Histone 4 Lysine 20 Methylation: A Case for Neurodevelopmental Disease


Abstract
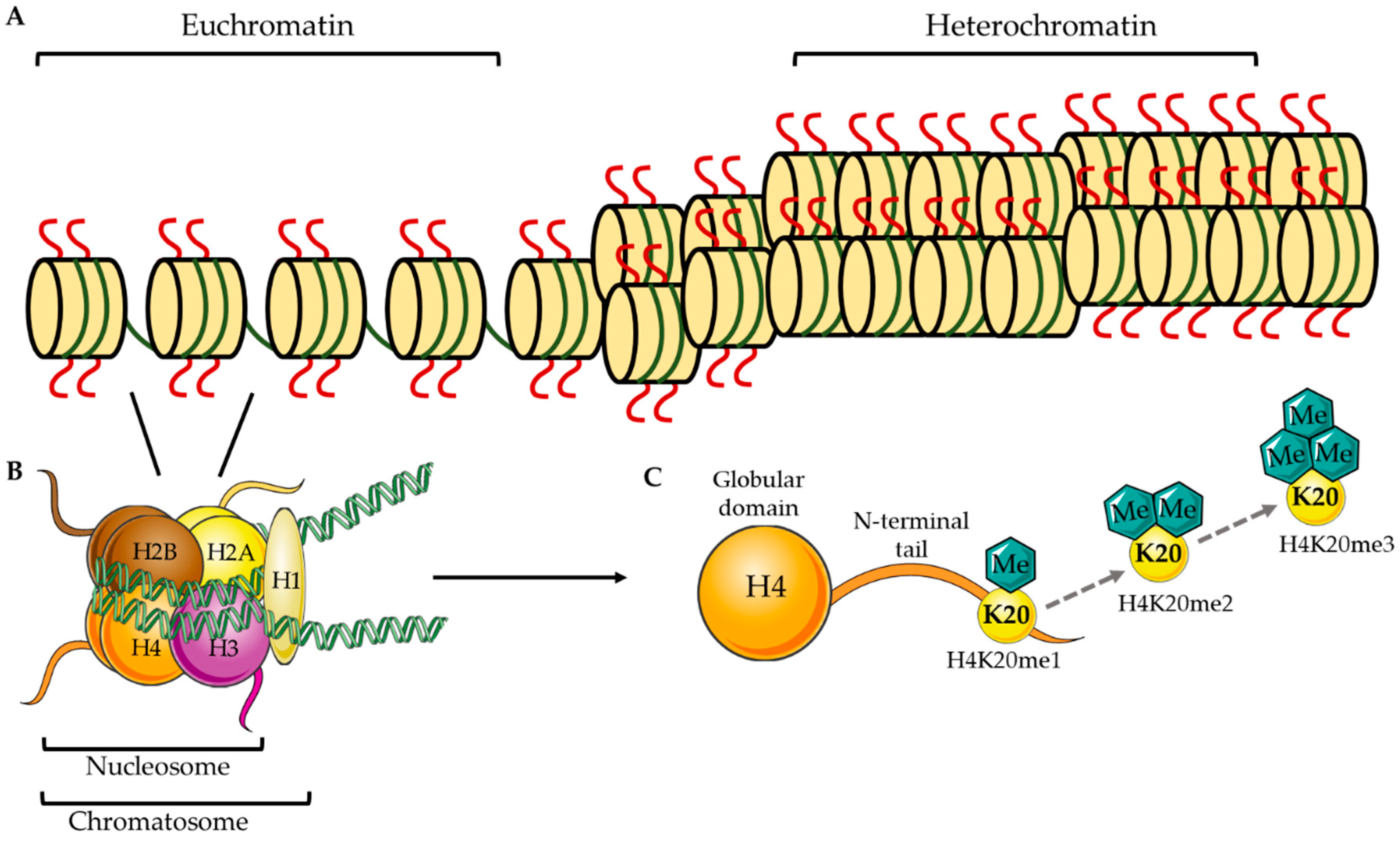
1. Epigenetic Methylation at H4K20
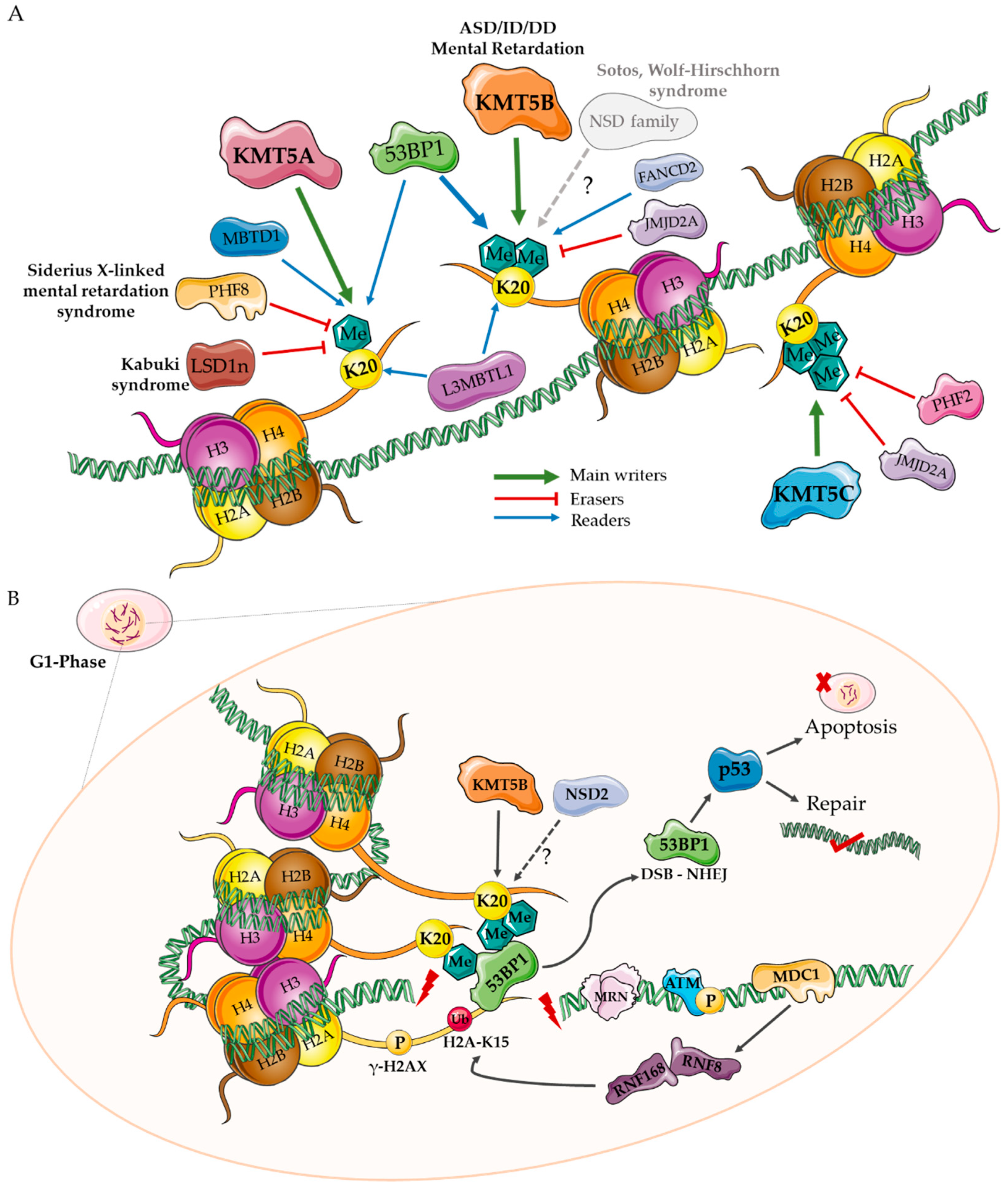
2. H4K20 Writers
2.1. KMT5A
2.2. KMT5B and KMT5C
2.3. NSD Family Writers
3. H4K20 Erasers
3.1. KDM4A
3.2. PHF8
3.3. LSD1/KDM1A
4. H4K20 Readers
4.1. MBT, L3MBTL1
4.2. TP53BP1
4.3. FANCD2
5. A Role for KMT Enzymes in Neurodevelopment?
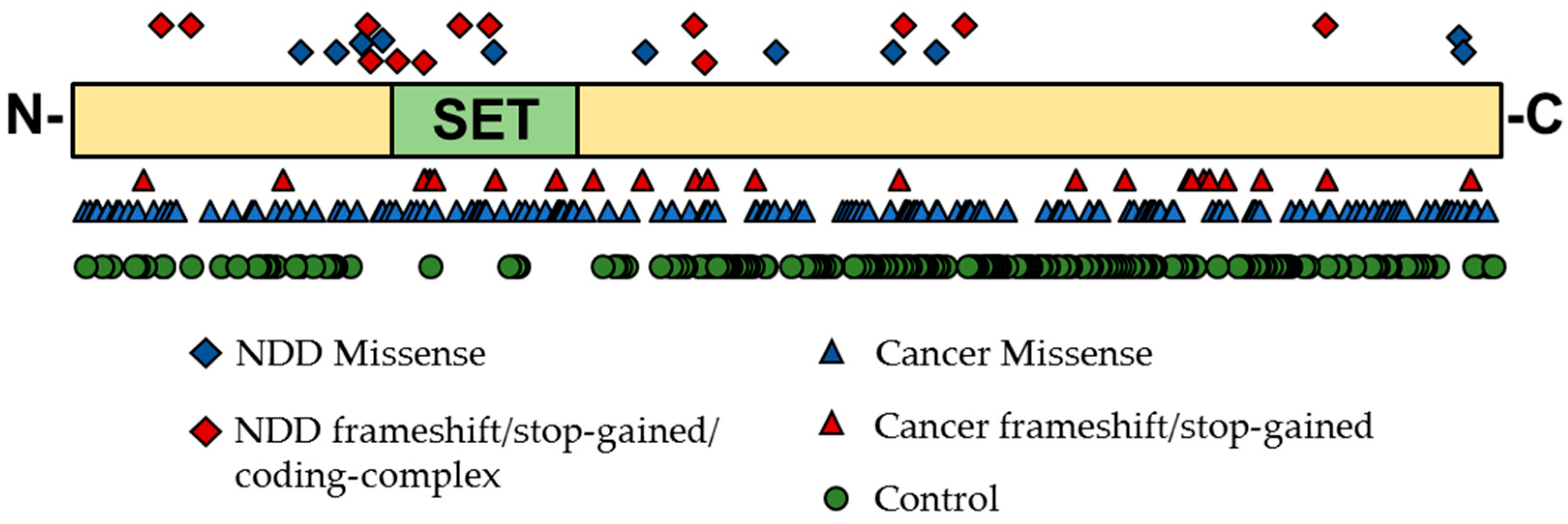
5.1. Genetic Evidence for KMT Genes
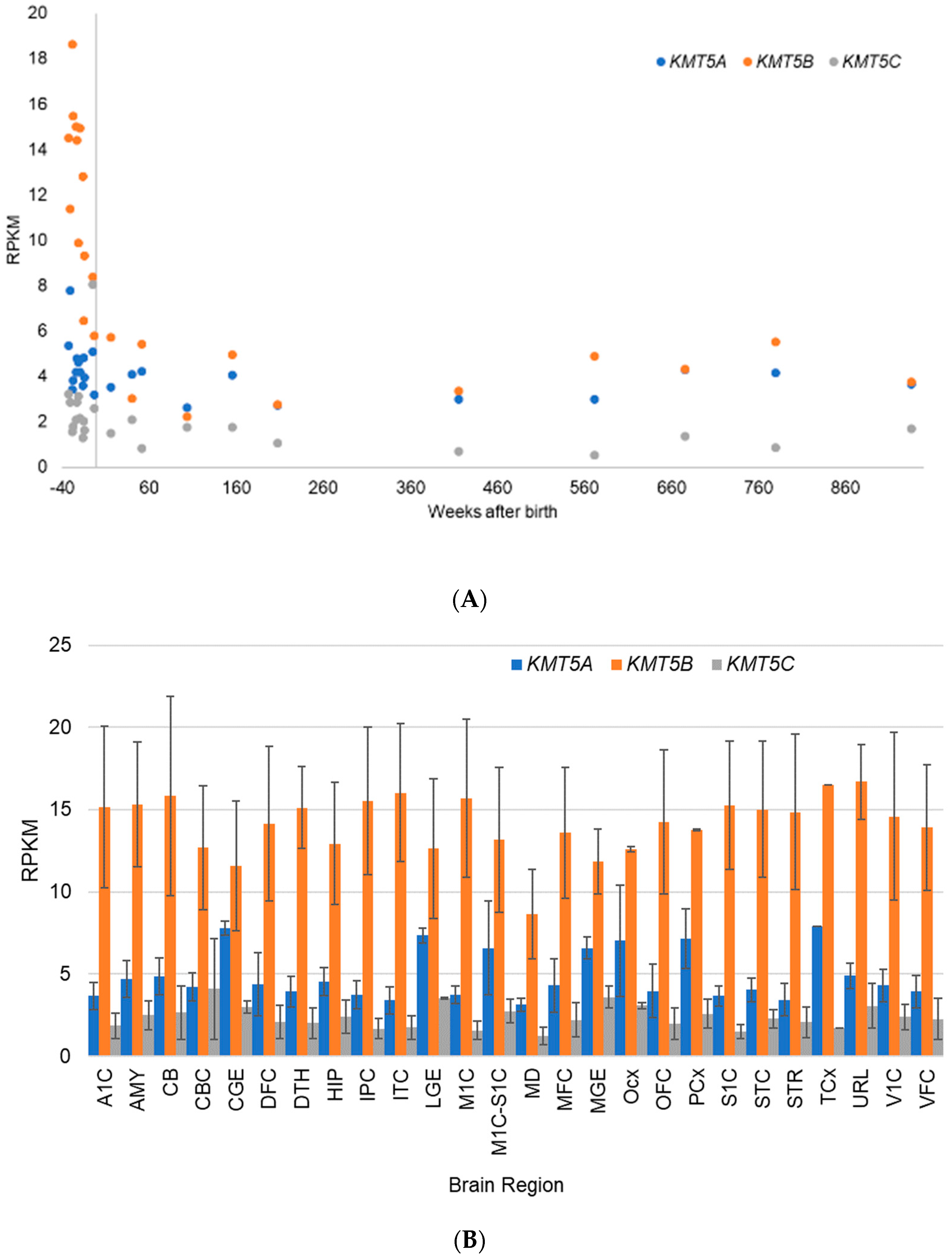
5.2. Gene Expression Evidence for KMT Genes
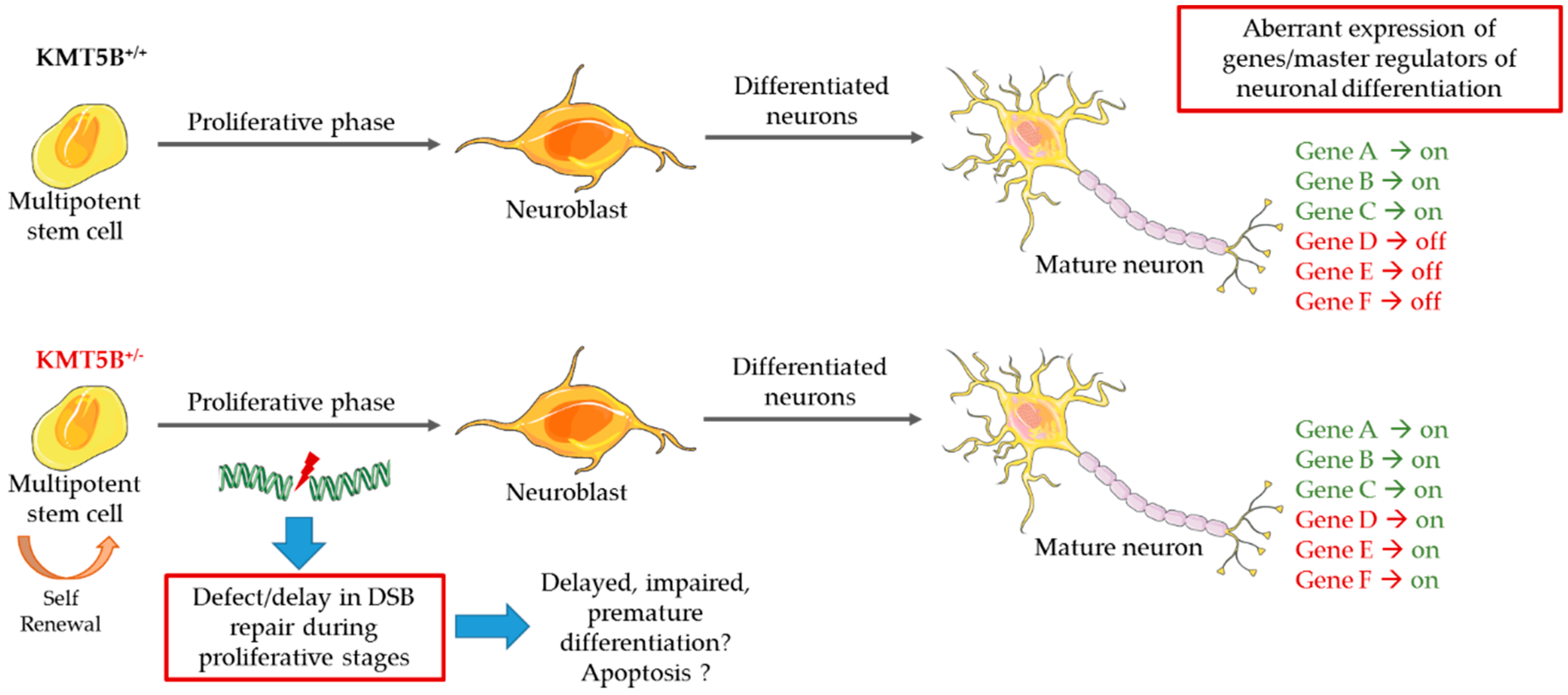
5.3. Model Systems Evidence for KMT Gene Involvement in Neurodevelopment
6. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, S.; Schotta, G.; Sorensen, C.S. Histone H4 lysine 20 methylation: Key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013, 41, 2797–2806. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.B.; Oda, H.; Shen, S.S.; Reinberg, D. PR-Set7 and H4K20me1: At the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 2012, 26, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wen, H.; Shi, X. Lysine methylation: Beyond histones. Acta Biochim. Biophys. Sin. (Shanghai) 2012, 44, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, L.; Milavetz, B. Decoding the histone H4 lysine 20 methylation mark. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Schotta, G.; Lachner, M.; Sarma, K.; Ebert, A.; Sengupta, R.; Reuter, G.; Reinberg, D.; Jenuwein, T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004, 18, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed]
- Simonetta, M.; de Krijger, I.; Serrat, J.; Moatti, N.; Fortunato, D.; Hoekman, L.; Bleijerveld, O.B.; Altelaar, A.F.M.; Jacobs, J.J.L. H4K20me2 distinguishes pre-replicative from post-replicative chromatin to appropriately direct DNA repair pathway choice by 53BP1-RIF1-MAD2L2. Cell Cycle 2018, 17, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, S.; Michelena, J.; Teloni, F.; Imhof, R.; Altmeyer, M. Replication-Coupled Dilution of H4K20me2 Guides 53BP1 to Pre-replicative Chromatin. Cell Rep. 2017, 19, 1819–1831. [Google Scholar] [CrossRef] [PubMed]
- Paquin, K.L.; Howlett, N.G. Understanding the Histone DNA Repair Code: H4K20me2 Makes Its Mark. Mol. Cancer Res. 2018, 16, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Mozzetta, C.; Boyarchuk, E.; Pontis, J.; Ait-Si-Ali, S. Sound of silence: The properties and functions of repressive Lys methyltransferases. Nat. Rev. Mol. Cell Biol. 2015, 16, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Couture, J.F.; Collazo, E.; Brunzelle, J.S.; Trievel, R.C. Structural and functional analysis of SET8, a histone H4 Lys-20 methyltransferase. Genes Dev. 2005, 19, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Southall, S.M.; Cronin, N.B.; Wilson, J.R. A novel route to product specificity in the Suv4-20 family of histone H4K20 methyltransferases. Nucleic Acids Res. 2014, 42, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Rice, J.C.; Sarma, K.; Erdjument-Bromage, H.; Werner, J.; Wang, Y.; Chuikov, S.; Valenzuela, P.; Tempst, P.; Steward, R.; et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 2002, 9, 1201–1213. [Google Scholar] [CrossRef]
