitochondrial toxicity. Glucophage (metformin), which causes much less mitochondrial toxicity is widely used in the clinic and only in rare occasions causes lactic acidosis in most likely already predisposed patients [55]. Table 1 provides additional examples of drugs and drug classes studied by our labs.

Table 1.
Each drug class contains drugs with more and less observed mitochondrial toxicity.
4. Polypharmacy in the Geriatric Population
Geriatric patients are not only predisposed by having lower mitochondrial function due to age but are often also experiencing one or more of the age-related diseases mentioned above (Figure 1). In addition, they are also often taking multiple medications (polypharmacy). In the United States, a 2010 and 2011 survey found that 87% of a representative sampling of 2206 adults aged 62 through 85, used at least one prescription medication and that more than one-third of the group were taking five or more prescription medications [62]. Additionally, it was found that 38% of those surveyed were using over-the-counter medications. A further study by Saraf et al. showed that following acute illness or injury, an average of 14 prescriptions were given to geriatric patients discharged from hospitals to skilled nursing facilities and that over one-third of these prescriptions included side-effects that could aggravate underlying geriatric conditions [63].
Of the most commonly used prescription and over-the-counter drugs in the US for older adults [64], many are known to cause mitochondrial toxicity such as the cholesterol lowering drugs (Zocor, Lipitor, Pravacol, Crestor), pain medication (Aspirin, Tylenol, Aleve) and heartburn medication (Prilosec). Table 2 lists references for mitochondrial dysfunction reported for these prescription and over-the-counter (OTC) medications.
Table 2.
Most Commonly Used Prescription and Over-the-Counter Medications (OTC) in US Older Adults. Modified from [64].
It is important to note that most studies have been conducted using isolated mitochondria and cell systems and often these systems are not of human nature. Also, it is important to understand that the safety margin for a particular drug will depend on exposure (Cmax) [54,97] as well as other contributing mechanistic toxicities [98]. For example, Trovan and Serzone (troglitazone and nefazodone) not only have inhibitory effects on mitochondrial function, but also cause additional toxicities through inhibition of the bile salt efflux pump (BSEP) and the formation of reactive metabolites.
Cholesterol lowering statins are the most commonly prescribed drugs in the geriatric population. Almost 50% of the geriatric population was taking a statin in 2011–2014 versus only 20% in 1999–2002. Statins can cause myopathy in 10–15% of the patients. The adverse events range from mild myalgia and fatigue, to life threatening rhabdomyolysis. Baycol was removed from the market in 2001 because it caused death in 52 patients by rhabdomyolysis, which led to kidney failure [99]. Women, frail individuals and those with low body index as well as patients with increased alcohol consumption are at higher risk. Harper and Jacobson, 2010, noted that Zocor (simvastatin) has greater muscle toxicity and drug interactions and should be avoided if the patient has had adverse events and should be substituted with Lescol (fluvastatin) or Crestor (rosuvastatin) [100].
Mitochondrial toxicity has been postulated as a contributing, if not causal, factor of the observed muscle toxicity. It has been postulated that a deficit in CoQ10 is the cause. CoQ10 is an essential electron carrier in the ETC and the pathway for its synthesis has commonality with that of the cholesterol pathway. A decrease in circulatory CoQ10 levels of 27–50% has been reported [101]. However, since circulating CoQ10 is carried by low-density lipoproteins (LDL) the observed decrease may simply reflect the decrease in circulating levels of LDL [101]. Very few studies have actually examined the level of CoQ10 in muscle, probably due to the invasive nature of human muscle biopsies. While these studies did demonstrate significant decreases (30%) in muscle CoQ10 levels, they failed to demonstrate a decrease in ATP synthesis and no myopathic side effects were reported [102,103]. Since muscle toxicity seems to be patient specific and somewhat rare (the same is true for liver toxicity caused by troglitazone and nefazodone), it is most likely that patients will have either underlying mitochondrial dysfunction (for example, a silent mitochondrial disease) or have different levels of transporter expression (MCT4 in the case of statin accumulation) due to polymorphism in enzymes involved in statin metabolism [104].
Beyond the effect on CoQ10, statins have been reported to have direct effects on the ETC, especially the lipophilic statins, simvastatin and fluvastatin. In addition, statins have the ability to uncouple oxidative phosphorylation, inhibit fatty acid beta oxidation, induce mitochondrial permeability transition, and cause oxidative stress and decrease mtDNA levels by thus far unidentified mechanisms [105].
