Two-Component FAD-Dependent Monooxygenases: Current Knowledge and Biotechnological Opportunities

 ,
,  ,
, 
Abstract
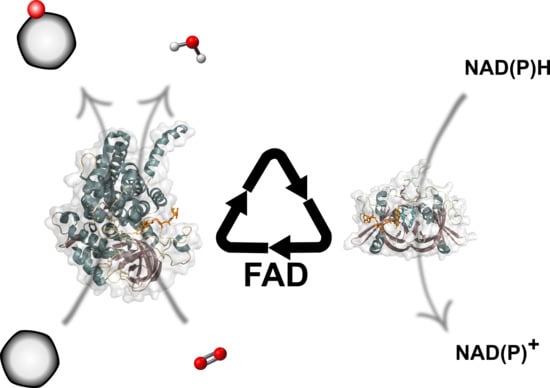
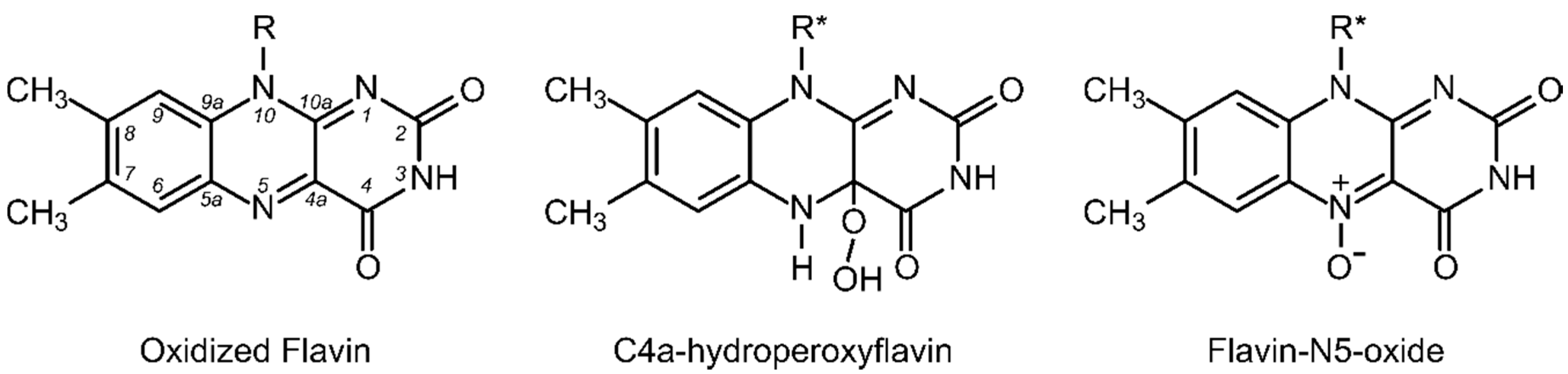
1. Introduction
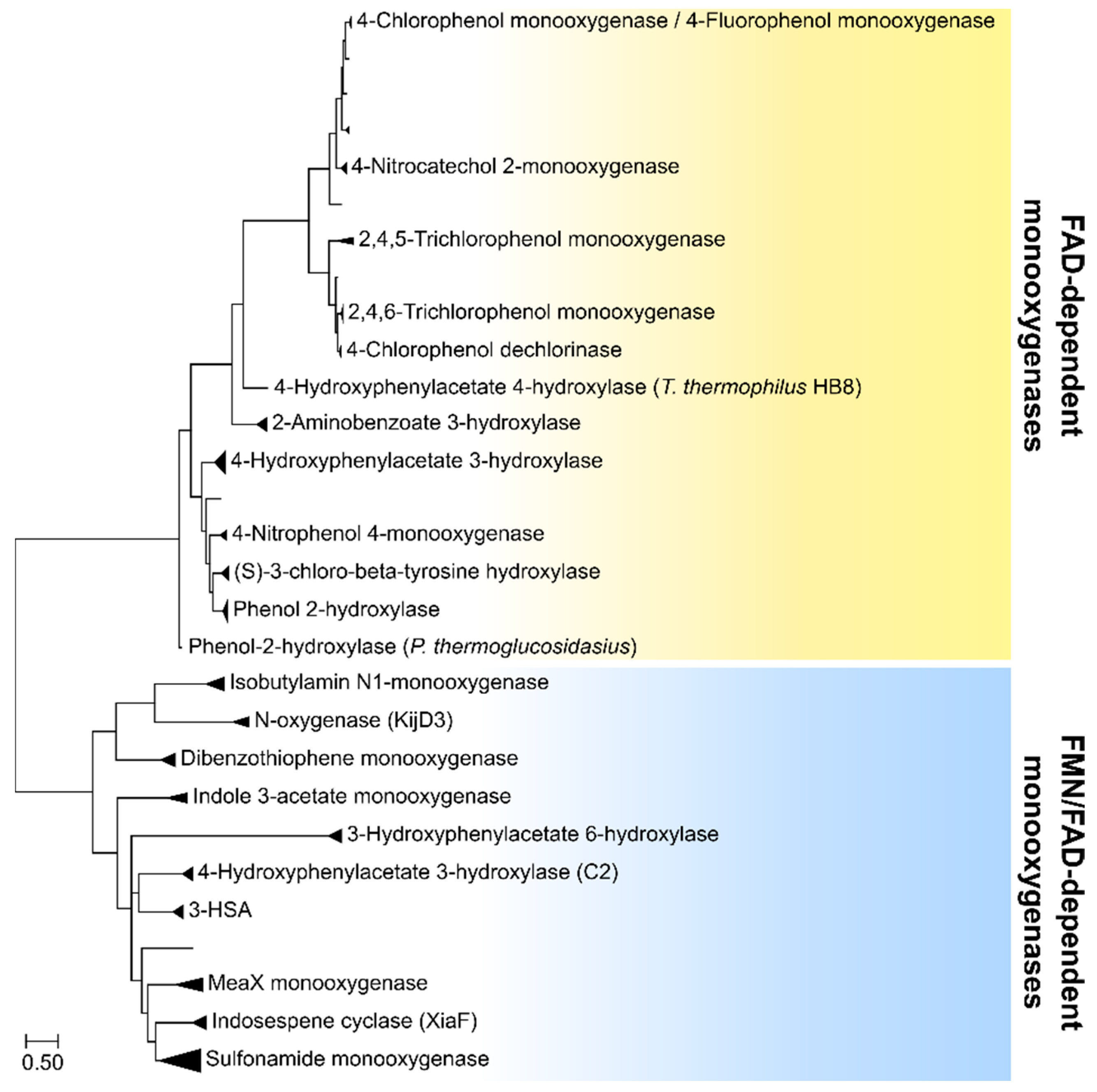
2. Two-Component FAD-Dependent Monooxygenase Systems
2.1. Group D Flavoprotein Monooxygenases
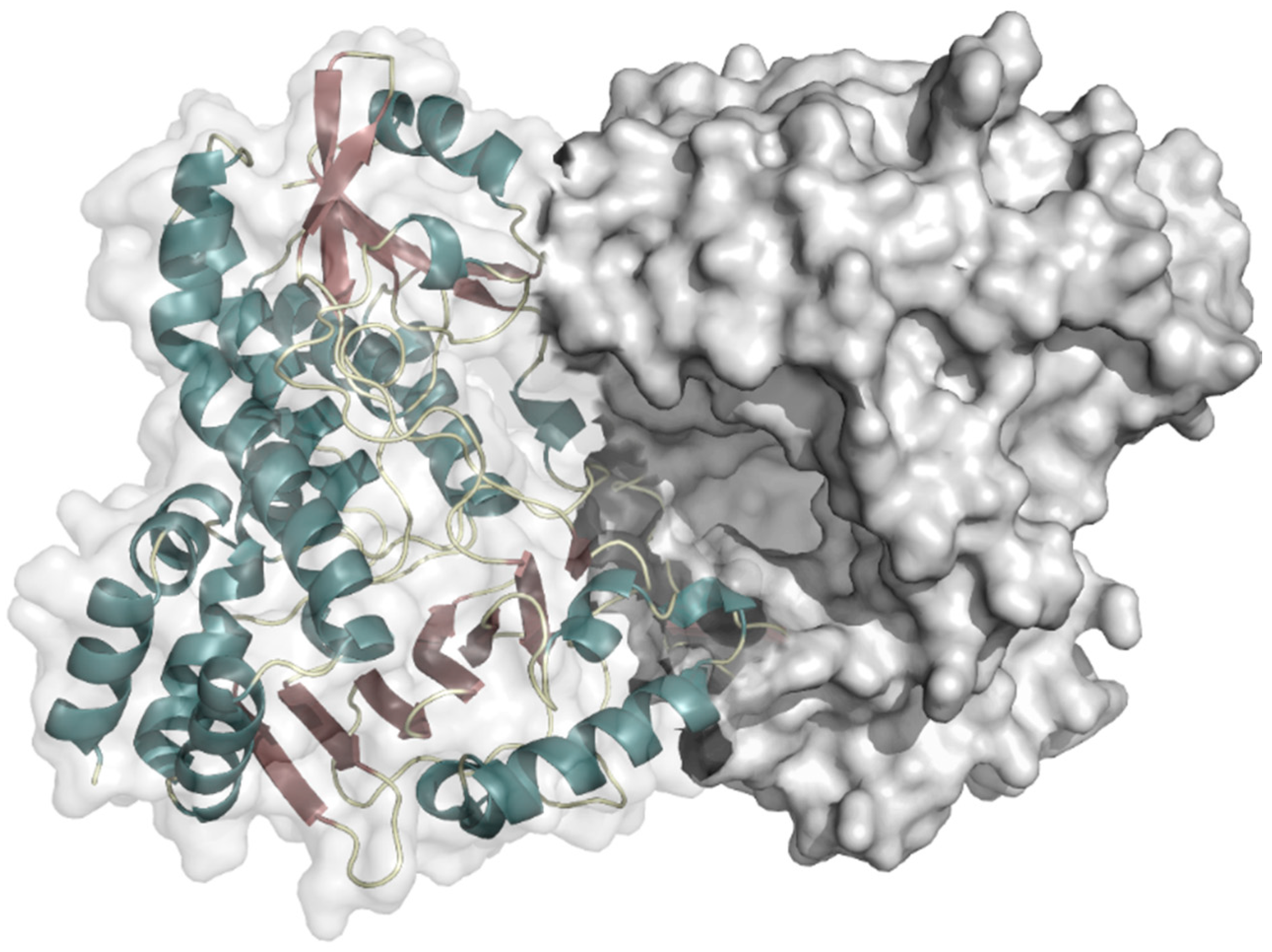
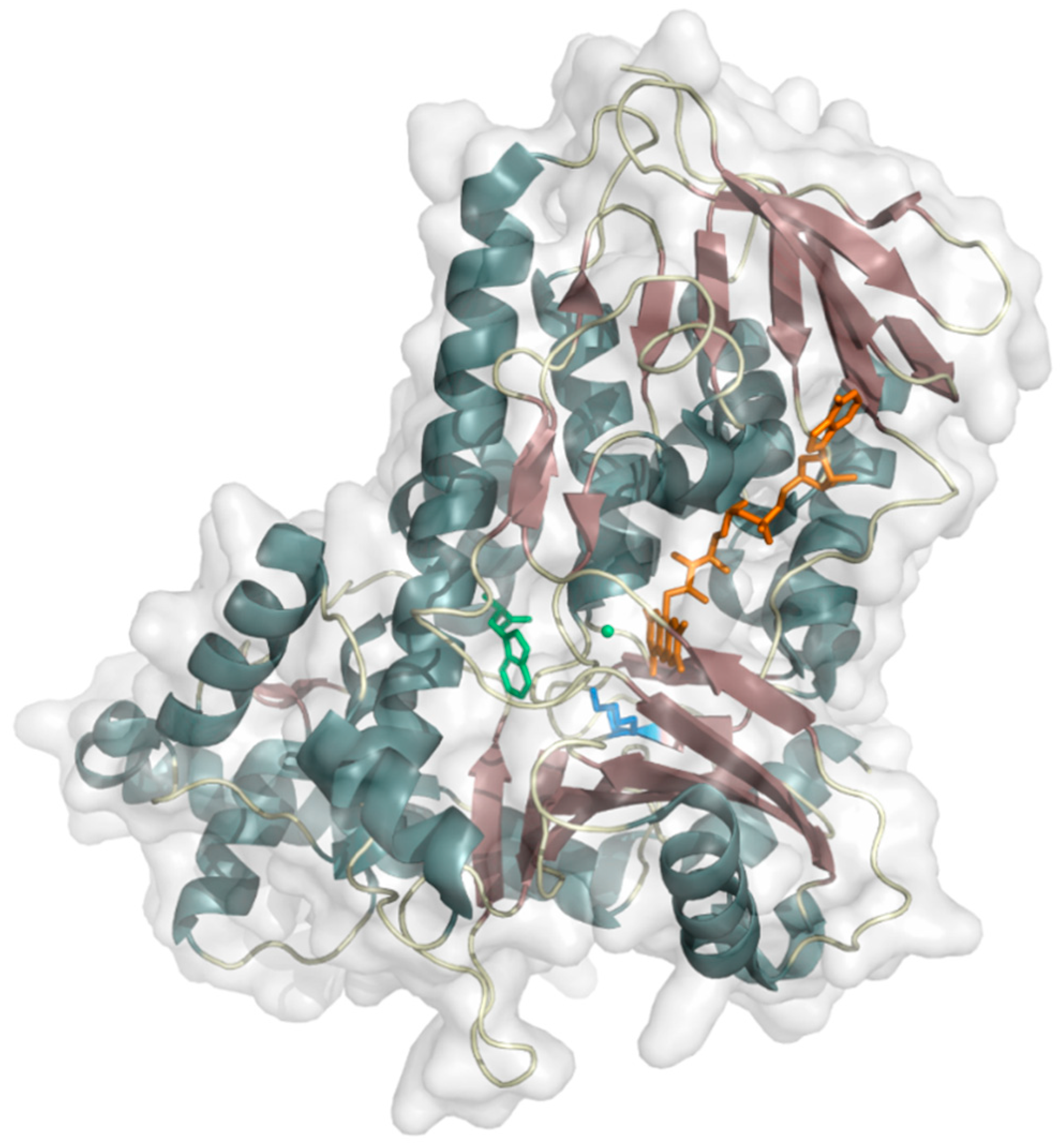
2.1.1. Phenol 2-Hydroxylase
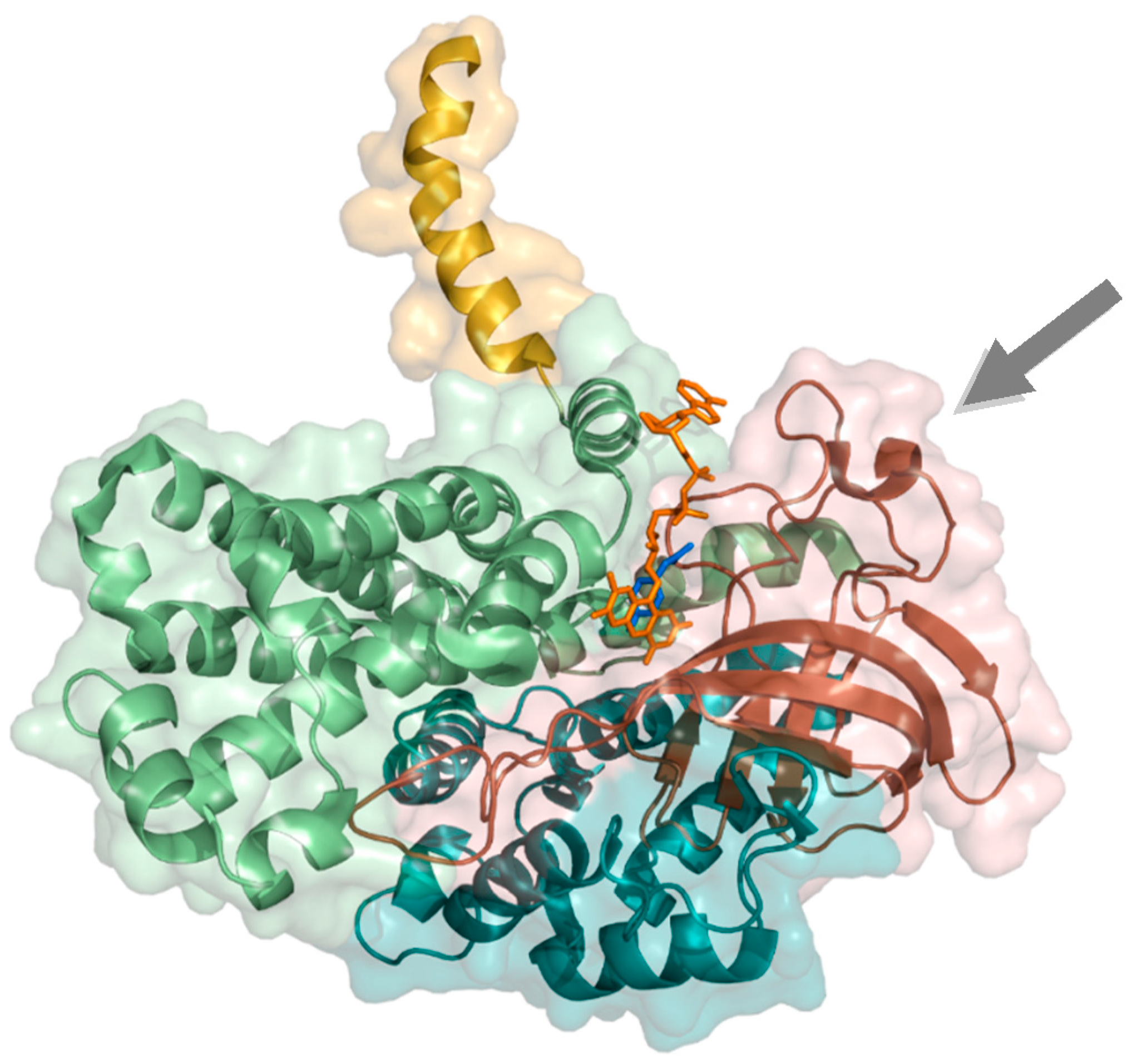
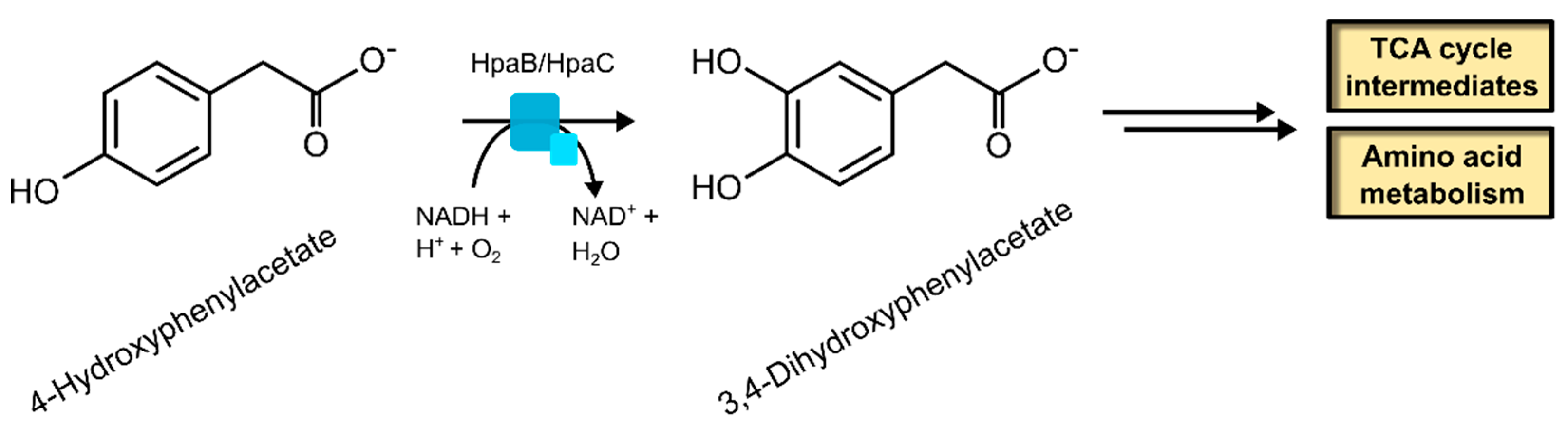
2.1.2. 4-Hydroxyphenylacetate 3-Hydroxylase
2.1.3. 4-Chlorophenol Dechlorinase
2.1.4. Biotechnological Relevance of Group D Flavoprotein Monooxygenases
2.2. Group E Flavoprotein Monooxygenases
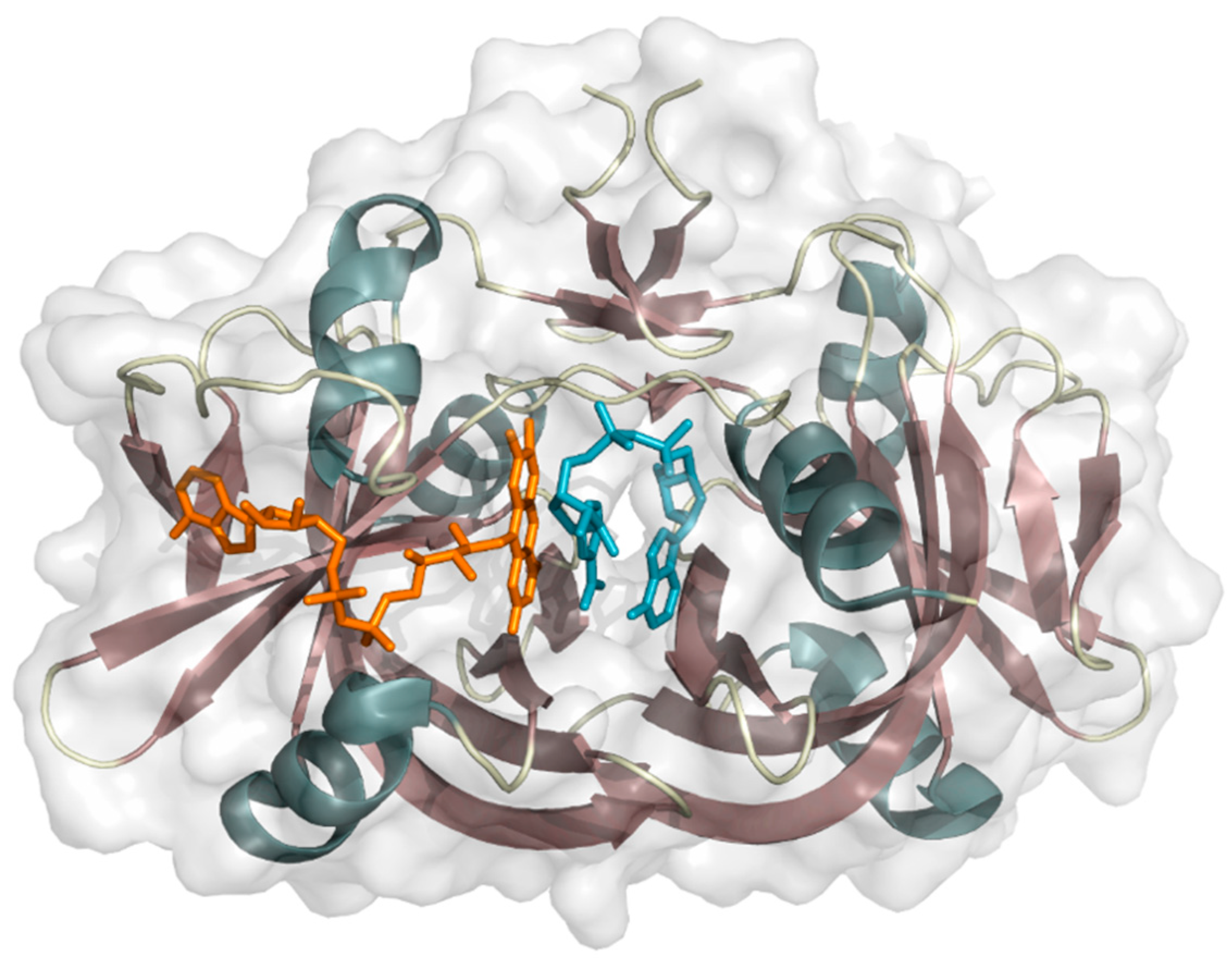
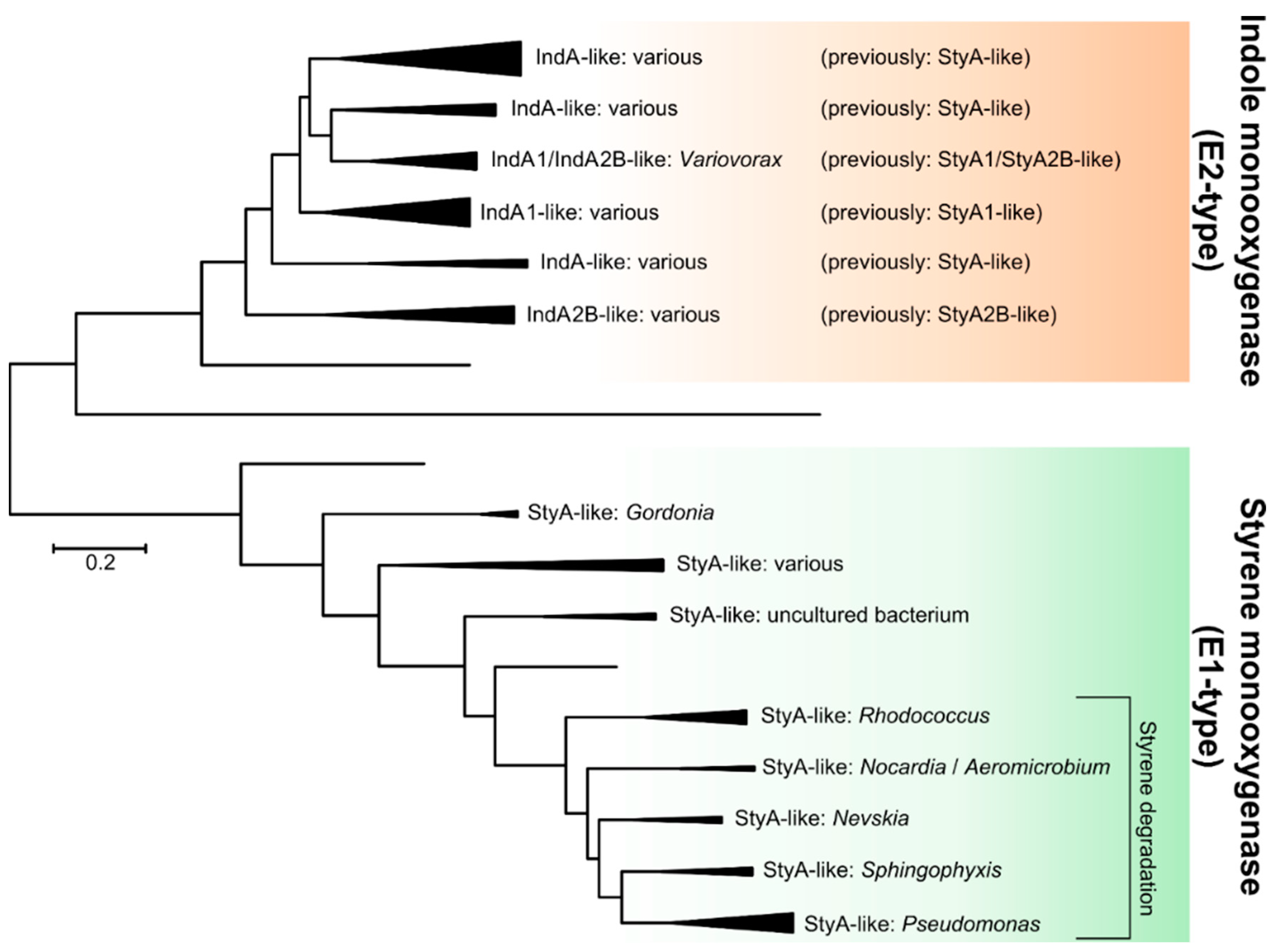
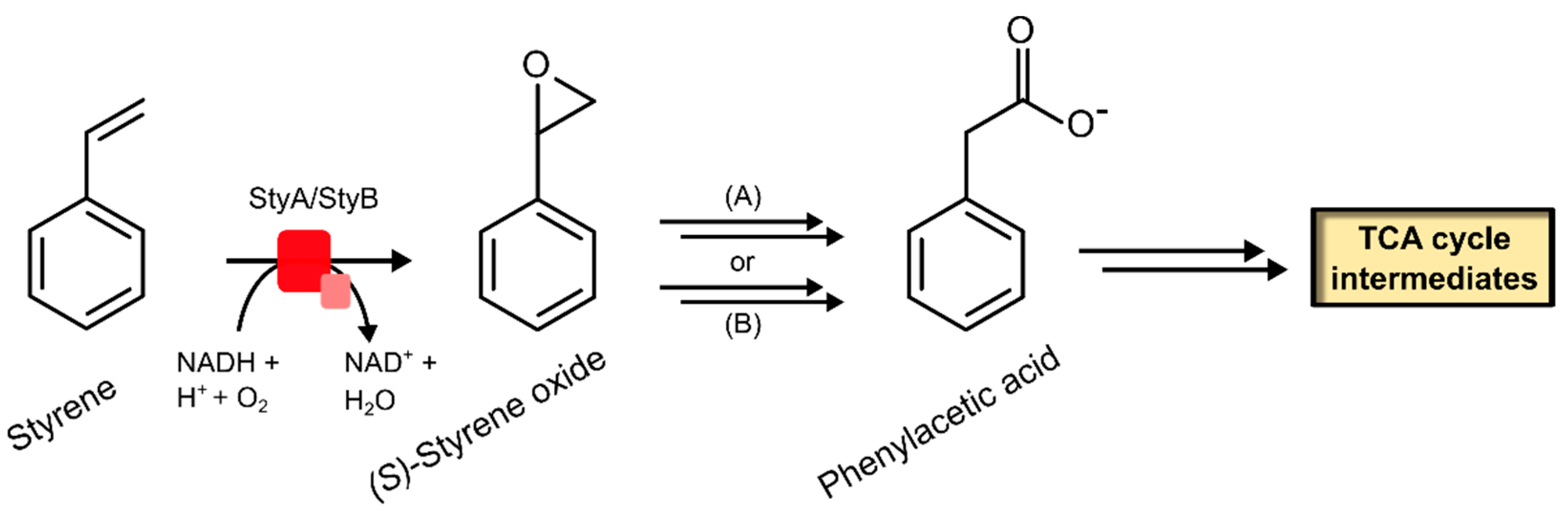
2.2.1. Styrene Monooxygenase
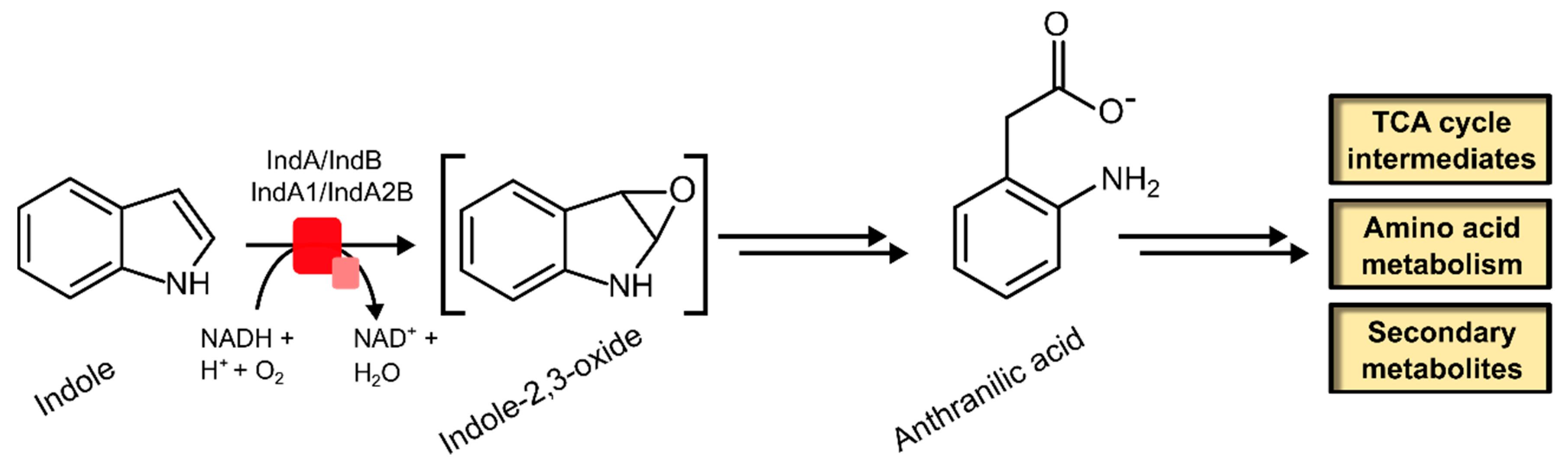
2.2.2. Indole Monooxygenase
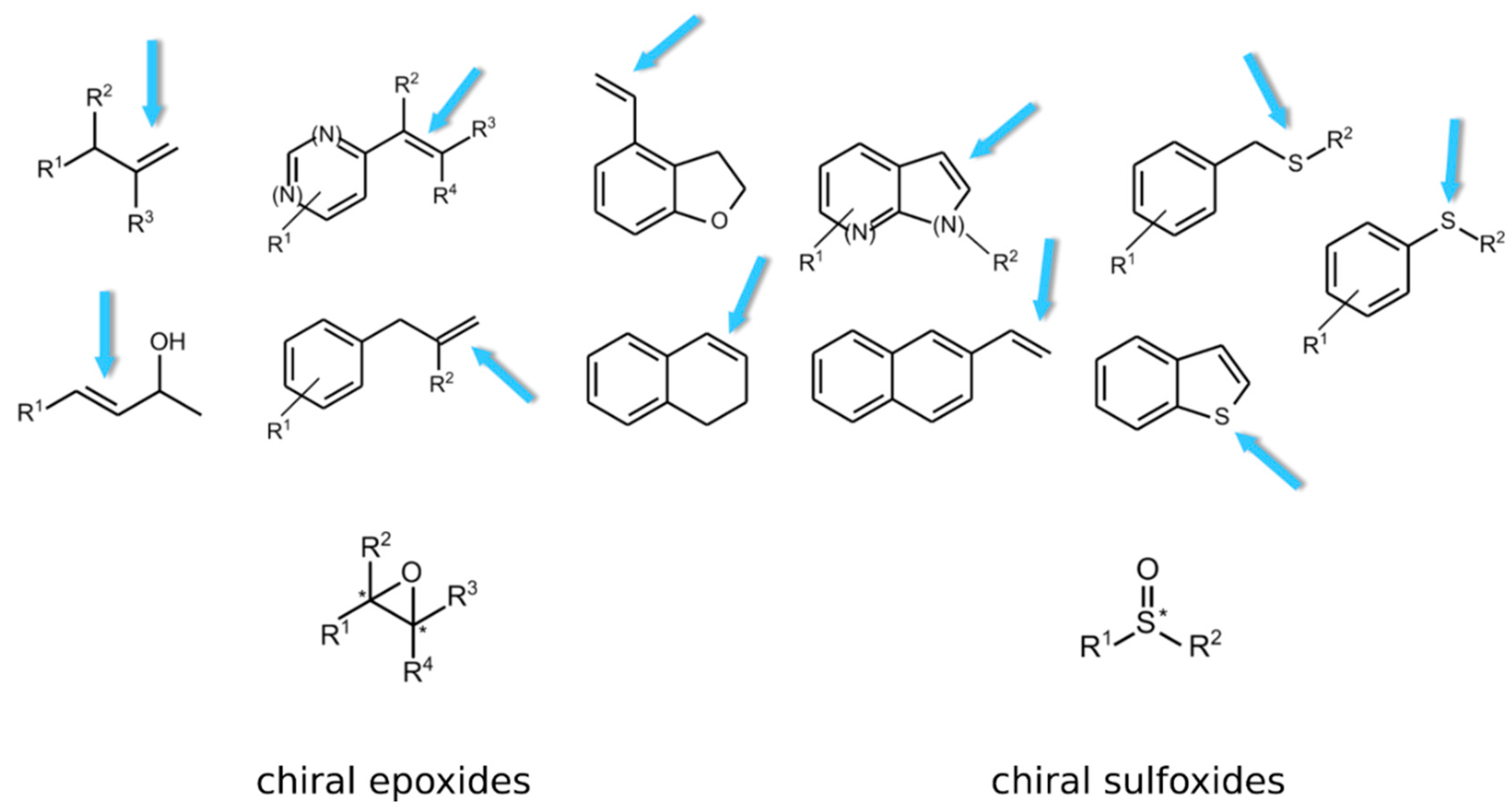
2.2.3. Biotechnological Relevance of Group E Flavoprotein Monooxygenases
2.3. Group F Flavoprotein Monooxygenases
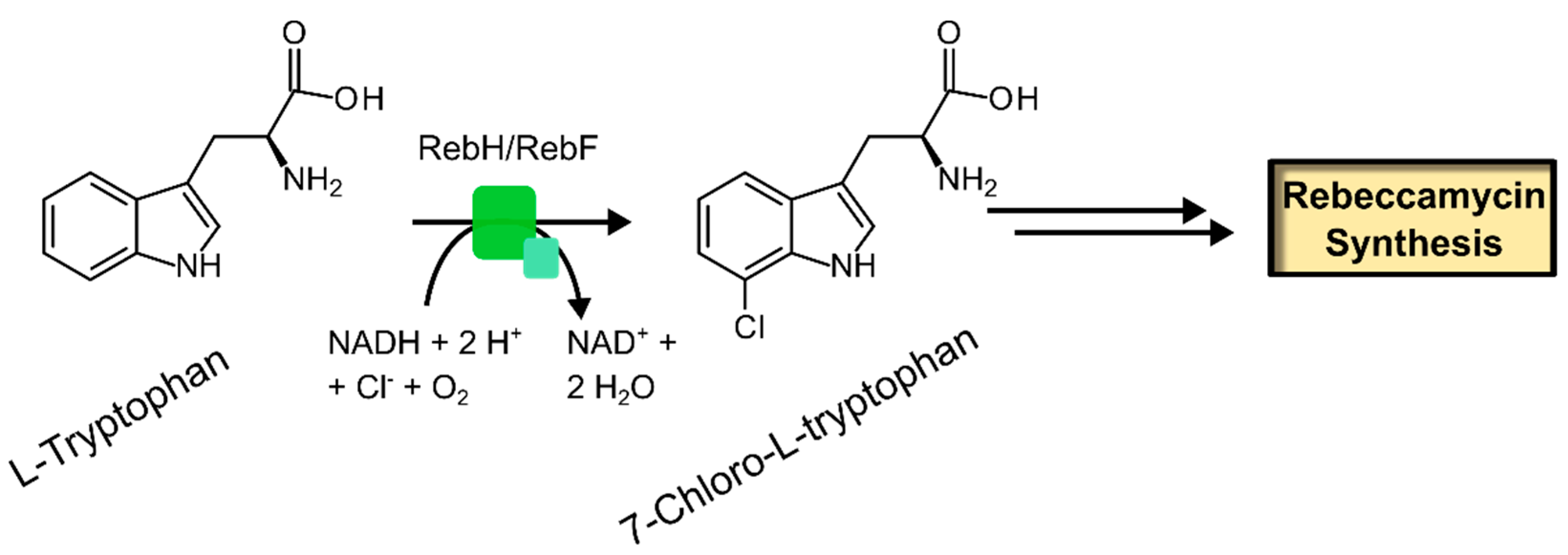
2.3.1. Free Substrate Halogenases
2.3.2. Carrier Protein-Bound Substrate Halogenases
2.3.3. Biotechnological Relevance of Group F Flavoprotein Monooxygenases
3. Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fagan, R.L.; Palfey, B.A. Flavin-dependent enzymes. In Comprehensive Natural Products II: Chemistry and Biology; Elsevier Ltd.: New York, NY, USA, 2010; pp. 37–113. [Google Scholar]
- Hefti, M.H.; Vervoort, J.; van Berkel, W.J.H. Deflavination and reconstitution of flavoproteins. Eur. J. Biochem. 2003, 270, 4227–4242. [Google Scholar] [CrossRef] [PubMed]
- Macheroux, P.; Kappes, B.; Ealick, S.E. Flavogenomics—A genomic and structural view of flavin-dependent proteins. FEBS J. 2011, 278, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Joosten, V.; van Berkel, W.J.H. Flavoenzymes. Curr. Opin. Chem. Biol. 2007, 11, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Van Berkel, W.J.H. Chemistry of Flavoenzymes. In Wiley Encyclopedia of Chemical Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Sobrado, P. Noncanonical reactions of flavoenzymes. Int. J. Mol. Sci. 2012, 13, 14219–14242. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, M.M.E.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J.H. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Van Berkel, W.J.H.; Kamerbeek, N.M.; Fraaije, M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006, 4, 670–689. [Google Scholar] [CrossRef] [PubMed]
- Chaiyen, P.; Fraaije, M.W.; Mattevi, A. The enigmatic reaction of flavins with oxygen. Trends Biochem. Sci. 2012, 37, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Romero, E.; Gómez Castellanos, J.R.; Gadda, G.; Fraaije, M.W.; Mattevi, A. Same substrate, many reactions: Oxygen activation in flavoenzymes. Chem. Rev. 2018, 118, 1742–1769. [Google Scholar] [CrossRef] [PubMed]
- Massey, V. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 1994, 269, 22459–22462. [Google Scholar] [PubMed]
- Drees, S.L.; Ernst, S.; Belviso, B.D.; Jagmann, N.; Hennecke, U.; Fetzner, S. PqsL uses reduced flavin to produce 2-hydroxylaminobenzoylacetate, a preferred PqsBC substrate in alkyl quinolone biosynthesis in Pseudomonas aeruginosa. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bučko, M.; Gemeiner, P.; Schenkmayerová, A.; Krajčovič, T.; Rudroff, F.; Mihovilovič, M.D. Baeyer-Villiger oxidations: Biotechnological approach. Appl. Microbiol. Biotechnol. 2016, 100, 6585–6599. [Google Scholar] [CrossRef] [PubMed]
- Mascotti, M.L.; Lapadula, W.J.; Juri Ayub, M. The origin and evolution of Baeyer-Villiger monooxygenases (BVMOs): An ancestral family of flavin monooxygenases. PLoS ONE 2015, 10, e0132689. [Google Scholar] [CrossRef] [PubMed]
- Catucci, G.; Gao, C.; Sadeghi, S.J.; Gilardi, G. Chemical applications of Class B flavoprotein monooxygenases. Rend. Lincei. 2017, 28, 195–206. [Google Scholar] [CrossRef]
- Goncalves, L.C.P.; Kracher, D.; Milker, S.; Fink, M.J.; Rudroff, F.; Ludwig, R.; Bommarius, A.S.; Mihovilovic, M.D. Mutagenesis-independent stabilization of class B flavin monooxygenases in operation. Adv. Synth. Catal. 2017, 359, 2121–2131. [Google Scholar] [CrossRef]
- Sucharitakul, J.; Tinikul, R.; Chaiyen, P. Mechanisms of reduced flavin transfer in the two-component flavin-dependent monooxygenases. Arch. Biochem. Biophys. 2014, 555–556, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Sutton, W.B. Mechanism of action and crystallization of lactic oxidative decarboxylase from Mycobacterium phlei. J. Biol. Chem. 1957, 226, 395–405. [Google Scholar] [PubMed]
- Massey, V. The chemical and biological versatility of riboflavin. Biochem. Soc. Trans. 2000, 28, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Torres Pazmiño, D.E.; Dudek, H.M.; Fraaije, M.W. Baeyer–Villiger monooxygenases: Recent advances and future challenges. Curr. Opin. Chem. Biol. 2010, 14, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Torres Pazmiño, D.E.; Winkler, M.; Glieder, A.; Fraaije, M.W. Monooxygenases as biocatalysts: Classification, mechanistic aspects and biotechnological applications. J. Biotechnol. 2010, 146, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Montersino, S.; Tischler, D.; Gassner, G.T.; van Berkel, W.J.H. Catalytic and structural features of flavoprotein hydroxylases and epoxidases. Adv. Synth. Catal. 2011, 353, 2301–2319. [Google Scholar] [CrossRef]
- Palfey, B.A.; McDonald, C.A. Control of catalysis in flavin-dependent monooxygenases. Arch. Biochem. Biophys. 2010, 493, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Adak, S.; Begley, T.P. Dibenzothiophene catabolism proceeds via a flavin-N5-oxide intermediate. J. Am. Chem. Soc. 2016, 138, 6424–6426. [Google Scholar] [CrossRef] [PubMed]
- Adak, S.; Begley, T.P. Flavin-N5-oxide: A new, catalytic motif in flavoenzymology. Arch. Biochem. Biophys. 2017, 632, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Saleem-Batcha, R.; Stull, F.; Sanders, J.N.; Moore, B.S.; Palfey, B.A.; Houk, K.N.; Teufel, R. Enzymatic control of dioxygen binding and functionalization of the flavin cofactor. Proc. Natl. Acad. Sci. USA 2018, 115, 4909–4914. [Google Scholar] [CrossRef] [PubMed]
- Piano, V.; Palfey, B.A.; Mattevi, A. Flavins as covalent catalysts: New mechanisms emerge. Trends Biochem. Sci. 2017, 42, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Fernández-Fueyo, E.; Hollmann, F.; Paul, C.; Pasic, M.; Schmidt, S.; Wang, Y.; Younes, S.; Zhang, W. Biocatalytic oxidation reactions—A Chemist’s perspective. Angew. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Holtmann, D.; Hollmann, F. The oxygen dilemma: A severe challenge for the application of monooxygenases? ChemBioChem 2016, 17, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Panke, S.; Held, M.; Wubbolts, M.G.; Witholt, B.; Schmid, A. Pilot-scale production of (S)-styrene oxide from styrene by recombinant Escherichia coli synthesizing styrene monooxygenase. Biotechnol. Bioeng. 2002, 80, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Coulombel, L.; Nolan, L.C.; Nikodinovic, J.; Doyle, E.M.; O’Connor, K.E. Biotransformation of 4-halophenols to 4-halocatechols using Escherichia coli expressing 4-hydroxyphenylacetate 3-hydroxylase. Appl. Microbiol. Biotechnol. 2011, 89, 1867–1875. [Google Scholar] [CrossRef] [PubMed]
- Ceccoli, R.D.; Bianchi, D.A.; Rial, D.V. Flavoprotein monooxygenases for oxidative biocatalysis: Recombinant expression in microbial hosts and applications. Front. Microbiol. 2014, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Furuya, T.; Kino, K. Catalytic activity of the two-component flavin-dependent monooxygenase from Pseudomonas aeruginosa toward cinnamic acid derivatives. Appl. Microbiol. Biotechnol. 2014, 98, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Hossain, G.S.; Li, J.; Shin, H.-D.; Du, G.; Chen, J.; Liu, L. Metabolic engineering of cofactor flavin adenine dinucleotide (FAD) synthesis and regeneration in Escherichia coli for production of α-keto acids. Biotechnol. Bioeng. 2017, 114, 1928–1936. [Google Scholar] [CrossRef] [PubMed]
