1. Introduction
Dictyostelium discoideum is a haploid social amoeba that has proven to be an excellent model system for studying a number of processes including adhesion, cell–cell signaling, development, host pathogen interactions, and gene regulation among others. It has three life cycles consisting of a vegetative phase, a developmental phase, and a sexually reproductive phase [
1]. Under normal conditions,
D. discoideum are unicellular amoeba, but upon starvation, the cells begin to secrete cAMP in oscillating waves causing them to form multicellular aggregates that move toward the increasing concentration of cAMP. Within 24 h of the onset of development, the aggregates form mounds, slugs, and finally, a stalk of vacuolated cells and a fruiting body that contains thousands of spore cells; the stalk cells are a result of a type of programmed cell death, and the spores will germinate into amoeba [
2]. A variety of proteins such as adhesion proteins, G proteins, second messengers, surface receptors, prespore markers, and prestalk markers are involved in this process, and while much is known about the types of proteins that are secreted or are involved in adhesions during development, the mechanism of their secretion and delivery to the cell surface is unknown [
3]. The movement of these proteins appears to rely on vesicular trafficking suggesting the involvement of Rab GTPases [
3,
4,
5].
Rab proteins belong to the family of small molecular weight GTPases; they are evolutionarily conserved essential regulators of membrane trafficking [
6]. They act as molecular switches that cycle between an inactive GDP- and an active GTP-bound state [
7]. Rabs are essential for signaling and the control of cell proliferation and differentiation. Since they are found downstream in signaling cascades, they can impact gene expression and influence growth [
6] They serve as scaffolds to temporally and spatially integrate membrane trafficking and intracellular signaling [
8,
9]. Rabs are best known for their roles in exocytic and endocytic membrane trafficking, which encompasses the constitutive and regulated secretory routes, endocytosis via caveolae or clathrin-coated vesicles (CCVs), micropinocytosis, and phagocytosis. They can control anterograde and retrograde trafficking between cellular compartments to coordinate cargo delivery and membrane recycling [
6], and they have been implemented in the control of cell-type-specific functions, such as regulated secretion [
6].
Dictyostelium has proven to be a useful system in which to investigate endosomal and lysosomal membrane trafficking [
10,
11]. It is a professional phagocyte, and pinocytosis occurs at a very high rate in
Dictyostelium; fluid-phase nutrients are internalized by macropinocytosis, concentrated in endosomes, and degraded in lysosomes [
10,
11,
12]. This can easily be manipulated through the size of the particles used; fluid-phase endocytosis is involved with the up-take of non-particulate materials like RITC-dextran, while phagocytosis can be triggered by using larger particles, such as latex beads. Lysosomes connect at least three membrane trafficking pathways including the endocytic, biosynthetic, and phagocytic pathways [
10,
11,
13] all of which have been extensively studied. The biosynthetic pathway includes the biosynthesis of lysosomal enzymes; these are synthesized in the ER as membrane-bound, N-glycosylated precursor proteins, and then they are transported to the Golgi. Lysosomal enzymes are targeted to lysosomes. However, the recognition and sorting machinery involved in this part of the process is poorly characterized in
Dictyostelium. Unlike mammalian cells,
D. discoideum appear to lack the mannose 6-phosphate receptors (MPRs) used in the targeting of lysosomal enzymes suggesting the existence of another lysosomal enzyme receptor [
10,
11,
14].
It was previously observed that DdRabS localizes to ER-Golgi membranes and the contractile vacuole system [
15]. This protein is a Rab GTPase that is somewhat homologous to Rab1 in other organisms; as is commonly done in this organism, proteins with a lower degree of homology are typically given a letter as a name while those with higher homology use a number and additional letters for any existing isoforms. In this study we report an investigation into the role of RabS in
Dictyostelium development, endocytosis, endocytic recycling, and the secretion of lysosomal enzymes. We have found that these Rab proteins can play multiple roles in the cell, so numerous studies were undertaken. Overexpressing DdRabS cells showed an increase in the rate of phagocytosis and pinocytosis with a corresponding increase in growth rate. It was observed that DdRabS were unable to complete the developmental process as they could not develop fruiting bodies. Aggregation was severely defective as cells had decreased cell-cell cohesion and chemotactic response to cAMP. Furthermore, when incubated with DIF-1 to induce cell death, DdRabS over-expressing cells showed a significant increase in cell mortality. Interestingly, cells expressing a dominant negative form of the DdRabS protein behaved similarly to the wild type (WT) AX4 cells. This data suggests that overexpression of RabS causes a cascade of signaling defects crucial to normal development in
Dictyostelium.