It has been reported that elderly patients taking cholesterol lowering drugs (Zocor, Lipitor, etc.) have shown drug–drug interactions (DDI), particularly with the anti-lipidemic drug Lopid (gemfibrozil [53,106]), but also with certain antibiotic classes, such as macrolides [107,108] and fluoroquinolones [109,110] and the heart medication, Nexteron (amiodarone) [64,111]. All of these drugs have been shown to cause mitochondrial toxicity by targeting a variety of, as well as multiple, mitochondrial mechanisms/targets.
While it is well documented that this is mainly due to changes in the drug metabolizing function in elderly patients, we hypothesize that polypharmacy with multiple drugs affecting mitochondrial function could contribute to the toxicities observed. We already spoke of the general mitochondrial decay with age and in age-related diseases.
How do we imagine an individual’s lifestyle affects their mitochondrial health and energy production, or their response to medications that carry a potential mitochondrial liability (Table 3)? Differences in body mass index (BMI) have shown that obesity correlates with mitochondrial dysfunction [112]. Additionally, diets of high fat and high sucrose have been shown to be detrimental to mitochondrial function [113,114]. Mitochondria are also weakened by inactive lifestyles and can be revitalized with exercise [115,116]. Studies by DiNicolantonio et al. show deleterious effects on mitochondrial function when consuming large quantities of sugar-containing foods and drinks [117], while other labs have shown that imbibing red wine in moderation [118] and drinking green tea [119] have an advantageous effect on mitochondrial function. Even the choice of area of residency can lead to mitochondrial impairment. Lim et al. have studied the effects of incidental ingestion of pesticides/herbicides and have shown a correlation between the area of residence and increased exposure to exogenous agricultural chemicals which have adverse effects on mitochondrial function [120].
Table 3.
Comparison of two geriatric people.
Now, we imagine how different individuals with ideal vs. less than ideal lifestyles might tolerate mitochondrial insults from medications. “Patient 1” has a lifestyle that promotes mitochondrial health while “Patient 2’s” lifestyle predisposes them to mitochondrial decay and toxicity in a number of ways. Taking medications that impair mitochondria is far more concerning for Patient 2 than for Patient 1. Patient 2’s healthcare providers could examine their lifestyle and medication regiment and act in unison to come up with a medication plan that minimizes mitochondrial liabilities with the drugs they are prescribing for them, opting for drugs with the best safety profile. As mentioned previously, the combination of Zocor with Lopid should at least be substituted with Crestor. An improvement in Patient 2’s diet could eliminate the daily use of Prilosec and Patient 2’s pain medication of choice should be Aspirin or Tylenol in moderation.
The question arises if mitochondrial dysfunction/toxicity caused by these medications could be prevented or counteracted by natural compounds or drugs recently developed to treat mitochondrial diseases. A recent review by Rai et al., 2016, provides an overview of a variety of pharmaceuticals which either target mitochondria or maintain its homeostasis [121]. While the majority of these drugs target diseases caused by mutations (MELAS, LHON, MERFF, etc.), one therapy is being developed for improving skeletal muscle function in elderly patients. Elamipretide (MTP-131) has been shown to prevent peroxidation of cardiolipin by binding to the molecule and preventing its interaction with cytochorme c, which prevents the latter from functioning as a peroxidase rather than an electron carrier. The drug is currently in Phase III [121]. Another compound is Idebenone, which is based structurally upon CoQ10. Idebanone is currently being explored for LHON and MELAS [121], but not as a supplemental drug in statin therapy. There are a host of natural products and supplements that have been discussed to improve mitochondrial function such as resveratrol, curcumin, NAD and N-acetyl cysteine (NAC), but there is no clinical evidence of clear success.