- Kara, S.; Schrittwieser, J.H.; Hollmann, F.; Ansorge-Schumacher, M.B. Recent trends and novel concepts in cofactor-dependent biotransformations. Appl. Microbiol. Biotechnol. 2014, 98, 1517–1529. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, F.; Witholt, B.; Schmid, A. [Cp∗Rh(bpy)(H2O)]2+: A versatile tool for efficient and non-enzymatic regeneration of nicotinamide and flavin coenzymes. J. Mol. Catal. B Enzym. 2002, 19–20, 167–176. [Google Scholar] [CrossRef]
- Hollmann, F.; Hofstetter, K.; Habicher, T.; Hauer, B.; Schmid, A. Direct electrochemical regeneration of monooxygenase subunits for biocatalytic asymmetric epoxidation. J. Am. Chem. Soc. 2005, 127, 6540–6541. [Google Scholar] [CrossRef] [PubMed]
- Burai, T.N.; Panay, A.J.; Zhu, H.; Lian, T.; Lutz, S. Light-driven, quantum dot-mediated regeneration of FMN to drive reduction of ketoisophorone by old yellow enzyme. ACS Catal. 2012, 2, 667–670. [Google Scholar] [CrossRef]
- Paul, C.E.; Gargiulo, S.; Opperman, D.J.; Lavandera, I.; Gotor-Fernández, V.; Gotor, V.; Taglieber, A.; Arends, I.W.C.E.; Hollmann, F. Mimicking nature: Synthetic nicotinamide cofactors for C=C bioreduction using enoate reductases. Org. Lett. 2013, 15, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Knaus, T.; Paul, C.E.; Levy, C.W.; de Vries, S.; Mutti, F.G.; Hollmann, F.; Scrutton, N.S. Better than nature: Nicotinamide biomimetics that outperform natural coenzymes. J. Am. Chem. Soc. 2016, 138, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Köhler, V.; Paul, C.E.; Hollmann, F.; Ward, T.R. Efficient in situ regeneration of NADH mimics by an artificial metalloenzyme. ACS Catal. 2016, 6, 3553–3557. [Google Scholar] [CrossRef]
- Paul, C.E.; Hollmann, F. A survey of synthetic nicotinamide cofactors in enzymatic processes. Appl. Microbiol. Biotechnol. 2016, 100, 4773–4778. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.E.; Tischler, D.; Riedel, A.; Heine, T.; Itoh, N.; Hollmann, F. Nonenzymatic regeneration of styrene monooxygenase for catalysis. ACS Catal. 2015, 5, 2961–2965. [Google Scholar] [CrossRef]
- Ellis, H.R. The FMN-dependent two-component monooxygenase systems. Arch. Biochem. Biophys. 2010, 497, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mascotti, M.L.; Ayub, M.J.; Furnham, N.; Thornton, J.M.; Laskowski, R.A. Chopping and changing: The evolution of the flavin-dependent monooxygenases. J. Mol. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Moonen, M.J.H.; Fraaije, M.W.; Rietjens, I.M.C.M.; Laane, C.; van Berkel, W.J.H. Flavoenzyme-catalyzed oxygenations and oxidations of phenolic compounds. Adv. Synth. Catal. 2002, 344, 1023–1035. [Google Scholar] [CrossRef]
- Ullrich, R.; Hofrichter, M. Enzymatic hydroxylation of aromatic compounds. Cell. Mol. Life Sci. 2007, 64, 271–293. [Google Scholar] [CrossRef] [PubMed]
- Kugel, S.; Baunach, M.; Baer, P.; Ishida-Ito, M.; Sundaram, S.; Xu, Z.; Groll, M.; Hertweck, C. Cryptic indole hydroxylation by a non-canonical terpenoid cyclase parallels bacterial xenobiotic detoxification. Nat. Commun. 2017, 8, 15804. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.J.; Li, W. Purification and characterization of isobutylamine N-hydroxylase from the valanimycin producer Streptomyces viridifaciens MG456-hF10. Arch. Biochem. Biophys. 1997, 339, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.; Schräder, T.; Andreesen, J.R. Two-component flavin-dependent pyrrole-2-carboxylate monooxygenase from Rhodococcus sp. FEBS J. 1997, 249, 739–747. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Liu, H.; Xiao, Y.; Zhang, X.-E.; Zhou, N.-Y. Identification and characterization of catabolic para-nitrophenol 4-monooxygenase and para-benzoquinone reductase from Pseudomonas sp. strain WBC-3. J. Bacteriol. 2009, 191, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dong, Y.; Li, X.; Ren, Y.; Li, Y.; Wang, W.; Wang, L.; Feng, L. Characterization of the anthranilate degradation pathway in Geobacillus thermodenitrificans NG80-2. Microbiology 2010, 156, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Xun, L.; Sandvik, E.R. Characterization of 4-hydroxyphenylacetate 3-hydroxylase (HpaB) of Escherichia coli as a reduced flavin adenine dinucleotide-utilizing monooxygenase. Appl. Environ. Microbiol. 2000, 66, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Duffner, F.M.; Kirchner, U.; Bauer, M.P.; Müller, R. Phenol/cresol degradation by the thermophilic Bacillus thermoglucosidasius A7: Cloning and sequence analysis of five genes involved in the pathway. Gene 2000, 256, 215–221. [Google Scholar] [CrossRef]
- Hayes, R.P.; Webb, B.N.; Subramanian, A.K.; Nissen, M.; Popchock, A.; Xun, L.; Kang, C. Structural and catalytic differences between two FADH2-dependent monooxygenases: 2,4,5-TCP 4-monooxygenase (TftD) from Burkholderia cepacia AC1100 and 2,4,6-TCP 4-monooxygenase (TcpA) from Cupriavidus necator JMP134. Int. J. Mol. Sci. 2012, 13, 9769–9784. [Google Scholar] [CrossRef] [PubMed]
- Louie, T.M.; Webster, C.M.; Xun, L. Genetic and biochemical characterization of a 2,4,6-trichlorophenol degradation pathway in Ralstonia eutropha JMP134. J. Bacteriol. 2002, 184, 3492–3500. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Yang, J.W.; Cho, W.; Kwak, S.; Park, S.; Lim, Y.; Choe, J.W.; Kim, H.S. Oxidative biodegradation of 4-chlorophenol by using recombinant monooxygenase cloned and overexpressed from Arthrobacter chlorophenolicus A6. Bioresour. Technol. 2017, 240, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Pimviriyakul, P.; Thotsaporn, K.; Sucharitakul, J.; Chaiyen, P. Kinetic mechanism of the dechlorinating flavin-dependent monooxygenase HadA. J. Biol. Chem. 2017, 292, 4818–4832. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; van Lanen, S.G.; Shen, B. Characterization of the two-component, FAD-dependent monooxygenase SgcC that requires carrier protein-tethered substrates for the biosynthesis of the enediyne antitumor antibiotic C-1027. J. Am. Chem. Soc. 2008, 130, 6616–6623. [Google Scholar] [CrossRef] [PubMed]
- Dresen, C.; Lin, L.Y.-C.; D’Angelo, I.; Tocheva, E.I.; Strynadka, N.; Eltis, L.D. A flavin-dependent monooxygenase from Mycobacterium tuberculosis involved in cholesterol catabolism. J. Biol. Chem. 2010, 285, 22264–22275. [Google Scholar] [CrossRef] [PubMed]
- Ricken, B.; Kolvenbach, B.A.; Bergesch, C.; Benndorf, D.; Kroll, K.; Strnad, H.; Vlček, Č.; Adaixo, R.; Hammes, F.; Shahgaldian, P.; et al. FMNH2-dependent monooxygenases initiate catabolism of sulfonamides in Microbacterium sp. strain BR1 subsisting on sulfonamide antibiotics. Sci. Rep. 2017, 7, 15783. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Meng, Q.; Yang, Y.; Chu, C.; Chen, Q.; Li, Y.; Cheng, D.; Hong, Q.; Yan, X.; He, J. The two-component monooxygenase MeaXY initiates the downstream pathway of chloroacetanilide herbicide catabolism in sphingomonads. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Osorio, L.; Luong, K.; Jirde, S.; Palfey, B.A.; Vey, J.L. Initial investigations of C4a-(hydro)peroxyflavin intermediate formation by dibenzothiophene monooxygenase. Biochem. Biophys. Res. Commun. 2016, 481, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.I.M.; Marchesi, J.R.; Janssen, D.B. Degradation of 4-fluorophenol by Arthrobacter sp. strain IF1. Appl. Microbiol. Biotechnol. 2008, 78, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.-H.; Chen, H.-P.; Huang, J.-H.; Liu, T.-T.; Lin, T.-K.; Wang, S.-J.; Tseng, C.-H.; Shu, H.-Y. Identification and characterization of an indigo-producing oxygenase involved in indole 3-acetic acid utilization by Acinetobacter baumannii. Antonie Leeuwenhoek 2012, 101, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Arias-Barrau, E.; Sandoval, A.; Naharro, G.; Olivera, E.R.; Luengo, J.M. A two-component hydroxylase involved in the assimilation of 3-hydroxyphenyl acetate in Pseudomonas putida. J. Biol. Chem. 2005, 280, 26435–26447. [Google Scholar] [CrossRef] [PubMed]