2. Materials and Methods
2.1. Cells and Culture Conditions
All
D. discoideum cell lines, WT strain AX4, pDneo2a-GFP, DdRabS-GFP, and DdRabS(DN)-GFP were grown axenically at 21 °C in shaking culture at 150 r.p.m. in HL5 medium:1% oxoid proteose peptone, 1% glucose, 0.5% yeast extract (Fisher Biotech, Fair lawn, NJ, USA), 2.4 mM Na
2HPO
4, and 8.8 mM KH
2PO
4, pH 6.5, and the media was supplemented with 300 mg/mL of streptomycin sulfate and 100 mg/mL of ampicillin (Sigma, St. Louis, MO, USA). Transfected cells also received 10 mg/mL of G418 (Invitrogen, Grand Island, NY, USA). To minimize autofluorescence from the media, cells were incubated in LoFlo media : 0.1% FM salts 1 (500 mM NH
4Cl 200 mM MgCl
2 10 mM CaCl
2), 0.1% FM salts 2 (FeCl
3 50 mM), 0.01% FM trace elements (8.56 mM Na
2-EDTA·2H
2O, 13 mM ZnSO
4·H
2O, 14.5 mM H
3BO
4, 2.6 mM MnCl
2·4H
2O, 0.7 mM CoCl
2·6H
2O, 0.6 mM CuSO
4·5H
2O, 0.1 mM (NH
4)6Mo
7O
24·4H
2O pH 6.5), 63 mM glucose, 5 mM K
2HPO
4, 1% casein peptone, pH 6.5 for 24 h before fluorescent experiments (
www.dictybase.org).
2.2. Creation of GFP Tagged DdRabS Cell Lines
The DdRabS overexpressing cell line was created by amplifying the gene from genomic DNA using polymerase chain reaction. The resulting amplificon was ligated into the pDneo2A-GFP vector and transformed into the AX4 strain of
Dictyostelium discoideum using electroporation; transformants were selected for using the antibiotic G418 (Invitrogen, Grand Island, NY, USA. The DdRabS(DN) cell line was created using the Stratagene QuickChange
® II Site-Directed Mutagenesis Kit (Stratagene, Santa Clara, CA, USA). The resulting plasmid was also transformed into AX4 and selected for with G418 [
15].
2.3. Endocytosis Assays
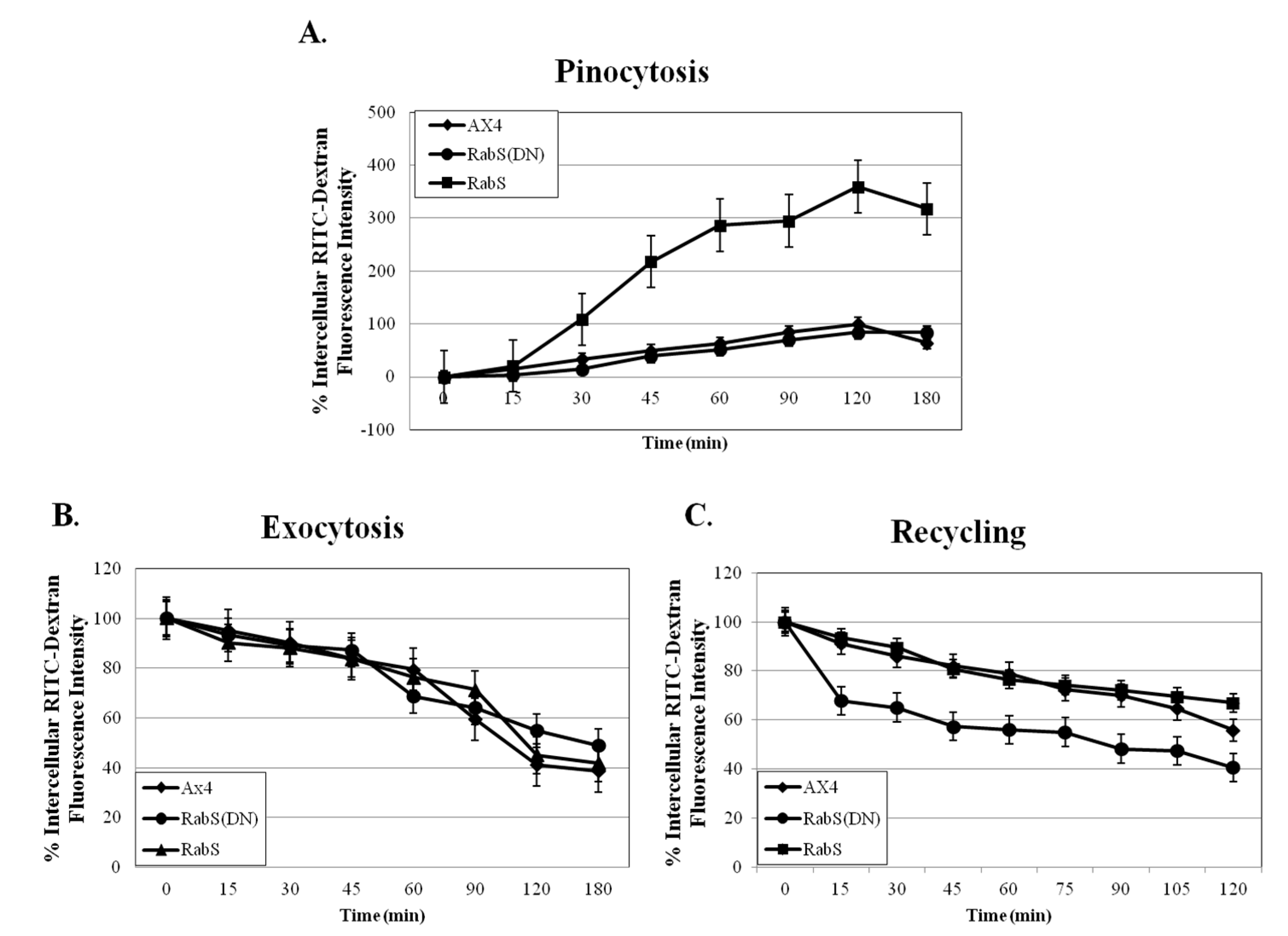
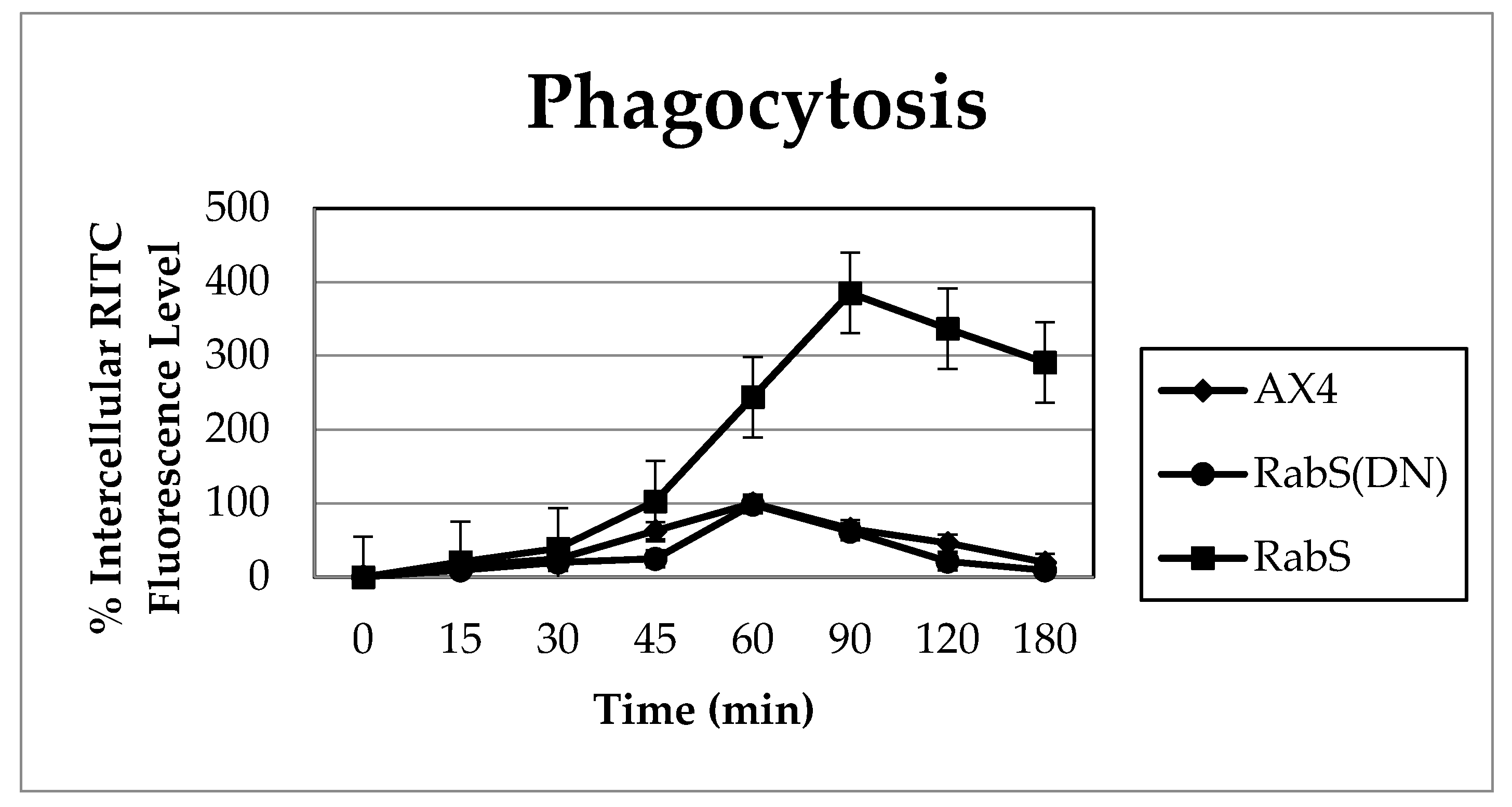