5. Conclusions
As patient treatment is often essential, we suggest using medication(s) with the best safety profile and the avoidance of concurrent usage of multiple medications that carry mitochondrial liabilities. Our hope is that future therapies will be devoid of mitochondrial liabilities as screening paradigms can be deployed preclinically. In addition, we also recommend lifestyle changes to further improve one’s mitochondrial function, such as weight loss, exercise and nutrition. Another major point is that the cause for mitochondrial dysfunction with age appears to be one of molecular homeostatic control of mitochondrial integrity or functional capacity, not simply an acute depletion of bioenergetic substrates, such as ATP. Consequently, bolus supplementation with mitochondrial cofactors has less potential than long-term lifestyle changes in extending vitality and drug tolerance in aging populations.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dimauro, S.; Davidzon, G. Mitochondrial DNA and disease. Ann. Med. 2005, 37, 222–232. [Google Scholar] [CrossRef]
- Blake, R.; Trounce, I.A. Mitochondrial dysfunction and complications associated with diabetes. Biochim. Biophys. Acta 2014, 1840, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in non-alcoholic fatty liver disease progression. J. Lipid Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Hoppel, C.L.; Lesnefsky, E.J.; Chen, Q.; Tandler, B. Mitochondrial Dysfunction in Cardiovascular Aging. Adv. Exp. Med. Biol. 2017, 982, 451–464. [Google Scholar] [PubMed]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Anupama, N.; Sindhu, G.; Raghu, K.G. Significance of mitochondria on cardiometabolic syndromes. Fundam. Clin. Pharmacol. 2018, 32, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef]
- Falah, M.; Houshmand, M.; Najafi, M.; Balali, M.; Mahmoudian, S.; Asghari, A.; Emamdjomeh, H.; Farhadi, M. The potential role for use of mitochondrial DNA copy number as predictive biomarker in presbycusis. Ther. Clin. Risk Manag. 2016, 12, 1573–1578. [Google Scholar] [CrossRef]
- Leruez, S.; Marill, A.; Bresson, T.; de Saint Martin, G.; Buisset, A.; Muller, J.; Tessier, L.; Gadras, C.; Verny, C.; Gohier, P.; et al. A Metabolomics Profiling of Glaucoma Points to Mitochondrial Dysfunction, Senescence, and Polyamines Deficiency. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4355–4361. [Google Scholar] [CrossRef]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive Oxygen Species, Aging and Articular Cartilage Homeostasis. Free Radic. Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef]
- Ng Kee Kwong, F.; Nicholson, A.G.; Harrison, C.L.; Hansbro, P.M.; Adcock, I.M.; Chung, K.F. Is mitochondrial dysfunction a driving mechanism linking COPD to nonsmall cell lung carcinoma? Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef]
- Zhu, Y.; Dean, A.E.; Horikoshi, N.; Heer, C.; Spitz, D.R.; Gius, D. Emerging evidence for targeting mitochondrial metabolic dysfunction in cancer therapy. J. Clin. Investig. 2018, 128, 3682–3691. [Google Scholar] [CrossRef]
- Always, S.E.; Mohamed, J.S.; Myers, M.J. Mitochondria Initiate and Regulate Sarcopenia. Exerc. Sport Sci. Rev. 2017, 45, 58–69. [Google Scholar] [CrossRef]
- Hara, H.; Kuwano, K.; Araya, J. Mitochondrial Quality Control in COPD and IPF. Cells 2018, 7, 86. [Google Scholar] [CrossRef]
- Raymond, P. The Rate of Living; Johns Hopkins University: New York, NY, USA, 1928. [Google Scholar]
- Lanza, I.R.; Nair, K.S. Mitochondrial function as a determinant of life span. Pflugers Arch. 2010, 459, 277–289. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Conley, K.E.; Jubrias, S.A.; Esselman, P.C. Oxidative capacity and ageing in human muscle. J. Physiol. 2000, 526, 203–210. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef]
- Harman, D. The biological clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Harman, D. Free radical theory of aging: Consequences of mitochondrial aging. Age 1983, 6, 86–94. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed. Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef]