- Filisetti, L.; Fontecave, M.; Niviere, V. Mechanism and substrate specificity of the flavin reductase ActVB from Streptomyces coelicolor. J. Biol. Chem. 2003, 278, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, U.; Westphal, A.H.; Müller, R.; van Berkel, W.J.H. Phenol hydroxylase from Bacillus thermoglucosidasius A7, a two-protein component monooxygenase with a dual role for FAD. J. Biol. Chem. 2003, 278, 47545–47553. [Google Scholar] [CrossRef] [PubMed]
- Omokoko, B.; Jäntges, U.K.; Zimmermann, M.; Reiss, M.; Hartmeier, W. Isolation of the phe-operon from G. stearothermophilus comprising the phenol degradative meta-pathway genes and a novel transcriptional regulator. BMC Microbiol. 2008, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Van Schie, P.M.; Young, L.Y. Biodegradation of phenol: Mechanisms and applications. Bioremediat. J. 2000, 4, 1–18. [Google Scholar] [CrossRef]
- Duffner, F.M.; Reinscheid, U.M.; Bauer, M.P.; Mutzel, A.; Müller, R. Strain differentiation and taxonomic characterisation of a thermophilic group of phenol-degrading bacilli. Syst. Appl. Microbiol. 1997, 20, 602–611. [Google Scholar] [CrossRef]
- Saa, L.; Jaureguibeitia, A.; Largo, E.; Llama, M.J.; Serra, J.L. Cloning, purification and characterization of two components of phenol hydroxylase from Rhodococcus erythropolis UPV-1. Appl. Microbiol. Biotechnol. 2010, 86, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Gröning, J.A.D.; Eulberg, D.; Tischler, D.; Kaschabek, S.R.; Schlömann, M. Gene redundancy of two-component (chloro)phenol hydroxylases in Rhodococcus opacus 1CP. FEMS Microbiol. Lett. 2014, 361, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Szőköl, J.; Rucká, L.; Šimčíková, M.; Halada, P.; Nešvera, J.; Pátek, M. Induction and carbon catabolite repression of phenol degradation genes in Rhodococcus erythropolis and Rhodococcus jostii. Appl. Microbiol. Biotechnol. 2014, 98, 8267–8279. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel, R.H.H.; Westphal, A.H.; Heck, A.J.R.; Walsh, M.A.; Rovida, S.; van Berkel, W.J.H.; Mattevi, A. Structural studies on flavin reductase PheA2 reveal binding of NAD in an unusual folded conformation and support novel mechanism of action. J. Biol. Chem. 2004, 279, 12860–12867. [Google Scholar] [CrossRef] [PubMed]
- Heine, T.; Scholtissek, A.; Westphal, A.H.; van Berkel, W.J.H.; Tischler, D. N-terminus determines activity and specificity of styrene monooxygenase reductases. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- Prieto, M.A.; Perez-Aranda, A.; Garcia, J.L. Characterization of an Escherichia coli aromatic hydroxylase with a broad substrate range. J. Bacteriol. 1993, 175, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Pornsuwan, S.; Maenpuen, S.; Kamutira, P.; Watthaisong, P.; Thotsaporn, K.; Tongsook, C.; Juttulapa, M.; Nijvipakul, S.; Chaiyen, P. 3,4-Dihydroxyphenylacetate 2,3-dioxygenase from Pseudomonas aeruginosa: An Fe(II)-containing enzyme with fast turnover. PLoS ONE 2017, 12, e0171135. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, U.; Massey, V.; Vaidyanathan, C.S. p-Hydroxyphenylacetate-3-hydroxylase. A two-protein component enzyme. J. Biol. Chem. 1992, 267, 25848–25855. [Google Scholar] [PubMed]
- Chaiyen, P.; Suadee, C.; Wilairat, P. A novel two-protein component flavoprotein hydroxylase. Eur. J. Biochem. 2001, 268, 5550–5561. [Google Scholar] [CrossRef] [PubMed]
- Thotsaporn, K.; Sucharitakul, J.; Wongratana, J.; Suadee, C.; Chaiyen, P. Cloning and expression of p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii: Evidence of the divergence of enzymes in the class of two-protein component aromatic hydroxylases. Biochim. Biophys. Acta Gene Struct. Expr. 2004, 1680, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Sucharitakul, J.; Chaiyen, P.; Entsch, B.; Ballou, D.P. The reductase of p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii requires p-hydroxyphenylacetate for effective catalysis. Biochemistry 2005, 44, 10434–10442. [Google Scholar] [CrossRef] [PubMed]
- Phongsak, T.; Sucharitakul, J.; Thotsaporn, K.; Oonanant, W.; Yuvaniyama, J.; Svasti, J.; Ballou, D.P.; Chaiyen, P. The C-terminal domain of 4-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii is an autoinhibitory domain. J. Biol. Chem. 2012, 287, 26213–26222. [Google Scholar] [CrossRef] [PubMed]
- Prieto, M.A.; Garcia, J.L. Molecular characterization of 4-hydroxyphenylacetate 3-hydroxylase of Escherichia coli. A two-protein component enzyme. J. Biol. Chem. 1994, 269, 22823–22829. [Google Scholar] [PubMed]
- Gibello, A.; Suarez, M.; Allende, J.L.; Martin, M. Molecular cloning and analysis of the genes encoding the 4-hydroxyphenylacetate hydroxylase from Klebsiella pneumoniae. Arch. Microbiol. 1997, 167, 160–166. [Google Scholar] [CrossRef]
- Hawumba, J.F.; Brözel, V.S.; Theron, J. Cloning and characterization of a 4-hydroxyphenylacetate 3-hydroxylase from the thermophile Geobacillus sp. PA-9. Curr. Microbiol. 2007, 55, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Soulimane, T.; O’Kane, S.R.; Kolaj, O. Isolation and purification of Thermus thermophilus HpaB by a crystallization approach. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Ortiz-Maldonado, M.; Entsch, B.; Ballou, D.P. Studies on the mechanism of p-hydroxyphenylacetate 3-hydroxylase from Pseudomonas aeruginosa: A system composed of a small flavin reductase and a large flavin-dependent oxygenase. Biochemistry 2010, 49, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Galán, B.; Díaz, E.; Prieto, M.A.; García, J.L. Functional analysis of the small component of the 4-hydroxyphenylacetate 3-monooxygenase of Escherichia coli W: A prototype of a new Flavin:NAD(P)H reductase subfamily. J. Bacteriol. 2000, 182, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, U.; Massey, V.; Miller, S.M. Mechanism of p-hydroxyphenylacetate-3-hydroxylase. A two-protein enzyme. J. Biol. Chem. 1994, 269, 150–155. [Google Scholar] [PubMed]
- Furuya, T.; Kino, K. Regioselective synthesis of piceatannol from resveratrol: Catalysis by two-component flavin-dependent monooxygenase HpaBC in whole cells. Tetrahedron Lett. 2014, 55, 2853–2855. [Google Scholar] [CrossRef]
- Louie, T.M.; Xie, X.S.; Xun, L. Coordinated production and utilization of FADH2 by NAD(P)H-flavin oxidoreductase and 4-hydroxyphenylacetate 3-monooxygenase. Biochemistry 2003, 42, 7509–7517. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Hisano, T.; Takeda, K.; Iwasaki, W.; Ebihara, A.; Miki, K. Crystal structure of the oxygenase component (HpaB) of the 4-hydroxyphenylacetate 3-monooxygenase from Thermus thermophilus HB8. J. Biol. Chem. 2007, 282, 33107–33117. [Google Scholar] [CrossRef] [PubMed]
- Thotsaporn, K.; Chenprakhon, P.; Sucharitakul, J.; Mattevi, A.; Chaiyen, P. Stabilization of C4a-hydroperoxyflavin in a two-component flavin-dependent monooxygenase is achieved through interactions at flavin N5 and C4a atoms. J. Biol. Chem. 2011, 286, 28170–28180. [Google Scholar] [CrossRef] [PubMed]
- Okai, M.; Kudo, N.; Lee, W.C.; Kamo, M.; Nagata, K.; Tanokura, M. Crystal structures of the short-chain flavin reductase HpaC from Sulfolobus tokodaii strain 7 in its three states: NAD(P)+-free, NAD+-bound, and NADP+-bound. Biochemistry 2006, 45, 5103–5110. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Hisano, T.; Iwasaki, W.; Ebihara, A.; Miki, K. Crystal structure of the flavin reductase component (HpaC) of 4-hydroxyphenylacetate 3-monooxygenase from Thermus thermophilus HB8: Structural basis for the flavin affinity. Proteins Struct. Funct. Bioinform. 2008, 70, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.N.; Ballinger, J.W.; Kim, E.; Belchik, S.M.; Lam, K.-S.; Youn, B.; Nissen, M.S.; Xun, L.; Kang, C. Characterization of chlorophenol 4-monooxygenase (TftD) and NADH:FAD oxidoreductase (TftC) of Burkholderia cepacia AC1100. J. Biol. Chem. 2010, 285, 2014–2027. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.; Cole, L.J.; Barr, E.W.; Bollinger, J.M.; Ballou, D.P.; Walsh, C.T. Flavin redox chemistry precedes substrate chlorination during the reaction of the flavin-dependent halogenase RebH. Biochemistry 2006, 45, 7904–7912. [Google Scholar] [CrossRef] [PubMed]