- Fang, J.; Feng, Q.; Ketel, C.S.; Wang, H.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Simon, J.A.; Zhang, Y. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr. Biol. 2002, 12, 1086–1099. [Google Scholar] [CrossRef]
- Xiao, B.; Jing, C.; Kelly, G.; Walker, P.A.; Muskett, F.W.; Frenkiel, T.A.; Martin, S.R.; Sarma, K.; Reinberg, D.; Gamblin, S.J.; et al. Specificity and mechanism of the histone methyltransferase Pr-Set. Genes Dev. 2005, 19, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Shoaib, M.; Walter, D.; Gillespie, P.J.; Izard, F.; Fahrenkrog, B.; Lleres, D.; Lerdrup, M.; Johansen, J.V.; Hansen, K.; Julien, E.; et al. Histone H4K20 methylation mediated chromatin compaction threshold ensures genome integrity by limiting DNA replication licensing. Nat. Commun. 2018, 9, 3704. [Google Scholar] [CrossRef] [PubMed]
- Milite, C.; Feoli, A.; Viviano, M.; Rescigno, D.; Cianciulli, A.; Balzano, A.L.; Mai, A.; Castellano, S.; Sbardella, G. The emerging role of lysine methyltransferase SETD8 in human diseases. Clin. Epigenetics 2016, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wang, W.; Kong, X.; Congdon, L.M.; Yokomori, K.; Kirschner, M.W.; Rice, J.C. Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 2010, 24, 2531–2542. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rice, J.C. A new regulator of the cell cycle: The PR-Set7 histone methyltransferase. Cell Cycle 2011, 10, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Shibata, E.; Park, J.; Jha, S.; Karnani, N.; Dutta, A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell 2010, 40, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Centore, R.C.; Havens, C.G.; Manning, A.L.; Li, J.M.; Flynn, R.L.; Tse, A.; Jin, J.; Dyson, N.J.; Walter, J.C.; Zou, L. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell 2010, 40, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.; Sy, S.M.; van Deursen, J.M.; Chen, J. Direct interaction between SET8 and proliferating cell nuclear antigen couples H4-K20 methylation with DNA replication. J. Biol. Chem. 2008, 283, 11073–11077. [Google Scholar] [CrossRef] [PubMed]
- Takawa, M.; Cho, H.S.; Hayami, S.; Toyokawa, G.; Kogure, M.; Yamane, Y.; Iwai, Y.; Maejima, K.; Ueda, K.; Masuda, A.; et al. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 2012, 72, 3217–3227. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Hubner, M.R.; Beck, D.B.; Vermeulen, M.; Hurwitz, J.; Spector, D.L.; Reinberg, D. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol. Cell 2010, 40, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Kachirskaia, I.; Yamaguchi, H.; West, L.E.; Wen, H.; Wang, E.W.; Dutta, S.; Appella, E.; Gozani, O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 2007, 27, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Dhami, G.K.; Liu, H.; Galka, M.; Voss, C.; Wei, R.; Muranko, K.; Kaneko, T.; Cregan, S.P.; Li, L.; Li, S.S. Dynamic methylation of Numb by Set8 regulates its binding to p53 and apoptosis. Mol. Cell 2013, 50, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Veschi, V.; Liu, Z.; Voss, T.C.; Ozbun, L.; Gryder, B.; Yan, C.; Hu, Y.; Ma, A.; Jin, J.; Mazur, S.J.; et al. Epigenetic siRNA and Chemical Screens Identify SETD8 Inhibition as a Therapeutic Strategy for p53 Activation in High-Risk Neuroblastoma. Cancer Cell 2017, 31, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Schotta, G.; Sengupta, R.; Kubicek, S.; Malin, S.; Kauer, M.; Callen, E.; Celeste, A.; Pagani, M.; Opravil, S.; De La Rosa-Velazquez, I.A.; et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008, 22, 2048–2061. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Siarheyeva, A.; Zeng, H.; Lam, R.; Dong, A.; Wu, X.H.; Li, Y.; Schapira, M.; Vedadi, M.; Min, J. Crystal structures of the human histone H4K20 methyltransferases SUV420H1 and SUV420H. FEBS Lett. 2013, 587, 3859–3868. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; de Lange, T. 53BP1: Pro choice in DNA repair. Trends Cell Biol. 2014, 24, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Tuzon, C.T.; Spektor, T.; Kong, X.; Congdon, L.M.; Wu, S.; Schotta, G.; Yokomori, K.; Rice, J.C. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep. 2014, 8, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Baldock, R.A.; Day, M.; Wilkinson, O.J.; Cloney, R.; Jeggo, P.A.; Oliver, A.W.; Watts, F.Z.; Pearl, L.H. ATM Localization and Heterochromatin Repair Depend on Direct Interaction of the 53BP1-BRCT2 Domain with gammaH2AX. Cell Rep. 2015, 13, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.L.; Swaroop, A.; Troche, C.; Licht, J.D. The Role of Nuclear Receptor-Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.; Milne, E.; Freeth, M. The cognitive profile of Sotos syndrome. J. Neuropsychol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bergemann, A.D.; Cole, F.; Hirschhorn, K. The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 2005, 21, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Trojer, P.; Xu, C.F.; Cheung, P.; Kuo, A.; Drury, W.J., 3rd; Qiao, Q.; Neubert, T.A.; Xu, R.M.; Gozani, O.; et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 2009, 284, 34283–34295. [Google Scholar] [CrossRef] [PubMed]
- Marango, J.; Shimoyama, M.; Nishio, H.; Meyer, J.A.; Min, D.J.; Sirulnik, A.; Martinez-Martinez, Y.; Chesi, M.; Bergsagel, P.L.; Zhou, M.M.; et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood 2008, 111, 3145–3154. [Google Scholar] [CrossRef] [PubMed]