- Tauchi, H.; Sato, T. Age changes in size and number of mitochondria of human hepatic cells. J. Gerontol. 1968, 23, 454–461. [Google Scholar] [CrossRef]
- Lopez-Lluch, G. Mitochondrial activity and dynamics changes regarding metabolism in ageing and obesity. Mech. Ageing Dev. 2017, 162, 108–121. [Google Scholar] [CrossRef]
- Welle, S.; Bhatt, K.; Shah, B.; Needler, N.; Delehanty, J.M.; Thornton, C.A. Reduced amount of mitochondrial DNA in aged human muscle. J. Appl. Physiol. (1985) 2003, 94, 1479–1484. [Google Scholar] [CrossRef][Green Version]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Lanza, I.R.; Short, D.K.; Short, K.R.; Raghavakaimal, S.; Basu, R.; Joyner, M.J.; McConnell, J.P.; Nair, K.S. Endurance exercise as a countermeasure for aging. Diabetes 2008, 57, 2933–2942. [Google Scholar] [CrossRef]
- Bailey, P.J.; Webster, G.C. Lowered rates of protein synthesis by mitochondria isolated from organisms of increasing age. Mech. Ageing Dev. 1984, 24, 233–241. [Google Scholar] [CrossRef]
- Marcus, D.L.; Ibrahim, N.G.; Freedman, M.L. Age-related decline in the biosynthesis of mitochondrial inner membrane proteins. Exp. Gerontol. 1982, 17, 333–341. [Google Scholar] [CrossRef]
- Rooyackers, O.E.; Adey, D.B.; Ades, P.A.; Nair, K.S. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc. Natl. Acad. Sci. USA 1996, 93, 15364–15369. [Google Scholar] [CrossRef]
- Rooyackers, O.E.; Kersten, A.H.; Wagenmakers, A.J. Mitochondrial protein content and in vivo synthesis rates in skeletal muscle from critically ill rats. Clin. Sci. (Lond.) 1996, 91, 475–481. [Google Scholar] [CrossRef]
- Mammucari, C.; Rizzuto, R. Signaling pathways in mitochondrial dysfunction and aging. Mech. Ageing Dev. 2010, 131, 536–543. [Google Scholar] [CrossRef]
- Vendelbo, M.H.; Nair, K.S. Mitochondrial longevity pathways. Biochim. Biophys. Acta 2011, 1813, 634–644. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Tatsuta, T.; Langer, T. Quality control of mitochondria: Protection against neurodegeneration and ageing. EMBO J. 2008, 27, 306–314. [Google Scholar] [CrossRef]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Karin, M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 2013, 18, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Introduction: Sirtuins in aging and diseases. Methods Mol. Biol. 2013, 1077, 3–10. [Google Scholar] [PubMed]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.A.; Reichert, A.S. Impaired quality control of mitochondria: Aging from a new perspective. Exp. Gerontol. 2010, 45, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Will, Y.; Dykens, J. Mitochondrial toxicity assessment in industry--a decade of technology development and insight. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1061–1067. [Google Scholar] [CrossRef]
- Swiss, R.; Will, Y. Assessment of mitochondrial toxicity in HepG2 cells cultured in high-glucose- or galactose-containing media. Curr. Protoc. Toxicol. 2011, 49, 2.20.1–2.20.14. [Google Scholar] [CrossRef]
- Kamalian, L.; Chadwick, A.E.; Bayliss, M.; French, N.S.; Monshouwer, M.; Snoeys, J.; Park, B.K. The utility of HepG2 cells to identify direct mitochondrial dysfunction in the absence of cell death. Toxicol. In Vitro 2015, 29, 732–740. [Google Scholar] [CrossRef]
- Hynes, J.; Swiss, R.L.; Will, Y. High-Throughput Analysis of Mitochondrial Oxygen Consumption. Methods Mol. Biol. 2018, 1782, 71–87. [Google Scholar]
- Nadanaciva, S.; Rana, P.; Beeson, G.C.; Chen, D.; Ferrick, D.A.; Beeson, C.C.; Will, Y. Assessment of drug-induced mitochondrial dysfunction via altered cellular respiration and acidification measured in a 96-well platform. J. Bioenerg. Biomembr. 2012, 44, 421–437. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Dykens, J.A.; Bernal, A.; Capaldi, R.A.; Will, Y. Mitochondrial impairment by PPAR agonists and statins identified via immunocaptured OXPHOS complex activities and respiration. Toxicol. Appl. Pharmacol. 2007, 223, 277–287. [Google Scholar] [CrossRef]