- Hatta, T.; Fujii, E.; Takizawa, N. Analysis of two gene clusters involved in 2,4,6-trichlorophenol degradation by Ralstonia pickettii DTP0602. Biosci. Biotechnol. Biochem. 2012, 76, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Fiege, H.; Voges, H.-W.; Hamamoto, T.; Umemura, S.; Iwata, T.; Miki, H.; Fujita, Y.; Buysch, H.-J.; Garbe, D.; Paulus, W. Phenol derivatives. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar]
- Tinikul, R.; Chenprakhon, P.; Maenpuen, S.; Chaiyen, P. Biotransformation of plant-derived phenolic acids. Biotechnol. J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Yan, Y. Biotechnological production of plant-specific hydroxylated phenylpropanoids. Biotechnol. Bioeng. 2014, 111, 1895–1899. [Google Scholar] [CrossRef] [PubMed]
- Furuya, T.; Sai, M.; Kino, K. Efficient monooxygenase-catalyzed piceatannol production: Application of cyclodextrins for reducing product inhibition. J. Biosci. Bioeng. 2018. [Google Scholar] [CrossRef] [PubMed]
- Orenes-Piñero, E.; García-Carmona, F.; Sánchez-Ferrer, A. A new process for obtaining hydroxytyrosol using transformed Escherichia coli whole cells with phenol hydroxylase gene from Geobacillus thermoglucosidasius. Food Chem. 2013, 139, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Van Lanen, S.G.; Oh, T.-J.; Liu, W.; Wendt-Pienkowski, E.; Shen, B. Characterization of the maduropeptin biosynthetic gene cluster from Actinomadura madurae ATCC 39144 supporting a unifying paradigm for enediyne biosynthesis. J. Am. Chem. Soc. 2007, 129, 13082–13094. [Google Scholar] [CrossRef] [PubMed]
- Lohman, J.R.; Huang, S.-X.; Horsman, G.P.; Dilfer, P.E.; Huang, T.; Chen, Y.; Wendt-Pienkowski, E.; Shen, B. Cloning and sequencing of the kedarcidin biosynthetic gene cluster from Streptoalloteichus sp. ATCC 53650 revealing new insights into biosynthesis of the enediyne family of antitumor antibiotics. Mol. BioSyst. 2013, 9, 478–491. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.P.; Nett, M.; Moore, B.S. Unraveling the biosynthesis of the sporolide cyclohexenone building block. J. Am. Chem. Soc. 2008, 130, 2406–2407. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Lohman, J.R.; Cao, H.; Tan, K.; Rudolf, J.D.; Ma, M.; Xu, W.; Bingman, C.A.; Yennamalli, R.M.; Bigelow, L.; et al. Crystal structures of SgcE6 and SgcC, the two-component monooxygenase that catalyzes hydroxylation of a carrier protein-tethered substrate during the biosynthesis of the enediyne antitumor antibiotic C-1027 in Streptomyces globisporus. Biochemistry 2016, 55, 5142–5154. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, T.; Shen, B. Tailoring enzymes acting on carrier protein-tethered substrates in natural product biosynthesis. Methods Enzymol. 2012, 516, 321–343. [Google Scholar] [CrossRef] [PubMed]
- Brünke, P.; Sterner, O.; Bailey, J.E.; Minas, W. Heterologous expression of the naphthocyclinone hydroxylase gene from Streptomyces arenae for production of novel hybrid polyketides. Antonie Leeuwenhoek 2001, 79, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Thoden, J.B.; Branch, M.C.; Zimmer, A.L.; Bruender, N.A.; Holden, H.M. Active site architecture of a sugar N-oxygenase. Biochemistry 2013, 52, 3191–3193. [Google Scholar] [CrossRef] [PubMed]
- Al-Mestarihi, A.; Romo, A.; Liu, H.-W.; Bachmann, B.O. Nitrososynthase-triggered oxidative carbon-carbon bond cleavage in baumycin biosynthesis. J. Am. Chem. Soc. 2013, 135, 11457–11460. [Google Scholar] [CrossRef] [PubMed]
- Hartmans, S.; van der Werf, M.J.; de Bont, J.A. Bacterial degradation of styrene involving a novel flavin adenine dinucleotide-dependent styrene monooxygenase. Appl. Environ. Microbiol. 1990, 56, 1347–1351. [Google Scholar] [PubMed]
- Tischler, D. Microbial Styrene Degradation; Springer International Publishing: Basel, Switzerland, 2015. [Google Scholar]
- Itoh, N.; Hayashi, K.; Okada, K.; Ito, T.; Mizuguchi, N. Characterization of styrene oxide isomerase, a key enzyme of styrene and styrene oxide metabolism in Corynebacterium sp. Biosci. Biotechnol. Biochem. 1997, 61, 2058–2062. [Google Scholar] [CrossRef]
- Beltrametti, F.; Marconi, A.M.; Bestetti, G.; Colombo, C.; Galli, E.; Ruzzi, M.; Zennaro, E. Sequencing and functional analysis of styrene catabolism genes from Pseudomonas fluorescens ST. Appl. Environ. Microbiol. 1997, 63, 2232–2239. [Google Scholar] [PubMed]
- Velasco, A.; Alonso, S.; Garcia, J.L.; Perera, J.; Diaz, E. Genetic and functional analysis of the styrene catabolic cluster of Pseudomonas sp. strain Y2. J. Bacteriol. 1998, 180, 1063–1071. [Google Scholar] [PubMed]
- Toda, H.; Itoh, N. Isolation and characterization of styrene metabolism genes from styrene-assimilating soil bacteria Rhodococcus sp. ST-5 and ST-10. J. Biosci. Bioeng. 2012, 113, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Oelschlägel, M.; Zimmerling, J.; Schlömann, M.; Tischler, D. Styrene oxide isomerase of Sphingopyxis sp. Kp5.2. Microbiology 2014, 160, 2481–2491. [Google Scholar] [CrossRef] [PubMed]
- Riedel, A.; Heine, T.; Westphal, A.H.; Conrad, C.; Rathsack, P.; van Berkel, W.J.H.; Tischler, D. Catalytic and hydrodynamic properties of styrene monooxygenases from Rhodococcus opacus 1CP are modulated by cofactor binding. AMB Express 2015, 5, 112. [Google Scholar] [CrossRef] [PubMed]
- Heine, T.; Zimmerling, J.; Ballmann, A.; Kleeberg, S.B.; Rückert, C.; Busche, T.; Winkler, A.; Kalinowski, J.; Poetsch, A.; Scholtissek, A.; et al. On the enigma of glutathione-dependent styrene degradation in Gordonia rubripertincta CWB2. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Eulberg, D.; Lakner, S.; Kaschabek, S.R.; van Berkel, W.J.H.; Schlömann, M. Identification of a novel self-sufficient styrene monooxygenase from Rhodococcus opacus 1CP. J. Bacteriol. 2009, 191, 4996–5009. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Kermer, R.; Gröning, J.A.D.; Kaschabek, S.R.; van Berkel, W.J.H.; Schlömann, M. StyA1 and StyA2B from Rhodococcus opacus 1CP: A Multifunctional styrene monooxygenase system. J. Bacteriol. 2010, 192, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Gröning, J.A.D.; Kaschabek, S.R.; Schlömann, M. One-component styrene monooxygenases: An evolutionary view on a rare class of flavoproteins. Appl. Biochem. Biotechnol. 2012, 167, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Schwabe, R.; Siegel, L.; Joffroy, K.; Kaschabek, S.; Scholtissek, A.; Heine, T. VpStyA1/VpStyA2B of Variovorax paradoxus EPS: An aryl alkyl sulfoxidase rather than a styrene epoxidizing monooxygenase. Molecules 2018, 23, 809. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.-H.; Chen, H.-P.; Shu, H.-Y.; Lee, S.-W. Detoxification of indole by an indole-induced flavoprotein oxygenase from Acinetobacter baumannii. PLoS ONE 2015, 10, e0138798. [Google Scholar] [CrossRef] [PubMed]
- Sadauskas, M.; Vaitekūnas, J.; Gasparavičiūtė, R.; Meškys, R. Genetic and biochemical characterization of indole biodegradation in Acinetobacter sp. strain O153. Appl. Environ. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Otto, K.; Hofstetter, K.; Röthlisberger, M.; Witholt, B.; Schmid, A. Biochemical characterization of StyAB from Pseudomonas sp. strain VLB120 as a two-component flavin-diffusible monooxygenase. J. Bacteriol. 2004, 186, 5292–5302. [Google Scholar] [CrossRef] [PubMed]
- Kantz, A.; Chin, F.; Nallamothu, N.; Nguyen, T.; Gassner, G.T. Mechanism of flavin transfer and oxygen activation by the two-component flavoenzyme styrene monooxygenase. Arch. Biochem. Biophys. 2005, 442, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Kantz, A.; Gassner, G.T. Nature of the reaction intermediates in the flavin adenine dinucleotide-dependent epoxidation mechanism of styrene monooxygenase. Biochemistry 2011, 50, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Toda, H.; Imae, R.; Komio, T.; Itoh, N. Expression and characterization of styrene monooxygenases of Rhodococcus sp. ST-5 and ST-10 for synthesizing enantiopure (S)-epoxides. Appl. Microbiol. Biotechnol. 2012, 96, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Di Gennaro, P.; Colmegna, A.; Galli, E.; Sello, G.; Pelizzoni, F.; Bestetti, G. A new biocatalyst for production of optically pure aryl epoxides by styrene monooxygenase from Pseudomonas fluorescens ST. Appl. Environ. Microbiol. 1999, 65, 2794–2797. [Google Scholar] [PubMed]