Phagocytosis was measured by incubating 10 mL of 2 × 106 cells/mL with 100 µL of fluorescent 2 µm rhodamine isothiocynate labeled latex beads (200:1 beads per cell) (RITC- latex beads, Sigma Aldrich, St. Louis, MO, USA) on a rotary shaker, and 1 mL samples were taken at the indicated time points (0, 15, 30, 45, 60 min); 100 µL were taken at the 0 time point and tested for protein content by Bradford assay. Samples were placed into 2 mL ice-cold Soerensen buffer (SB): 14.5 mM KH2PO4, 2.5 mM Na2HPO4, pH 6, and layered on top of 10 mL 20% w/v PEG 8000. Then they were centrifuged at 800 g and 4 °C for 10 min; the cell pellet was washed twice with cold SB, resuspended in 1 mL lysis buffer (50 mM Na2HPO4, pH 9.3, 0.2% Triton X-100), and fluorescence was measured with a Horiba Jobin Yvon analytical grade FluorMax3 Spectrofluorimeter, excitation 544 nm, emission 574 nm. Relative fluorescence was plotted in comparison to the WT as a function of time after correcting for differences in protein content. Experiments were performed in triplicate.
Fluid phase pinocytosis was measured by incubating 5 mL cells at 5 × 106 cells/mL with 100 µL of 100 mg/mL fluorescent rhodamine isothiocynate dextran, molecular weight 10,000, (RITC- dextran, Sigma Aldrich, St. Louis, MO, USA) on a rotary shaker, and 500 µL samples were taken at the indicated time points (0, 15, 30, 45, 60, 90, 120, 180 min); 100 µL of the cell suspension was tested for protein content by Bradford assay. Samples were placed into 50 µL of Trypan blue, inverted once, and centrifuged at 800 g 4 °C for 2 min. The supernatant was discarded, the pellet was washed with 1 mL SB, and then resuspended with 1 mL SB. Fluorescence was measured with a Horiba Jobin Yvon analytical grade FluorMax3 Spectrofluorimeter, excitation 544 nm, emission 574 nm. Fluorescence was plotted against time after subtracting the fluorescence at the 0 min time point and correcting for differences in protein concentration. Experiments were performed in triplicate.