- Dykens, J.A.; Jamieson, J.D.; Marroquin, L.D.; Nadanaciva, S.; Xu, J.J.; Dunn, M.C.; Smith, A.R.; Will, Y. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol. Sci. 2008, 103, 335–345. [Google Scholar] [CrossRef]
- Dykens, J.A.; Jamieson, J.; Marroquin, L.; Nadanaciva, S.; Billis, P.A.; Will, Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol. Appl. Pharmacol. 2008, 233, 203–210. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Aleo, M.D.; Strock, C.J.; Stedman, D.B.; Wang, H.; Will, Y. Toxicity assessments of nonsteroidal anti-inflammatory drugs in isolated mitochondria, rat hepatocytes, and zebrafish show good concordance across chemical classes. Toxicol. Appl. Pharmacol. 2013, 272, 272–280. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Dillman, K.; Gebhard, D.F.; Shrikhande, A.; Will, Y. High-content screening for compounds that affect mtDNA-encoded protein levels in eukaryotic cells. J. Biomol. Screen. 2010, 15, 937–948. [Google Scholar] [CrossRef]
- Solem, L.E.; Henry, T.R.; Wallace, K.B. Disruption of mitochondrial calcium homeostasis following chronic doxorubicin administration. Toxicol. Appl. Pharmacol. 1994, 129, 214–222. [Google Scholar] [CrossRef]
- Palmeira, C.M.; Serrano, J.; Kuehl, D.W.; Wallace, K.B. Preferential oxidation of cardiac mitochondrial DNA following acute intoxication with doxorubicin. Biochim. Biophys. Acta 1997, 1321, 101–106. [Google Scholar] [CrossRef]
- Wallace, K.B. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol. Toxicol. 2003, 93, 105–115. [Google Scholar] [CrossRef]
- Sardao, V.A.; Oliveira, P.J.; Holy, J.; Oliveira, C.R.; Wallace, K.B. Doxorubicin-induced mitochondrial dysfunction is secondary to nuclear p53 activation in H9c2 cardiomyoblasts. Cancer Chemother. Pharmacol. 2009, 64, 811–827. [Google Scholar] [CrossRef]
- Qato, D.M.; Wilder, J.; Schumm, L.P.; Gillet, V.; Alexander, G.C. Changes in Prescription and Over-the-Counter Medication and Dietary Supplement Use among Older Adults in the United States, 2005 vs 2011. JAMA Intern. Med. 2016, 176, 473–482. [Google Scholar] [CrossRef]
- Saraf, A.A.; Petersen, A.W.; Simmons, S.F.; Schnelle, J.F.; Bell, S.P.; Kripalani, S.; Myers, A.P.; Mixon, A.S.; Long, E.A.; Jacobsen, J.M.; et al. Medications associated with geriatric syndromes and their prevalence in older hospitalized adults discharged to skilled nursing facilities. J. Hosp. Med. 2016, 11, 694–700. [Google Scholar] [CrossRef]
- Merel, S.E.; Paauw, D.S. Common Drug Side Effects and Drug-Drug Interactions in Elderly Adults in Primary Care. J. Am. Geriatr. Soc. 2017, 65, 1578–1585. [Google Scholar] [CrossRef]
- Chatterjee, S.S.; Stefanovich, V. Influence of anti-inflammatory agents on rat liver mitochondrial ATPase. Arzneimittel-Forschung 1976, 26, 499–502. [Google Scholar]
- Nulton-Persson, A.C.; Szweda, L.I.; Sadek, H.A. Inhibition of cardiac mitochondrial respiration by salicylic acid and acetylsalicylate. J. Cardiovasc. Pharmacol. 2004, 44, 591–595. [Google Scholar] [CrossRef]
- Raza, H.; John, A. Implications of altered glutathione metabolism in aspirin-induced oxidative stress and mitochondrial dysfunction in HepG2 cells. PLoS ONE 2012, 7, e36325. [Google Scholar] [CrossRef]
- Deschamps, D.; Fisch, C.; Fromenty, B.; Berson, A.; Degott, C.; Pessayre, D. Inhibition by salicylic acid of the activation and thus oxidation of long chain fatty acids. Possible role in the development of Reye’s syndrome. J. Pharmacol. Exp. Ther. 1991, 259, 894–904. [Google Scholar]
- Oh, K.W.; Qian, T.; Brenner, D.A.; Lemasters, J.J. Salicylate enhances necrosis and apoptosis mediated by the mitochondrial permeability transition. Toxicol. Sci. 2003, 73, 44–52. [Google Scholar] [CrossRef]
- Zimmerman, H.J. Effects of aspirin and acetaminophen on the liver. Arch. Intern. Med. 1981, 141, 333–342. [Google Scholar] [CrossRef]
- Bonifacio, A.; Mullen, P.J.; Mityko, I.S.; Navegantes, L.C.; Bouitbir, J.; Krahenbuhl, S. Simvastatin induces mitochondrial dysfunction and increased atrogin-1 expression in H9c2 cardiomyocytes and mice in vivo. Arch. Toxicol. 2016, 90, 203–215. [Google Scholar] [CrossRef]