- Morishita, M.; Mevius, D.; di Luccio, E. In vitro histone lysine methylation by NSD1, NSD2/MMSET/WHSC1 and NSD3/WHSC1L. BMC Struct. Biol. 2014, 14, 25. [Google Scholar]
- Rayasam, G.V.; Wendling, O.; Angrand, P.O.; Mark, M.; Niederreither, K.; Song, L.; Lerouge, T.; Hager, G.L.; Chambon, P.; Losson, R. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J. 2003, 22, 3153–3163. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Pesavento, J.J.; Starnes, T.W.; Cryderman, D.E.; Wallrath, L.L.; Kelleher, N.L.; Mizzen, C.A. Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J. Biol. Chem. 2008, 283, 12085–12092. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Zhang, L.; Luo, K.; Qin, Y.; Chesi, M.; Fei, F.; Bergsagel, P.L.; Wang, L.; You, Z.; Lou, Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011, 470, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.Y.; Mizzen, C.A. Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J. Mol. Cell Biol. 2013, 5, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Fortschegger, K.; Shiekhattar, R. Plant homeodomain fingers form a helping hand for transcription. Epigenetics 2011, 6, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Thompson, J.R.; Botuyan, M.V.; Mer, G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct. Mol. Biol. 2008, 15, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Mallette, F.A.; Mattiroli, F.; Cui, G.; Young, L.C.; Hendzel, M.J.; Mer, G.; Sixma, T.K.; Richard, S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012, 31, 1865–1878. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Pascual, G.; Liu, W.; Kaikkonen, M.U.; Do, K.; Spann, N.J.; Boutros, M.; Perrimon, N.; Rosenfeld, M.G.; Glass, C.K. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol. Cell 2012, 48, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.H.; Sarkissian, M.; Hu, G.Q.; Wang, Z.; Bhattacharjee, A.; Gordon, D.B.; Gonzales, M.; Lan, F.; Ongusaha, P.P.; Huarte, M.; et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 2010, 466, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Laumonnier, F.; Holbert, S.; Ronce, N.; Faravelli, F.; Lenzner, S.; Schwartz, C.E.; Lespinasse, J.; Van Esch, H.; Lacombe, D.; Goizet, C.; et al. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. J. Med. Genet. 2005, 42, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.M.; Shen, E.Y.; Bagot, R.C.; Anselmo, A.; Jiang, Y.; Javidfar, B.; Wojtkiewicz, G.J.; Cloutier, J.; Chen, J.W.; Sadreyev, R.; et al. Phf8 loss confers resistance to depression-like and anxiety-like behaviors in mice. Nat. Commun. 2017, 8, 15142. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, S.; Zhou, Y.; Han, Y.; Li, S.; Xu, Q.; Xu, L.; Zhu, Z.; Deng, Y.; Yu, L.; et al. Phf8 histone demethylase deficiency causes cognitive impairments through the mTOR pathway. Nat. Commun. 