- Bernasconi, S.; Orsini, F.; Sello, G.; Colmegna, A.; Galli, E.; Bestetti, G. Bioconversion of substituted styrenes to the corresponding enantiomerically pure epoxides by a recombinant Escherichia coli strain. Tetrahedron Lett. 2000, 41, 9157–9161. [Google Scholar] [CrossRef]
- Hollmann, F.; Lin, P.-C.; Witholt, B.; Schmid, A. Stereospecific biocatalytic epoxidation: The first example of direct regeneration of a FAD-dependent monooxygenase for catalysis. J. Am. Chem. Soc. 2003, 125, 8209–8217. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, S.; Orsini, F.; Sello, G.; Di Gennaro, P. Bacterial monooxygenase mediated preparation of nonracemic chiral oxiranes: Study of the effects of substituent nature and position. Tetrahedron Asymmetry 2004, 15, 1603–1606. [Google Scholar] [CrossRef]
- Dunn, H.D.; Curtin, T.; O’riordan, M.A.; Coen, P.; Kieran, P.M.; Malone, D.M.; O’Connor, K.E. Aromatic and aliphatic hydrocarbon consumption and transformation by the styrene degrading strain Pseudomonas putida CA-3. FEMS Microbiol. Lett. 2005, 249, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Van Hellemond, E.W.; Janssen, D.B.; Fraaije, M.W.; Janssen, D.B.; Fraaije, M.W. Discovery of a novel styrene monooxygenase originating from the metagenome. Appl. Environ. Microbiol. 2007, 73, 5832–5839. [Google Scholar] [CrossRef] [PubMed]
- Gursky, L.J.; Nikodinovic-Runic, J.; Feenstra, K.A.; O’Connor, K.E. In vitro evolution of styrene monooxygenase from Pseudomonas putida CA-3 for improved epoxide synthesis. Appl. Microbiol. Biotechnol. 2010, 85, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Qiao, J.; Liu, Y.; Wu, Z.-L. Styrene monooxygenase from Pseudomonas sp. LQ26 catalyzes the asymmetric epoxidation of both conjugated and unconjugated alkenes. J. Mol. Catal. B Enzym. 2010, 67, 236–241. [Google Scholar] [CrossRef]
- Qaed, A.A.; Lin, H.; Tang, D.-F.; Wu, Z.-L. Rational design of styrene monooxygenase mutants with altered substrate preference. Biotechnol. Lett. 2011, 33, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Boyd, D.R.; Sharma, N.D.; McMurray, B.; Haughey, S.A.; Allen, C.C.R.; Hamilton, J.T.G.; McRoberts, W.C.; O’Ferrall, R.A.M.; Nikodinovic-Runic, J.; Coulombel, L.A.; et al. Bacterial dioxygenase- and monooxygenase-catalysed sulfoxidation of benzobthiophenes. Org. Biomol. Chem. 2012, 10, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Tang, D.-F.; Ahmed, A.A.Q.; Liu, Y.; Wu, Z.-L. Mutations at the putative active cavity of styrene monooxygenase: Enhanced activity and reversed enantioselectivity. J. Biotechnol. 2012, 161, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Toda, H.; Imae, R.; Itoh, N. Efficient biocatalysis for the production of enantiopure (S)-epoxides using a styrene monooxygenase (SMO) and Leifsonia alcohol dehydrogenase (LSADH) system. Tetrahedron Asymmetry 2012, 23, 1542–1549. [Google Scholar] [CrossRef]
- Nikodinovic-Runic, J.; Coulombel, L.; Francuski, D.; Sharma, N.D.; Boyd, D.R.; Ferrall, R.M.O.; O’Connor, K.E. The oxidation of alkylaryl sulfides and benzobthiophenes by Escherichia coli cells expressing wild-type and engineered styrene monooxygenase from Pseudomonas putida CA-3. Appl. Microbiol. Biotechnol. 2013, 97, 4849–4858. [Google Scholar] [CrossRef] [PubMed]
- Toda, H.; Imae, R.; Itoh, N. Bioproduction of chiral epoxyalkanes using styrene monooxygenase from Rhodococcus sp. ST-10 (RhSMO). Adv. Synth. Catal. 2014, 356, 3443–3450. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Liu, Y.; Wu, Z.-L. Synthesis of enantiopure glycidol derivatives via a one-pot two-step enzymatic cascade. Org. Biomol. Chem. 2015, 13, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Toda, H.; Ohuchi, T.; Imae, R.; Itoh, N. Microbial production of aliphatic (S)-epoxyalkanes by using Rhodococcus sp. strain ST-10 styrene monooxygenase expressed in organic-solvent-tolerant Kocuria rhizophila DC2201. Appl. Environ. Microbiol. 2015, 81, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Y.-C.; Wu, Z.-L. Asymmetric bio-epoxidation catalyzed with the styrene monooxygenase from Pseudomonas sp. LQ26. Bioresour. Bioprocess. 2016, 3, 147. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Guo, C.; Liu, Y.; Wang, H.-B.; Wu, Z.-L. Enzymatic cascades for the stereo-complementary epimerisation of in situ generated epoxy alcohols. Org. Biomol. Chem. 2017, 15, 2562–2568. [Google Scholar] [CrossRef] [PubMed]
- Ukaegbu, U.E.; Kantz, A.; Beaton, M.; Gassner, G.T.; Rosenzweig, A.C. Structure and ligand binding properties of the epoxidase component of styrene monooxygenase. Biochemistry 2010, 49, 1678–1688. [Google Scholar] [CrossRef] [PubMed]
- Morrison, E.; Kantz, A.; Gassner, G.T.; Sazinsky, M.H. Structure and mechanism of styrene monooxygenase reductase: New insight into the FAD-transfer reaction. Biochemistry 2013, 52, 6063–6075. [Google Scholar] [CrossRef] [PubMed]
- Yeo, Y.-J.; Shin, S.; Lee, S.-G.; Park, S.; Jeong, Y.-J. Production, purification, and characterization of soluble NADH-flavin oxidoreductase (StyB) from Pseudomonas putida SN1. J. Microbiol. Biotechnol. 2009, 19, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Eppink, M.H.; Schreuder, H.A.; van Berkel, W.J.H. Identification of a novel conserved sequence motif in flavoprotein hydroxylases with a putative dual function in FAD/NAD(P)H binding. Protein Sci. 1997, 6, 2454–2458. [Google Scholar] [CrossRef] [PubMed]
- Schreuder, H.A.; van der Laan, J.M.; Hol, W.G.J.; Drenth, J. Crystal structure of p-hydroxybenzoate hydroxylase complexed with its reaction product 3,4-dihydroxybenzoate. J. Mol. Biol. 1988, 199, 637–648. [Google Scholar] [CrossRef]
- Entsch, B.; Cole, L.J.; Ballou, D.P. Protein dynamics and electrostatics in the function of p-hydroxybenzoate hydroxylase. Arch. Biochem. Biophys. 2005, 433, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Heine, T.; Tucker, K.; Okonkwo, N.; Assefa, B.; Conrad, C.; Scholtissek, A.; Schlömann, M.; Gassner, G.; Tischler, D. Engineering styrene monooxygenase for biocatalysis: Reductase-epoxidase fusion proteins. Appl. Biochem. Biotechnol. 2017, 181, 1590–1610. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.; Ju, J.; Borlee, B.R.; Williamson, L.L.; Shen, B.; Raffa, K.F.; Handelsman, J.; Guan, C.; Ju, J.; Borlee, B.R.; et al. Signal mimics derived from a metagenomic analysis of the gypsy moth gut microbiota. Appl. Environ. Microbiol. 2007, 73, 3669–3676. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Zhang, J.; Zhou, S.N. Identification and production of indigo retrieved from a deep-sea sediment metagenomic library: Submitted to GenBank: ABZ79366. 2008; article unpublished. [Google Scholar]
- Gröning, J.A.D.; Kaschabek, S.R.; Schlömann, M.; Tischler, D. A mechanistic study on SMOB-ADP1: An NADH:flavin oxidoreductase of the two-component styrene monooxygenase of Acinetobacter baylyi ADP1. Arch. Microbiol. 2014, 196, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Schlömann, M.; van Berkel, W.J.H.; Gassner, G.T. FAD C(4a)-hydroxide stabilized in a naturally fused styrene monooxygenase. FEBS Lett. 2013, 587, 3848–3852. [Google Scholar] [CrossRef] [PubMed]
- Holtmann, D.; Fraaije, M.W.; Arends, I.W.C.E.; Opperman, D.J.; Hollmann, F. The taming of oxygen: Biocatalytic oxyfunctionalisations. Chem. Commun. 2014, 50, 13180–13200. [Google Scholar] [CrossRef] [PubMed]
- Domínguez de María, P.; Hollmann, F. On the (un)greenness of biocatalysis: Some challenging figures and some promising options. Front. Microbiol. 2015, 6, 1257. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.-T.; Mattevi, A.; Fraaije, M.W. Expanding the repertoire of flavoenzyme-based biocatalysis. In Future Directions in Biocatalysis; Matsuda, T., Ed.; Elsevier Science: San Diego, CA, USA, 2017; pp. 119–133. [Google Scholar]
- Matsui, T.; Dekishima, Y.; Ueda, M. Biotechnological production of chiral organic sulfoxides: Current state and perspectives. Appl. Microbiol. Biotechnol. 2014, 98, 7699–7706. [Google Scholar] [CrossRef] [PubMed]