- Fisar, Z.; Hroudova, J.; Singh, N.; Koprivova, A.; Maceckova, D. Effect of Simvastatin, Coenzyme Q10, Resveratrol, Acetylcysteine and Acetylcarnitine on Mitochondrial Respiration. Folia Biol. 2016, 62, 53–66. [Google Scholar]
- van Diemen, M.P.J.; Berends, C.L.; Akram, N.; Wezel, J.; Teeuwisse, W.M.; Mik, B.G.; Kan, H.E.; Webb, A.; Beenakker, J.W.M.; Groeneveld, G.J. Validation of a pharmacological model for mitochondrial dysfunction in healthy subjects using simvastatin: A randomized placebo-controlled proof-of-pharmacology study. Eur. J. Pharmacol. 2017, 815, 290–297. [Google Scholar] [CrossRef]
- Busanello, E.N.B.; Figueira, T.R.; Marques, A.C.; Navarro, C.D.C.; Oliveira, H.C.F.; Vercesi, A.E. Facilitation of Ca(2+) -induced opening of the mitochondrial permeability transition pore either by nicotinamide nucleotide transhydrogenase deficiency or statins treatment. Cell Biol. Int. 2018, 42, 742–746. [Google Scholar] [CrossRef]
- Sirvent, P.; Bordenave, S.; Vermaelen, M.; Roels, B.; Vassort, G.; Mercier, J.; Raynaud, E.; Lacampagne, A. Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem. Biophys. Res. Commun. 2005, 338, 1426–1434. [Google Scholar] [CrossRef]
- Sirvent, P.; Mercier, J.; Vassort, G.; Lacampagne, A. Simvastatin triggers mitochondria-induced Ca2+ signaling alteration in skeletal muscle. Biochem. Biophys. Res. Commun. 2005, 329, 1067–1075. [Google Scholar] [CrossRef]
- Wagner, B.K.; Kitami, T.; Gilbert, T.J.; Peck, D.; Ramanathan, A.; Schreiber, S.L.; Golub, T.R.; Mootha, V.K. Large-scale chemical dissection of mitochondrial function. Nat. Biotechnol. 2008, 26, 343–351. [Google Scholar] [CrossRef]
- Tonazzi, A.; Giangregorio, N.; Console, L.; Indiveri, C. Mitochondrial carnitine/acylcarnitine translocase: Insights in structure/function relationships. Basis for drug therapy and side effects prediction. Mini Rev. Med. Chem. 2015, 15, 396–405. [Google Scholar] [CrossRef]
- Urbano, F.; Bugliani, M.; Filippello, A.; Scamporrino, A.; Di Mauro, S.; Di Pino, A.; Scicali, R.; Noto, D.; Rabuazzo, A.M.; Averna, M.; et al. Atorvastatin but Not Pravastatin Impairs Mitochondrial Function in Human Pancreatic Islets and Rat beta-Cells. Direct Effect of Oxidative Stress. Sci. Rep. 2017, 7, 11863. [Google Scholar] [CrossRef]
- Godoy, J.C.; Niesman, I.R.; Busija, A.R.; Kassan, A.; Schilling, J.M.; Schwarz, A.; Alvarez, E.A.; Dalton, N.D.; Drummond, J.C.; Roth, D.M.; et al. Atorvastatin, but not pravastatin, inhibits cardiac Akt/mTOR signaling and disturbs mitochondrial ultrastructure in cardiac myocytes. FASEB J. 2019, 33, 1209–1225. [Google Scholar] [CrossRef]
- Broniarek, I.; Jarmuszkiewicz, W. Atorvastatin affects negatively respiratory function of isolated endothelial mitochondria. Arch. Biochem. Biophys. 2018, 637, 64–72. [Google Scholar] [CrossRef]
- Hinson, J.A.; Reid, A.B.; McCullough, S.S.; James, L.P. Acetaminophen-induced hepatotoxicity: Role of metabolic activation, reactive oxygen/nitrogen species, and mitochondrial permeability transition. Drug Metab. Rev. 2004, 36, 805–822. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Suda, C.; Horie, T. Involvement of mitochondrial permeability transition in acetaminophen-induced liver injury in mice. J. Hepatol. 2005, 42, 110–116. [Google Scholar] [CrossRef]
- Prill, S.; Bavli, D.; Levy, G.; Ezra, E.; Schmalzlin, E.; Jaeger, M.S.; Schwarz, M.; Duschl, C.; Cohen, M.; Nahmias, Y. Real-time monitoring of oxygen uptake in hepatic bioreactor shows CYP450-independent mitochondrial toxicity of acetaminophen and amiodarone. Arch. Toxicol. 2016, 90, 1181–1191. [Google Scholar] [CrossRef]
- Moles, A.; Torres, S.; Baulies, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial-Lysosomal Axis in Acetaminophen Hepatotoxicity. Front. Pharmacol. 2018, 9, 453. [Google Scholar] [CrossRef]
- Manuel, M.A.; Weiner, M.W. Effects of ethacrynic acid and furosemide on isolated rat kidney mitochondria: Inhibition of electron transport in the region of phosphorylation site II. J. Pharmacol. Exp. Ther. 1976, 198, 209. [Google Scholar]