2018, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.X.; Yu, J.H.; Lorentzen, P.; Park, K.M.; Jamal, S.M.; Tabor, H.K.; Rauch, A.; Saenz, M.S.; Boltshauser, E.; Patterson, K.E.; et al. Gene discovery for Mendelian conditions via social networking: De novo variants in KDM1A cause developmental delay and distinctive facial features. Genet. Med. 2016, 18, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Tunovic, S.; Barkovich, J.; Sherr, E.H.; Slavotinek, A.M. De novo ANKRD11 and KDM1A gene mutations in a male with features of KBG syndrome and Kabuki syndrome. Am. J. Med. Genet. A 2014, 164A, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Pilotto, S.; Speranzini, V.; Marabelli, C.; Rusconi, F.; Toffolo, E.; Grillo, B.; Battaglioli, E.; Mattevi, A. LSD1/KDM1A mutations associated to a newly described form of intellectual disability impair demethylase activity and binding to transcription factors. Hum. Mol. Genet. 2016, 25, 2578–2587. [Google Scholar] [CrossRef] [PubMed]
- Laurent, B.; Ruitu, L.; Murn, J.; Hempel, K.; Ferrao, R.; Xiang, Y.; Liu, S.; Garcia, B.A.; Wu, H.; Wu, F.; et al. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol. Cell 2015, 57, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Telese, F.; Tan, Y.; Li, W.; Jin, C.; He, X.; Basnet, H.; Ma, Q.; Merkurjev, D.; Zhu, X.; et al. LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat. Neurosci. 2015, 18, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.S.; Nam, H.J.; Lee, J.; Kim, D.; Choi, J.E.; Kang, S.J.; Kim, S.; Kim, H.; Kwak, C.; Shim, K.W.; et al. PKCalpha-mediated phosphorylation of LSD1 is required for presynaptic plasticity and hippocampal learning and memory. Sci. Rep. 2017, 7, 4912. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.B.; Ma, J.Y.; Zhang, Q.H.; Lin, F.; Wang, Z.W.; Huang, L.; Schatten, H.; Sun, Q.Y. MBTD1 is associated with Pr-Set7 to stabilize H4K20me1 in mouse oocyte meiotic maturation. Cell Cycle 2013, 12, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Allali-Hassani, A.; Nady, N.; Qi, C.; Ouyang, H.; Liu, Y.; MacKenzie, F.; Vedadi, M.; Arrowsmith, C.H. L3MBTL1 recognition of mono- and dimethylated histones. Nat. Struct. Mol. Biol. 2007, 14, 1229–1230. [Google Scholar] [CrossRef] [PubMed]
- Trojer, P.; Li, G.; Sims, R.J., 3rd; Vaquero, A.; Kalakonda, N.; Boccuni, P.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Nimer, S.D.; et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 2007, 129, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Acs, K.; Luijsterburg, M.S.; Ackermann, L.; Salomons, F.A.; Hoppe, T.; Dantuma, N.P. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 2011, 18, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Shen, E.Y.; Jiang, Y.; Mao, W.; Futai, K.; Hock, H.; Akbarian, S. Cognition and mood-related behaviors in L3mbtl1 null mutant mice. PLoS ONE 2015, 10, e0121252. [Google Scholar] [CrossRef] [PubMed]
- Scoumanne, A.; Chen, X. Protein methylation: A new mechanism of p53 tumor suppressor regulation. Histol. Histopathol. 2008, 23, 1143–1149. [Google Scholar] [PubMed]