- Brondani, P.B.; Fraaije, M.W.; de Gonzalo, G. Recent developments in flavin-based catalysis: Enzymatic sulfoxidation. In Green Biocatalysis, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; pp. 149–164. [Google Scholar]
- De Souza, R.O.M.A.; Miranda, L.S.M.; Bornscheuer, U.T. A retrosynthesis approach for biocatalysis in organic synthesis. Chem. Eur. J. 2017, 23, 12040–12063. [Google Scholar] [CrossRef] [PubMed]
- McKenna, R.; Pugh, S.; Thompson, B.; Nielsen, D.R. Microbial production of the aromatic building-blocks (S)-styrene oxide and (R)-1,2-phenylethanediol from renewable resources. Biotechnol. J. 2013, 8, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Goundry, W.R.F.; Adams, B.; Benson, H.; Demeritt, J.; McKown, S.; Mulholland, K.; Robertson, A.; Siedlecki, P.; Tomlin, P.; Vare, K. Development and scale-up of a biocatalytic process to form a chiral sulfoxide. Org. Process Res. Dev. 2017, 21, 107–113. [Google Scholar] [CrossRef]
- Cheng, L.; Yin, S.; Chen, M.; Sun, B.; Hao, S.; Wang, C. Enhancing indigo production by over-expression of the styrene monooxygenase in Pseudomonas putida. Curr. Microbiol. 2016, 73, 248–254. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, K.E.; Dobson, A.D.; Hartmans, S. Indigo formation by microorganisms expressing styrene monooxygenase activity. Appl. Environ. Microbiol. 1997, 63, 4287–4291. [Google Scholar] [PubMed]
- Heine, T.; Großmann, C.; Hofmann, S.; Tischler, D. Enzymgesteuerte Indigoproduktion. BIOspektrum 2018, 24, 446–448. [Google Scholar] [CrossRef]
- McClay, K.; Boss, C.; Keresztes, I.; Steffan, R.J. Mutations of toluene-4-monooxygenase that alter regiospecificity of indole oxidation and lead to production of novel indigoid pigments. Appl. Environ. Microbiol. 2005, 71, 5476–5483. [Google Scholar] [CrossRef] [PubMed]
- Farias-Silva, E.; Cola, M.; Calvo, T.R.; Barbastefano, V.; Ferreira, A.L.; de Paula Michelatto, D.; Alves de Almeida, A.C.; Hiruma-Lima, C.A.; Vilegas, W.; Brito, A.R.M.S. Antioxidant activity of indigo and its preventive effect against ethanol-induced DNA damage in rat gastric mucosa. Planta Med. 2007, 73, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Pathak, H.; Madamwar, D. Biosynthesis of indigo dye by newly isolated naphthalene-degrading strain Pseudomonas sp. HOB1 and its application in dyeing cotton fabric. Appl. Biochem. Biotechnol. 2010, 160, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Głowacki, E.D.; Voss, G.; Leonat, L.; Irimia-Vladu, M.; Bauer, S.; Sariciftci, N.S. Indigo and tyrian purple - From ancient natural dyes to modern organic semiconductors. Isr. J. Chem. 2012, 52, 540–551. [Google Scholar] [CrossRef]
- Irimia-Vladu, M.; Głowacki, E.D.; Troshin, P.A.; Schwabegger, G.; Leonat, L.; Susarova, D.K.; Krystal, O.; Ullah, M.; Kanbur, Y.; Bodea, M.A.; et al. Indigo—A natural pigment for high performance ambipolar organic field effect transistors and circuits. Adv. Mater. 2012, 24, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Dua, A.; Chauhan, K.; Pathak, H. Biotransformation of indigo pigment by indigenously isolated Pseudomonas sp. HAV-1 and assessment of its antioxidant property. Biotechnol. Res. Int. 2014, 2014, 109249. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Pun, A.B.; Zherebetskyy, D.; Liu, Y.; Liu, F.; Klivansky, L.M.; McGough, A.M.; Zhang, B.A.; Lo, K.; Russell, T.P.; et al. New form of an old natural dye: Bay-annulated indigo (BAI) as an excellent electron accepting unit for high performance organic semiconductors. J. Am. Chem. Soc. 2014, 136, 15093–15101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hollmann, F. Nonconventional regeneration of redox enzymes—A practical approach for organic synthesis? Chem. Commun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.; Bühler, B.; Schmid, A. Production host selection for asymmetric styrene epoxidation: Escherichia coli vs. solvent-tolerant Pseudomonas. J. Ind. Microbiol. Biotechnol. 2012, 39, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Wu, S.; Praveen, P.; Loh, K.-C.; Li, Z. Enhancing productivity for cascade biotransformation of styrene to (S)-vicinal diol with biphasic system in hollow fiber membrane bioreactor. Appl. Microbiol. Biotechnol. 2017, 101, 1857–1868. [Google Scholar] [CrossRef] [PubMed]
- Volmer, J.; Schmid, A.; Bühler, B. The application of constitutively solvent-tolerant P. taiwanensis VLB120ΔCΔttgV for stereospecific epoxidation of toxic styrene alleviates carrier solvent use. Biotechnol. J. 2017, 12, 1600558. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.; Hofstetter, K.; Feiten, H.-J.; Hollmann, F.; Witholt, B. Integrated biocatalytic synthesis on gram scale: The highly enantioselective preparation of chiral oxiranes with styrene monooxygenase. Adv. Synth. Catal. 2001, 343, 732–737. [Google Scholar] [CrossRef]
- Oelschlägel, M.; Zimmerling, J.; Tischler, D. A review: The styrene metabolizing cascade of side-chain oxygenation as biotechnological basis to gain various valuable compounds. Front. Microbiol. 2018, 9, 4796. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhou, Y.; Wang, T.; Too, H.-P.; Wang, D.I.C.; Li, Z. Highly regio- and enantioselective multiple oxy- and amino-functionalizations of alkenes by modular cascade biocatalysis. Nat. Commun. 2016, 7, 11917. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wei, J.; Qiu, X.; Jiao, N. Homogeneous oxygenase catalysis. Chem. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Corrado, M.L.; Knaus, T.; Mutti, F. Chimeric styrene monooxygenase with increased efficiency in asymmetric biocatalytic epoxidation. ChemBioChem 2018. [Google Scholar] [CrossRef] [PubMed]
- Van Pée, K.-H.; Unversucht, S. Biological dehalogenation and halogenation reactions. Chemosphere 2003, 52, 299–312. [Google Scholar] [CrossRef]
- Payne, J.T.; Andorfer, M.C.; Lewis, J.C. Engineering flavin-dependent halogenases. Methods Enzymol. 2016, 575, 93–126. [Google Scholar] [CrossRef] [PubMed]
- Latham, J.; Brandenburger, E.; Shepherd, S.A.; Menon, B.R.K.; Micklefield, J. Development of halogenase enzymes for use in synthesis. Chem. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, P.R.; Widmann, C.; Wibberg, D.; Schröder, L.; Frese, M.; Kottke, T.; Kalinowski, J.; Niemann, H.H.; Sewald, N. A flavin-dependent halogenase from metagenomic analysis prefers bromination over chlorination. PLoS ONE 2018, 13, e0196797. [Google Scholar] [CrossRef] [PubMed]
- Weichold, V.; Milbredt, D.; van Pée, K.-H. Specific enzymatic halogenation-From the discovery of halogenated enzymes to their applications in vitro and in vivo. Angew. Chem. Int. Ed. Engl. 2016, 55, 6374–6389. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Miles, Z.D.; Winter, J.M.; Eustáquio, A.S.; El Gamal, A.A.; Moore, B.S. Enzymatic halogenation and dehalogenation reactions: Pervasive and mechanistically diverse. Chem. Rev. 2017, 117, 5619–5674. [Google Scholar] [CrossRef] [PubMed]
- Gkotsi, D.S.; Dhaliwal, J.; McLachlan, M.M.; Mulholand, K.R.; Goss, R.J. Halogenases: Powerful tools for biocatalysis (mechanisms applications and scope). Curr. Opin. Chem. Biol. 2018, 43, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Fraley, A.E.; Sherman, D.H. Halogenase engineering and its utility in medicinal chemistry. Bioorg. Med. Chem. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Flecks, S.; Unversucht, S.; Haupt, C.; van Pée, K.-H.; Naismith, J.H. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science 2005, 309, 2216–2219. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.T.; Andorfer, M.C.; Lewis, J.C. Regioselective arene halogenation using the FAD-dependent halogenase RebH. Angew. Chem. Int. Ed. Engl. 2013, 52, 5271–5274. [Google Scholar] [CrossRef] [PubMed]