- Orita, Y.; Fukuhara, Y.; Yanase, M.; Ando, A.; Okada, N.; Abe, H. Effect of furosemide on mitochondrial electron transport system and oxidative phosphorylation. Arzneimittel-Forschung 1983, 33, 1446–1450. [Google Scholar]
- Wong, S.G.W.; Card, J.W.; Racz, W.J. The role of mitochondrial injury in bromobenzene and furosemide induced hepatotoxicity. Toxicol. Lett. 2000, 116, 171–181. [Google Scholar] [CrossRef]
- Tai, Y.K.; Cheong, Y.M.; Almsherqi, Z.A.; Chia, S.H.; Deng, Y.; McLachlan, C.S. High dose clopidogrel decreases mice liver mitochondrial respiration function in vitro. Int. J. Cardiol. 2009, 133, 250–252. [Google Scholar] [CrossRef]
- Maseneni, S.; Donzelli, M.; Brecht, K.; Krahenbuhl, S. Toxicity of thienopyridines on human neutrophil granulocytes and lymphocytes. Toxicology 2013, 308, 11–19. [Google Scholar] [CrossRef]
- Zahno, A.; Bouitbir, J.; Maseneni, S.; Lindinger, P.W.; Brecht, K.; Krahenbuhl, S. Hepatocellular toxicity of clopidogrel: Mechanisms and risk factors. Free Radic. Biol. Med. 2013, 65, 208–216. [Google Scholar] [CrossRef]
- Felix, L.; Oliveira, M.M.; Videira, R.; Maciel, E.; Alves, N.D.; Nunes, F.M.; Alves, A.; Almeida, J.M.; Domingues, M.R.; Peixoto, F.P. Carvedilol exacerbate gentamicin-induced kidney mitochondrial alterations in adult rat. Exp. Toxicol. Pathol. 2017, 69, 83–92. [Google Scholar] [CrossRef]
- Busanello, E.N.B.; Marques, A.C.; Lander, N.; de Oliveira, D.N.; Catharino, R.R.; Oliveira, H.C.F.; Vercesi, A.E. Pravastatin Chronic Treatment Sensitizes Hypercholesterolemic Mice Muscle to Mitochondrial Permeability Transition: Protection by Creatine or Coenzyme Q10. Front. Pharmacol. 2017, 8, 185. [Google Scholar] [CrossRef]
- Westwood, F.R.; Scott, R.C.; Marsden, A.M.; Bigley, A.; Randall, K. Rosuvastatin: Characterization of induced myopathy in the rat. Toxicol. Pathol. 2008, 36, 345–352. [Google Scholar] [CrossRef]
- van Leeuwen, J.S.; Unlu, B.; Vermeulen, N.P.; Vos, J.C. Differential involvement of mitochondrial dysfunction, cytochrome P450 activity, and active transport in the toxicity of structurally related NSAIDs. Toxicol. In Vitro 2012, 26, 197–205. [Google Scholar] [CrossRef]
- Moreno-Sanchez, R.; Bravo, C.; Vasquez, C.; Ayala, G.; Silveira, L.H.; Martinez-Lavin, M. Inhibition and uncoupling of oxidative phosphorylation by nonsteroidal anti-inflammatory drugs: Study in mitochondria, submitochondrial particles, cells, and whole heart. Biochem. Pharmacol. 1999, 57, 743–752. [Google Scholar] [CrossRef]
- Porceddu, M.; Buron, N.; Roussel, C.; Labbe, G.; Fromenty, B.; Borgne-Sanchez, A. Prediction of liver injury induced by chemicals in human with a multiparametric assay on isolated mouse liver mitochondria. Toxicol. Sci. 2012, 129, 332–345. [Google Scholar] [CrossRef]
- Aleo, M.D.; Luo, Y.; Swiss, R.; Bonin, P.D.; Potter, D.M.; Will, Y. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology 2014, 60, 1015–1022. [Google Scholar] [CrossRef]
- Mendes, P.; Robles, P.G.; Mathur, S. Statin-induced rhabdomyolysis: A comprehensive review of case reports. Physiother. Can. 2014, 66, 124–132. [Google Scholar] [CrossRef]
- Harper, C.R.; Jacobson, T.A. Evidence-based management of statin myopathy. Curr. Atheroscler. Rep. 2010, 12, 322–330. [Google Scholar] [CrossRef]
- Hargreaves, I.P.; Duncan, A.J.; Heales, S.J.; Land, J.M. The effect of HMG-CoA reductase inhibitors on coenzyme Q10: Possible biochemical/clinical implications. Drug Saf. 2005, 28, 659–676. [Google Scholar] [CrossRef]
- Paiva, H.; Thelen, K.M.; Van Coster, R.; Smet, J.; De Paepe, B.; Mattila, K.M.; Laakso, J.; Lehtimaki, T.; von Bergmann, K.; Lutjohann, D.; et al. High-dose statins and skeletal muscle metabolism in humans: A randomized, controlled trial. Clin. Pharmacol. Ther. 2005, 78, 60–68. [Google Scholar] [CrossRef]
- Avis, H.J.; Hargreaves, I.P.; Ruiter, J.P.; Land, J.M.; Wanders, R.J.; Wijburg, F.A. Rosuvastatin lowers coenzyme Q10 levels, but not mitochondrial adenosine triphosphate synthesis, in children with familial hypercholesterolemia. J. Pediatr. 2011, 158, 458–462. [Google Scholar] [CrossRef]