- Chitale, S.; Richly, H. H4K20me2: Orchestrating the recruitment of DNA repair factors in nucleotide excision repair. Nucleus 2018, 9, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Li, C.; Yin, Z.; Wen, J.; Meng, H.; Xue, L.; Wang, J. Histone methylation in DNA repair and clinical practice: New findings during the past 5-years. J. Cancer 2018, 9, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Renaud, E.; Barascu, A.; Rosselli, F. Impaired TIP60-mediated H4K16 acetylation accounts for the aberrant chromatin accumulation of 53BP1 and RAP80 in Fanconi anemia pathway-deficient cells. Nucleic Acids Res. 2016, 44, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, J.H.; Lee, I.S.; Lee, S.B.; Cho, K.S. Histone Lysine Methylation and Neurodevelopmental Disorders. Int. J. Mol. Sci. 2017, 18, 1404. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Han, Y.; Petrovski, S.; Owzar, K.; Goldstein, D.B.; Allen, A.S. Incorporating Functional Information in Tests of Excess De Novo Mutational Load. Am. J. Hum. Genet. 2015, 97, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Stessman, H.A.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Sestan, N.; State, M.W. Lost in Translation: Traversing the Complex Path from Genomics to Therapeutics in Autism Spectrum Disorder. Neuron 2018, 100, 406–423. [Google Scholar] [CrossRef] [PubMed]
- Kleine-Kohlbrecher, D.; Christensen, J.; Vandamme, J.; Abarrategui, I.; Bak, M.; Tommerup, N.; Shi, X.; Gozani, O.; Rappsilber, J.; Salcini, A.E.; et al. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol. Cell 2010, 38, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Denovo-db, Seattle, WA, USA. Available online: Denovo-db.gs.washington.edu (accessed on 27 December 2018).
- Faundes, V.; Newman, W.G.; Bernardini, L.; Canham, N.; Clayton-Smith, J.; Dallapiccola, B.; Davies, S.J.; Demos, M.K.; Goldman, A.; Gill, H.; et al. Clinical Assessment of the Utility of Sequencing and Evaluation as a Service (CAUSES) Study; Deciphering Developmental Disorders (DDD) Study; Banka, S. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am. J. Hum. Genet. 2018, 102, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Ding, S.L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional landscape of the prenatal human brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.L.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Biron, V.L.; McManus, K.J.; Hu, N.; Hendzel, M.J.; Underhill, D.A. Distinct dynamics and distribution of histone methyl-lysine derivatives in mouse development. Dev. Biol. 2004, 276, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Stadler, F.; Kolb, G.; Rubusch, L.; Baker, S.P.; Jones, E.G.; Akbarian, S. Histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain. J. Neurochem. 