- Rachid, S.; Krug, D.; Kunze, B.; Kochems, I.; Scharfe, M.; Zabriskie, T.M.; Blöcker, H.; Müller, R. Molecular and biochemical studies of chondramide formation-highly cytotoxic natural products from Chondromyces crocatus Cm c5. Chem. Biol. 2006, 13, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Zehner, S.; Kotzsch, A.; Bister, B.; Süssmuth, R.D.; Méndez, C.; Salas, J.A.; van Pée, K.-H. A regioselective tryptophan 5-halogenase is involved in pyrroindomycin biosynthesis in Streptomyces rugosporus LL-42D005. Chem. Biol. 2005, 12, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Seibold, C.; Schnerr, H.; Rumpf, J.; Kunzendorf, A.; Hatscher, C.; Wage, T.; Ernyei, A.J.; Dong, C.; Naismith, J.H.; van Pée, K.-H. A flavin-dependent tryptophan 6-halogenase and its use in modification of pyrrolnitrin biosynthesis. Biocatal. Biotrans. 2009, 24, 401–408. [Google Scholar] [CrossRef]
- Zeng, J.; Zhan, J. Characterization of a tryptophan 6-halogenase from Streptomyces toxytricini. Biotechnol. Lett. 2011, 33, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, S.A.; Karthikeyan, C.; Latham, J.; Struck, A.-W.; Thompson, M.L.; Menon, B.R.K.; Styles, M.Q.; Levy, C.; Leys, D.; Micklefield, J. Extending the biocatalytic scope of regiocomplementary flavin-dependent halogenase enzymes. Chem. Sci. 2015, 6, 3454–3460. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.M.; Uria, A.R.; Helfrich, E.J.N.; Milbredt, D.; van Pée, K.-H.; Piel, J.; Goss, R.J.M. An unusual flavin-dependent halogenase from the metagenome of the marine sponge Theonella swinhoei WA. ACS Chem. Biol. 2017, 12, 1281–1287. [Google Scholar] [CrossRef] [PubMed]
- Milbredt, D.; Patallo, E.P.; van Pée, K.-H. A tryptophan 6-halogenase and an amidotransferase are involved in thienodolin biosynthesis. ChemBioChem 2014, 15, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhan, J. A novel fungal flavin-dependent halogenase for natural product biosynthesis. ChemBioChem 2010, 11, 2119–2123. [Google Scholar] [CrossRef] [PubMed]
- Menon, B.R.K.; Brandenburger, E.; Sharif, H.H.; Klemstein, U.; Shepherd, S.A.; Greaney, M.F.; Micklefield, J. RadH: A versatile halogenase for integration into synthetic pathways. Angew. Chem. Int. Ed. Engl. 2017, 56, 11841–11845. [Google Scholar] [CrossRef] [PubMed]
- Fraley, A.E.; Garcia-Borràs, M.; Tripathi, A.; Khare, D.; Mercado-Marin, E.V.; Tran, H.; Dan, Q.; Webb, G.P.; Watts, K.R.; Crews, P.; et al. Function and structure of MalA/MalA’, iterative halogenases for late-stage C-H functionalization of indole alkaloids. J. Am. Chem. Soc. 2017, 139, 12060–12068. [Google Scholar] [CrossRef] [PubMed]
- Dorrestein, P.C.; Yeh, E.; Garneau-Tsodikova, S.; Kelleher, N.L.; Walsh, C.T. Dichlorination of a pyrrolyl-S-carrier protein by FADH2-dependent halogenase PltA during pyoluteorin biosynthesis. Proc. Natl. Acad. Sci. USA 2005, 102, 13843–13848. [Google Scholar] [CrossRef] [PubMed]
- El Gamal, A.; Agarwal, V.; Diethelm, S.; Rahman, I.; Schorn, M.A.; Sneed, J.M.; Louie, G.V.; Whalen, K.E.; Mincer, T.J.; Noel, J.P.; et al. Biosynthesis of coral settlement cue tetrabromopyrrole in marine bacteria by a uniquely adapted brominase-thioesterase enzyme pair. Proc. Natl. Acad. Sci. USA 2016, 113, 3797–3802. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Ryan, K.S.; Gulder, T.A.M.; Hughes, C.C.; Moore, B.S. Flavoenzyme-catalyzed atropo-selective N,C-bipyrrole homocoupling in marinopyrrole biosynthesis. J. Am. Chem. Soc. 2012, 134, 12434–12437. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Van Lanen, S.G.; Shen, B. Regiospecific chlorination of (S)-beta-tyrosyl-S-carrier protein catalyzed by SgcC3 in the biosynthesis of the enediyne antitumor antibiotic C-1027. J. Am. Chem. Soc. 2007, 129, 12432–12438. [Google Scholar] [CrossRef] [PubMed]
- Buedenbender, S.; Rachid, S.; Müller, R.; Schulz, G.E. Structure and action of the myxobacterial chondrochloren halogenase CndH: A new variant of FAD-dependent halogenases. J. Mol. Biol. 2009, 385, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Podzelinska, K.; Latimer, R.; Bhattacharya, A.; Vining, L.C.; Zechel, D.L.; Jia, Z. Chloramphenicol biosynthesis: The structure of CmlS, a flavin-dependent halogenase showing a covalent flavin-aspartate bond. J. Mol. Biol. 2010, 397, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Teufel, R.; Agarwal, V.; Moore, B.S. Unusual flavoenzyme catalysis in marine bacteria. Curr. Opin. Chem. Biol. 2016, 31, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Van Pée, K.-H.; Patallo, E.P. Flavin-dependent halogenases involved in secondary metabolism in bacteria. Appl. Microbiol. Biotechnol. 2006, 70, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.; Garneau, S.; Walsh, C.T. Robust in vitro activity of RebF and RebH, a two-component reductase/halogenase, generating 7-chlorotryptophan during rebeccamycin biosynthesis. Proc. Natl. Acad. Sci. USA 2005, 102, 3960–3965. [Google Scholar] [CrossRef] [PubMed]
- Flecks, S.; Patallo, E.P.; Zhu, X.; Ernyei, A.J.; Seifert, G.; Schneider, A.; Dong, C.; Naismith, J.H.; van Pée, K.-H. New insights into the mechanism of enzymatic chlorination of tryptophan. Angew. Chem. Int. Ed. Engl. 2008, 47, 9533–9536. [Google Scholar] [CrossRef] [PubMed]
- Karabencheva-Christova, T.G.; Torras, J.; Mulholland, A.J.; Lodola, A.; Christov, C.Z. Mechanistic Insights into the reaction of chlorination of tryptophan catalyzed by tryptophan 7-halogenase. Sci. Rep. 2017, 7, 17395. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.; Blasiak, L.C.; Koglin, A.; Drennan, C.L.; Walsh, C.T. Chlorination by a long-lived intermediate in the mechanism of flavin-dependent halogenases. Biochemistry 2007, 46, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; de Laurentis, W.; Leang, K.; Herrmann, J.; Ihlefeld, K.; van Pée, K.-H.; Naismith, J.H. Structural insights into regioselectivity in the enzymatic chlorination of tryptophan. J. Mol. Biol. 2009, 391, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.; Polnick, S.; Nicke, T.; William, P.; Patallo, E.P.; Naismith, J.H.; van Pée, K.-H. Changing the regioselectivity of the tryptophan 7-halogenase PrnA by site-directed mutagenesis. Angew. Chem. Int. Ed. Engl. 2011, 50, 2951–2953. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, S.A.; Menon, B.R.K.; Fisk, H.; Struck, A.-W.; Levy, C.; Leys, D.; Micklefield, J. A structure-guided switch in the regioselectivity of a tryptophan halogenase. ChemBioChem 2016, 17, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Entsch, B.; van Berkel, W.J.H. Structure and mechanism of para-hydroxybenzoate hydroxylase. FASEB J. 1995, 9, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Hölzer, M.; Burd, W.; Reißig, H.-U.; van Pée, K.-H. Substrate specificity and regioselectivity of tryptophan 7-halogenase from Pseudomonas fluorescens BL915. Adv. Synth. Catal. 2001, 343, 591–595. [Google Scholar] [CrossRef]
- Lee, J.-K.; Zhao, H. Identification and characterization of the flavin:NADH reductase (PrnF) involved in a novel two-component arylamine oxygenase. J. Bacteriol. 2007, 189, 8556–8563. [Google Scholar] [CrossRef] [PubMed]
- Unversucht, S.; Hollmann, F.; Schmid, A.; van Pée, K.-H. FADH2-dependence of tryptophan 7-halogenase. Adv. Synth. Catal. 2005, 347, 1163–1167. [Google Scholar] [CrossRef]
- Bitto, E.; Huang, Y.; Bingman, C.A.; Singh, S.; Thorson, J.S.; Phillips, G.N. The structure of flavin-dependent tryptophan 7-halogenase RebH. Proteins Struct. Funct. Bioinform. 2008, 70, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.M.; Grüschow, S.; Goss, R.J.M. Scope and potential of halogenases in biosynthetic applications. Curr. Opin. Chem. Biol. 2013, 17, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Van Lanen, S.G.; Lin, S.; Horsman, G.P.; Shen, B. Characterization of SgcE6, the flavin reductase component supporting FAD-dependent halogenation and hydroxylation in the biosynthesis of the enediyne antitumor antibiotic C-1027. FEMS Microbiol. Lett. 2009, 300, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Christenson, S.D.; Standage, S.; Shen, B. Biosynthesis of the enediyne antitumor antibiotic C-1027. Science 2002, 297, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, M.C.; Grob, J.E.; Hajdin, C.E.; Chael, J.R.; Siuti, P.; Lilly, J.; Tan, K.L.; Lewis, J.C. Understanding flavin-dependent halogenase reactivity via substrate activity profiling. ACS Catal. 2017, 7, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Van Pée, K.-H. Transformation with tryptophan halogenase genes leads to the production of new chlorinated alkaloid metabolites by a medicinal plant. ChemBioChem 2011, 12, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Goss, R.J.M.; Shankar, S.; Fayad, A.A. The generation of “unnatural” products: Synthetic biology meets synthetic chemistry. Nat. Prod. Rep. 2012, 29, 870–889. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; O’Connor, S.E. Halogenase engineering for the generation of new natural product analogues. ChemBioChem 2015, 16, 2129–2135. [Google Scholar] [CrossRef] [PubMed]