- Neergheen, V.; Dyson, A.; Wainwright, L.; Hargreaves, I.P. Statin and Fibrate-induced Dichotomy of Mitochondrial Function. In Mitochondrial Dysfunction Caused by Drugs and Environmental Toxicants; Will, Y., Dykens, J., Eds.; Wiley: New York, NY, USA, 2018; pp. 459–473. [Google Scholar]
- Hargreaves, I.P.; Al Shahrani, M.; Wainwright, L.; Heales, S.J. Drug-Induced Mitochondrial Toxicity. Drug Saf. 2016, 39, 661–674. [Google Scholar] [CrossRef]
- Johnson, T.E.; Zhang, X.; Shi, S.; Umbenhauer, D.R. Statins and PPARalpha agonists induce myotoxicity in differentiated rat skeletal muscle cultures but do not exhibit synergy with co-treatment. Toxicol. Appl. Pharmacol. 2005, 208, 210–221. [Google Scholar] [CrossRef]
- Salimi, A.; Eybagi, S.; Seydi, E.; Naserzadeh, P.; Kazerouni, N.P.; Pourahmad, J. Toxicity of macrolide antibiotics on isolated heart mitochondria: A justification for their cardiotoxic adverse effect. Xenobiotica 2016, 46, 82–93. [Google Scholar] [CrossRef]
- Mathis, A.; Wild, P.; Boettger, E.C.; Kapel, C.M.; Deplazes, P. Mitochondrial ribosome as the target for the macrolide antibiotic clarithromycin in the helminth Echinococcus multilocularis. Antimicrob. Agents Chemother. 2005, 49, 3251–3255. [Google Scholar] [CrossRef]
- Ding, L.; Zang, L.; Zhang, Y.; Zhang, Y.; Wang, X.; Ai, W.; Ding, N.; Wang, H. Joint toxicity of fluoroquinolone and tetracycline antibiotics to zebrafish (Danio rerio) based on biochemical biomarkers and histopathological observation. J. Toxicol. Sci. 2017, 42, 267–280. [Google Scholar] [CrossRef]
- Kaur, K.; Fayad, R.; Saxena, A.; Frizzell, N.; Chanda, A.; Das, S.; Chatterjee, S.; Hegde, S.; Baliga, M.S.; Ponemone, V.; et al. Fluoroquinolone-related neuropsychiatric and mitochondrial toxicity: A collaborative investigation by scientists and members of a social network. J. Community Support. Oncol. 2016, 14, 54–65. [Google Scholar] [CrossRef]
- Finsterer, J.; Zarrouk-Mahjoub, S. Mitochondrial toxicity of cardiac drugs and its relevance to mitochondrial disorders. Expert Opin. Drug Metab. Toxicol. 2015, 11, 15–24. [Google Scholar] [CrossRef]
- De Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef]
- Miotto, P.M.; LeBlanc, P.J.; Holloway, G.P. High-Fat Diet Causes Mitochondrial Dysfunction as a Result of Impaired ADP Sensitivity. Diabetes 2018, 67, 2199–2205. [Google Scholar] [CrossRef]
- Jorgensen, W.; Rud, K.A.; Mortensen, O.H.; Frandsen, L.; Grunnet, N.; Quistorff, B. Your mitochondria are what you eat: A high-fat or a high-sucrose diet eliminates metabolic flexibility in isolated mitochondria from rat skeletal muscle. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Gram, M.; Dahl, R.; Dela, F. Physical inactivity and muscle oxidative capacity in humans. Eur. J. Sport Sci. 2014, 14, 376–383. [Google Scholar] [CrossRef]
- Kim, Y.; Triolo, M.; Hood, D.A. Impact of Aging and Exercise on Mitochondrial Quality Control in Skeletal Muscle. Oxid. Med. Cell. Longev. 2017, 2017, 3165396. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; Berger, A. Added sugars drive nutrient and energy deficit in obesity: A new paradigm. Open Heart 2016, 3, e000469. [Google Scholar] [CrossRef]
- Chen, W.J.; Du, J.K.; Hu, X.; Yu, Q.; Li, D.X.; Wang, C.N.; Zhu, X.Y.; Liu, Y.J. Protective effects of resveratrol on mitochondrial function in the hippocampus improves inflammation-induced depressive-like behavior. Physiol. Behav. 2017, 182, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Assuncao, M.; Andrade, J.P. Protective action of green tea catechins in neuronal mitochondria during aging. Front. Biosci. 2015, 20, 247–262. [Google Scholar]
- Lim, S.; Ahn, S.Y.; Song, I.C.; Chung, M.H.; Jang, H.C.; Park, K.S.; Lee, K.U.; Pak, Y.K.; Lee, H.K. Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS ONE 2009, 4, e5186. [Google Scholar] [CrossRef]
- Rai, P.K.; Russell, O.M.; Lightowlers, R.N.; Turnbull, D.M. Potential compounds for the treatment of mitochondrial disease. Br. Med. Bull. 2015, 116, 5–18. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).