2005, 94, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, A.; Steward, R. Aberrant monomethylation of histone H4 lysine 20 activates the DNA damage checkpoint in Drosophila melanogaster. J. Cell Biol. 2007, 176, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Okamoto, I.; Murphy, N.; Chu, J.; Price, S.M.; Shen, M.M.; Torres-Padilla, M.E.; Heard, E.; Reinberg, D. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol. Cell. Biol. 2009, 29, 2278–2295. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Xu, P.F.; Zhou, T.; Hu, M.; Fu, C.T.; Zhang, Y.; Jin, Y.; Chen, Y.; Chen, S.J.; Huang, Q.H.; et al. Genome-wide survey and developmental expression mapping of zebrafish SET domain-containing genes. PLoS ONE 2008, 3, e1499. [Google Scholar] [CrossRef] [PubMed]
- Thisse, B.; Heyer, V.; Lux, A.; Alunni, V.; Degrave, A.; Seiliez, I.; Kirchner, J.; Parkhill, J.P.; Thisse, C. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 2004, 77, 505–519. [Google Scholar] [PubMed]
- Nicetto, D.; Hahn, M.; Jung, J.; Schneider, T.D.; Straub, T.; David, R.; Schotta, G.; Rupp, R.A. Suv4-20h histone methyltransferases promote neuroectodermal differentiation by silencing the pluripotency-associated Oct-25 gene. PLoS Genet. 2013, 9, e1003188. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.T.; Sandstrom, R.S.; Huang, S.A.; Wang, Y.; Schotta, G.; Berger, M.S.; Lin, C.A. Cross-species Analyses Unravel the Complexity of H3K27me3 and H4K20me3 in the Context of Neural Stem Progenitor Cells. Neuroepigenetics 2016, 6, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.T.; Zunino, G.; Huang, S.A.; Cardona, S.M.; Cardona, A.E.; Berger, M.S.; Lemmon, V.P.; Lin, C.A. Region specific knock-out reveals distinct roles of chromatin modifiers in adult neurogenic niches. Cell Cycle 2018, 17, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Evertts, A.G.; Manning, A.L.; Wang, X.; Dyson, N.J.; Garcia, B.A.; Coller, H.A. H4K20 methylation regulates quiescence and chromatin compaction. Mol. Biol. Cell 2013, 24, 3025–3037. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Zhang, X.; Fang, Y.; Nie, Y.; Cai, S.; Chen, Y.; Mo, D. mir-127-3p inhibits the proliferation of myocytes by targeting KMT5a. Biochem. Biophys. Res. Commun. 2018, 503, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Tsang, L.W.; Hu, N.; Underhill, D.A. Comparative analyses of SUV420H1 isoforms and SUV420H2 reveal differences in their cellular localization and effects on myogenic differentiation. PLoS ONE 2010, 5, e14447. [Google Scholar] [CrossRef] [PubMed]
- Terranova, R.; Sauer, S.; Merkenschlager, M.; Fisher, A.G. The reorganisation of constitutive heterochromatin in differentiating muscle requires HDAC activity. Exp. Cell Res. 2005, 310, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Boonsanay, V.; Zhang, T.; Georgieva, A.; Kostin, S.; Qi, H.; Yuan, X.; Zhou, Y.; Braun, T. Regulation of Skeletal Muscle Stem Cell Quiescence by Suv4-20h1-Dependent Facultative Heterochromatin Formation. Cell Stem Cell 2016, 18, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Neguembor, M.V.; Xynos, A.; Onorati, M.C.; Caccia, R.; Bortolanza, S.; Godio, C.; Pistoni, M.; Corona, D.F.; Schotta, G.; Gabellini, D. FSHD muscular dystrophy region gene 1 binds Suv4-20h1 histone methyltransferase and impairs myogenesis. J. Mol. Cell Biol. 2013, 5, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Chekhun, V.F.; Lukyanova, N.Y.; Kovalchuk, O.; Tryndyak, V.P.; Pogribny, I.P. Epigenetic profiling of multidrug-resistant human MCF-7 breast adenocarcinoma cells reveals novel hyper- and hypomethylated targets. Mol. Cancer Ther. 2007, 6, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Tryndyak, V.P.; Kovalchuk, O.; Pogribny, I.P. Loss of DNA methylation and histone H4 lysine 20 trimethylation in human breast cancer cells is associated with aberrant expression of DNA methyltransferase 1, Suv4-20h2 histone methyltransferase and methyl-binding proteins. Cancer Biol. Ther. 2006, 5, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.; Burford, A.; Molinari, V.; Kessler, K.; Popov, S.; Clarke, M.; Taylor, K.R.; Pemberton, H.N.; Lord, C.J.; Gutteridge, A.; et al. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat. Med. 2018, 24, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kimball, S.; Liu, H.; Holowatyj, A.; Yang, Z.Q. Genetic alterations of histone lysine methyltransferases and their significance in breast cancer. Oncotarget 2015, 6, 2466–2482. [Google Scholar] [CrossRef] [PubMed]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cortez, V.C.; Martinez-Redondo, P.; Catala-Moll, F.; Rodriguez-Ubreva, J.; Garcia-Gomez, A.; Poorani-Subramani, G.; Ciudad, L.; Hernando, H.; Perez-Garcia, A.; Company, C.; et al. Activation-induced cytidine deaminase targets SUV4-20-mediated histone H4K20 trimethylation to class-switch recombination sites. Sci. Rep. 2017, 7, 7594. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (HUGO) | Alias | Synonymous (z) | Missense (z) | LoF (pLI) | Associated Disorder |
|---|---|---|---|---|---|
| KMT5A | SETD8 | 0.66 | 2.44 | 0.95 | - |
| KMT5B | SUV420H1 | −0.29 | 2.71 | 1.00 | MR, AD |
| KMT5C | SUV420H2 | −1.82 | 1.99 | 0.69 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wickramasekara, R.N.; Stessman, H.A.F. Histone 4 Lysine 20 Methylation: A Case for Neurodevelopmental Disease. Biology 2019, 8, 11. https://doi.org/10.3390/biology8010011
Wickramasekara RN, Stessman HAF. Histone 4 Lysine 20 Methylation: A Case for Neurodevelopmental Disease. Biology. 2019; 8(1):11. https://doi.org/10.3390/biology8010011
Chicago/Turabian StyleWickramasekara, Rochelle N., and Holly A. F. Stessman. 2019. "Histone 4 Lysine 20 Methylation: A Case for Neurodevelopmental Disease" Biology 8, no. 1: 11. https://doi.org/10.3390/biology8010011
APA StyleWickramasekara, R. N., & Stessman, H. A. F. (2019). Histone 4 Lysine 20 Methylation: A Case for Neurodevelopmental Disease. Biology, 8(1), 11. https://doi.org/10.3390/biology8010011