- Glenn, W.S.; Nims, E.; O’Connor, S.E. Reengineering a tryptophan halogenase to preferentially chlorinate a direct alkaloid precursor. J. Am. Chem. Soc. 2011, 133, 19346–19349. [Google Scholar] [CrossRef] [PubMed]
- Heide, L.; Westrich, L.; Anderle, C.; Gust, B.; Kammerer, B.; Piel, J. Use of a halogenase of hormaomycin biosynthesis for formation of new clorobiocin analogues with 5-chloropyrrole moieties. ChemBioChem 2008, 9, 1992–1999. [Google Scholar] [CrossRef] [PubMed]
- Eustáquio, A.S.; Gust, B.; Luft, T.; Li, S.-M.; Chater, K.F.; Heide, L. Clorobiocin biosynthesis in Streptomyces. Chem. Biol. 2003, 10, 279–288. [Google Scholar] [CrossRef]
- Sánchez, C.; Zhu, L.; Braña, A.F.; Salas, A.P.; Rohr, J.; Méndez, C.; Salas, J.A. Combinatorial biosynthesis of antitumor indolocarbazole compounds. Proc. Natl. Acad. Sci. USA 2005, 102, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Frese, M.; Sewald, N. Enzymatic halogenation of tryptophan on a gram scale. Angew. Chem. Int. Ed. Engl. 2015, 54, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, M.C.; Park, H.J.; Vergara-Coll, J.; Lewis, J.C. Directed evolution of RebH for catalyst-controlled halogenation of indole C-H bonds. Chem. Sci. 2016, 7, 3720–3729. [Google Scholar] [CrossRef] [PubMed]
- Van Pée, K.-H.; Milbredt, D.; Patallo, E.P.; Weichold, V.; Gajewi, M. Application and modification of flavin-dependent halogenases. Methods Enzymol. 2016, 575, 65–92. [Google Scholar] [CrossRef] [PubMed]
- Schnepel, C.; Sewald, N. Enzymatic halogenation: A timely strategy for regioselective C-H activation. Chem. Eur. J. 2017, 23, 12064–12086. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, M.C.; Lewis, J.C. Understanding and improving the activity of flavin-dependent halogenases via random and targeted mutagenesis. Annu. Rev. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, M.C.; Belsare, K.D.; Girlich, A.M.; Lewis, J.C. Aromatic halogenation by using bifunctional flavin reductase-halogenase fusion enzymes. ChemBioChem 2017, 18, 2099–2103. [Google Scholar] [CrossRef] [PubMed]
- Menon, B.R.K.; Latham, J.; Dunstan, M.S.; Brandenburger, E.; Klemstein, U.; Leys, D.; Karthikeyan, C.; Greaney, M.F.; Shepherd, S.A.; Micklefield, J. Structure and biocatalytic scope of thermophilic flavin-dependent halogenase and flavin reductase enzymes. Org. Biomol. Chem. 2016, 14, 9354–9361. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Vaidyanathan, C.S. A new 4-nitrophenol 2-hydroxylase from a Nocardia sp. Biochem. Int. 1984, 8, 609–615. [Google Scholar] [PubMed]
- Takeo, M.; Murakami, M.; Niihara, S.; Yamamoto, K.; Nishimura, M.; Kato, D.-I.; Negoro, S. Mechanism of 4-nitrophenol oxidation in Rhodococcus sp. strain PN1: Characterization of the two-component 4-nitrophenol hydroxylase and regulation of its expression. J. Bacteriol. 2008, 190, 7367–7374. [Google Scholar] [CrossRef] [PubMed]
- Kadiyala, V.; Spain, J.C. A two-component monooxygenase catalyzes both the hydroxylation of p-nitrophenol and the oxidative release of nitrite from 4-nitrocatechol in Bacillus sphaericus JS905. Appl. Environ. Microbiol. 1998, 64, 2479–2484. [Google Scholar] [PubMed]
- Huijbers, M.M.E.; Martínez-Júlvez, M.; Westphal, A.H.; Delgado-Arciniega, E.; Medina, M.; van Berkel, W.J.H. Proline dehydrogenase from Thermus thermophilus does not discriminate between FAD and FMN as cofactor. Sci. Rep. 2017, 7, 43880. [Google Scholar] [CrossRef] [PubMed]
- Rudroff, F.; Mihovilovic, M.D.; Gröger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and challenges for combining chemo- and biocatalysis. Nat. Catal. 2018, 1, 12–22. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.; Kroutil, W. Artificial biocatalytic linear cascades for preparation of organic molecules. Chem. Rev. 2018, 118, 270–348. [Google Scholar] [CrossRef] [PubMed]
- Sperl, J.M.; Sieber, V. Multienzyme cascade reactions—Status and recent advances. ACS Catal. 2018, 8, 2385–2396. [Google Scholar] [CrossRef]
- Muschiol, J.; Peters, C.; Oberleitner, N.; Mihovilovic, M.D.; Bornscheuer, U.T.; Rudroff, F. Cascade catalysis–strategies and challenges en route to preparative synthetic biology. Chem. Commun. 2015, 51, 5798–5811. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | EC Number | Reference |
|---|---|---|
| Pyrrole 2-carboxylate monooxygenase | 1.14.13.130 | [51] |
| 4-Nitrocatechol 4-monooxygenase | 1.14.13.166 | [52] |
| 4-Nitrophenol 4-monooxygenase | 1.14.13.167 | [52] |
| 2-Aminobenzoate 3-monooxygenase | 1.14.14.8 | [53] |
| 4-Hydroxyphenylacetate 3-hydroxylase | 1.14.14.9 | [54] |
| Phenol 2-hydroxylase | 1.14.14.20 | [55] |
| Isobutylamine N1-monooxygenase | 1.14.14.30 | [49] |
| 2,4,5-Trichlorophenol monooxygenase | - | [56] |
| 2,4,6-Trichlorophenol 4-monooxygenase | - | [57] |
| 4-Chlorophenol monooxygenase | - | [58] |
| 4-Chlorophenol dechlorinase | - | [59] |
| (S)-3-Chloro-β-tyrosine hydroxylase | - | [60] |
| Indosespene cyclase XiaF | - | [48] |
| 3-HSA | - | [61] |
| Sulfonamide monooxygenase | - | [62] |
| MeaX monooxygenase | - | [63] |
| Dibenzothiophene monooxygenase | - | [64] |
| 4-Fluorophenol monooxygenase | - | [65] |
| Indole 3-acetate monooxygenase | - | [66] |
| 3-Hydroxyphenylacetate 6-hydroxylase | - | [67] |
| Enzyme | EC Number | References |
|---|---|---|
| Styrene monooxygenase | 1.14.14.11 | [119,120,121,122,129,130,131] |
| Indole monooxygenase | - | [121,123,124,126,127] |
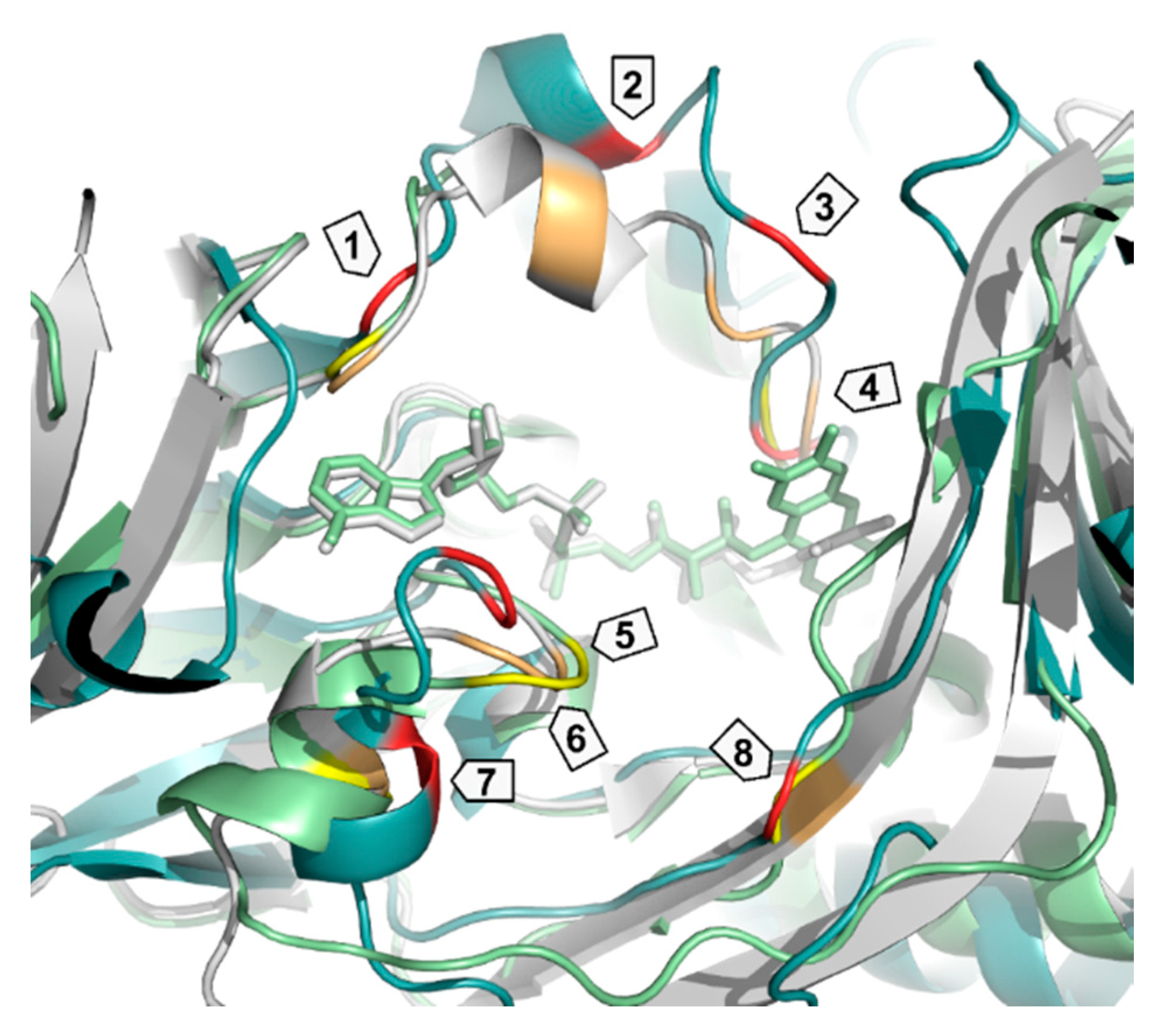
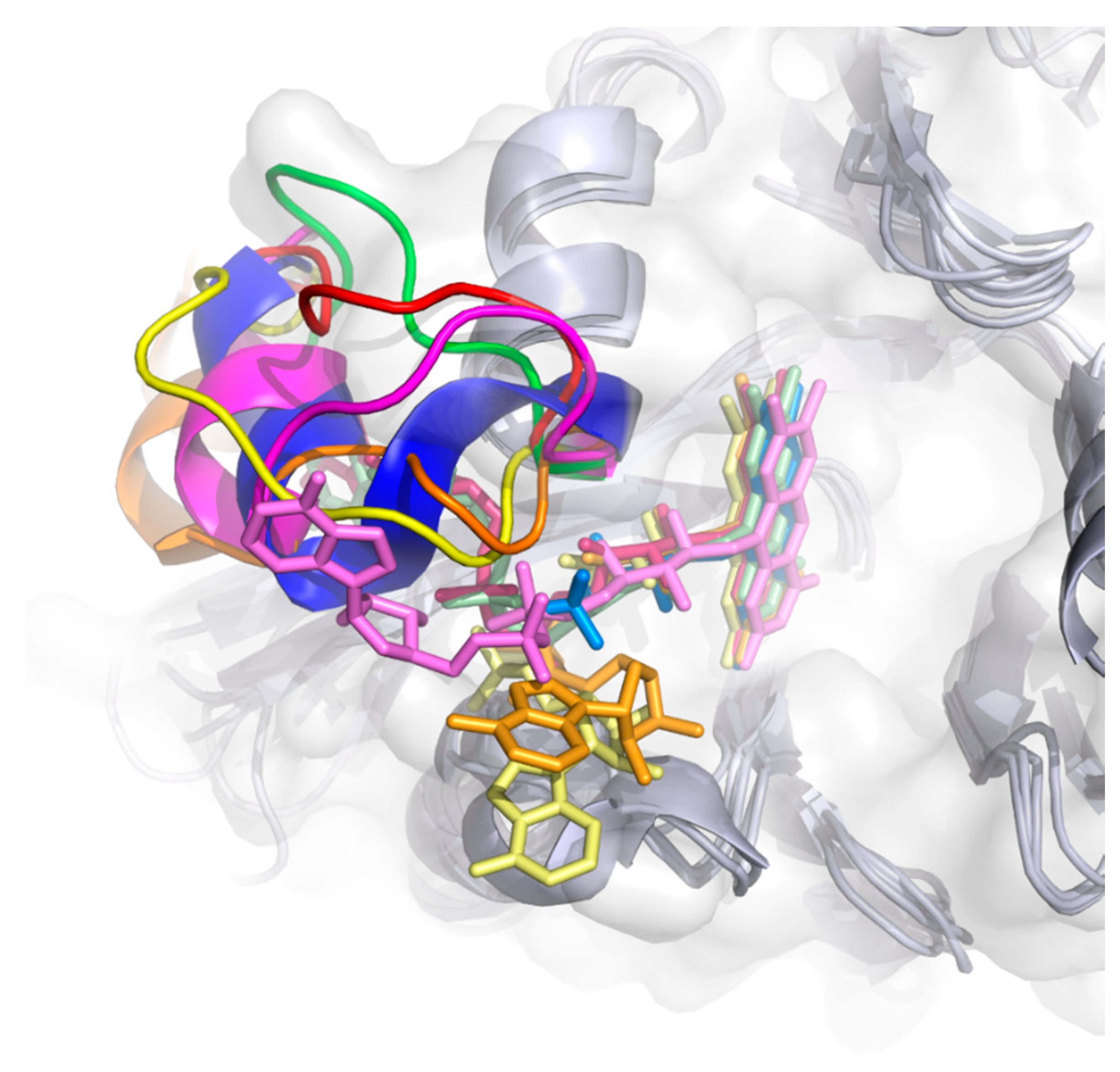
| Enzyme | PDB ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|---|
| PqsL | 2X3N | Q36 | R41 | I43 | G45 | I163 | A164 | R168 | G275 |
| SMOA | 3IHM | D33 | Y39 | R43 | N46 | G144 | K145 | G149 | V274 |
| PHBH | 1PBE | R33 | Y38 | R42 | R44 | F161 | H162 | R166 | R269 |
| Enzyme | EC Number | Reference |
|---|---|---|
| Free substrate halogenases | ||
| Typtophan 7-halogenase | 1.14.19.9 | [197,198] |
| Tryptophan 2-halogenase (a) | - | [199] |
| Tryptophan 4-halogenase (b) | - | - |
| Tryptophan 5-halogenase | - | [200] |
| Tryptophan 6-halogenase | - | [201,202,203,204,205] |
| Indole-3-halogenase | - | [192] |
| Fungal halogenases | - | [206,207,208] |
| Carrier protein-bound substrate halogenases | ||
| Pyrrolyl-S-PCP halogenases | - | [209,210,211] |
| (S)-β-Tyrosyl-S-PCP halogenase | - | [212] |
| Chondrochloren halogenase (a) | - | [213] |
| Chondramide halogenase (a) | - | [199] |
| Chloramphenicol halogenase (a) | - | [214] |
| Tiacumin halogenase (a) | - | - |
| Monodeoxypyoluteorin C3-halogenase (a) | - | [215] |
| Monodeoxypyoluteorin N-halogenase (a) | - | [215] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heine, T.; Van Berkel, W.J.H.; Gassner, G.; Van Pée, K.-H.; Tischler, D. Two-Component FAD-Dependent Monooxygenases: Current Knowledge and Biotechnological Opportunities. Biology 2018, 7, 42. https://doi.org/10.3390/biology7030042
Heine T, Van Berkel WJH, Gassner G, Van Pée K-H, Tischler D. Two-Component FAD-Dependent Monooxygenases: Current Knowledge and Biotechnological Opportunities. Biology. 2018; 7(3):42. https://doi.org/10.3390/biology7030042
Chicago/Turabian StyleHeine, Thomas, Willem J. H. Van Berkel, George Gassner, Karl-Heinz Van Pée, and Dirk Tischler. 2018. "Two-Component FAD-Dependent Monooxygenases: Current Knowledge and Biotechnological Opportunities" Biology 7, no. 3: 42. https://doi.org/10.3390/biology7030042
APA StyleHeine, T., Van Berkel, W. J. H., Gassner, G., Van Pée, K.-H., & Tischler, D. (2018). Two-Component FAD-Dependent Monooxygenases: Current Knowledge and Biotechnological Opportunities. Biology, 7(3), 42. https://doi.org/10.3390/biology7030042