Exocytosis was measured by preparing the cells as for the pinocytosis assay; after 3 h, the cells were centrifuged at 500 g for 3 min, washed twice with nutrient media, and resuspended in 5 mL fresh media (RITC-dextran, Sigma Aldrich, St. Louis, MO, USA). Then 500 µL samples were taken at the indicated time points and treated as in the pinocytosis assay (0, 15, 30, 45, 60, 90, 120, 180 min). Fluorescence was measured with a Horiba Jobin Yvon analytical grade FluorMax3 Spectrofluorimeter, excitation 544 nm, emission 574 nm. Fluorescence was plotted against time; the 0 min time point for each cell line was assigned 100% relative fluorescence, and the rest of the time points were calculated relative to them. Experiments were performed in triplicate.
Recycling rates were measured by incubating 10
8 cells in 4 mg Lucifer yellow (Lucifer yellow, Sigma Aldrich, St. Louis, MO, USA) dissolved in 10 mL of medium; then, the cells were centrifuged at 500
g for 3 min and resuspended in LoFlo medium. Then, 1 mL samples were taken at the indicated time points and added to 100 µL of Trypan blue, inverted, and centrifuged for 2 min at 800
g. The pellets were resuspended in 1 m SB, and the fluorescence was measured with a Horiba Jobin Yvon analytical grade FluorMax3 Spectrofluorimeter, excitation 470 nm, emission 515 nm. Again, the fluorescence at the 0 time point was subtracted from each data point and then plotted against time. The 0 min time point for each cell line was assigned 100% relative fluorescence, and the rest of the time points were calculated relative to them [
16]. Experiments were performed in triplicate.
2.4. Endosome Visualization: RITC-Dextran Loading
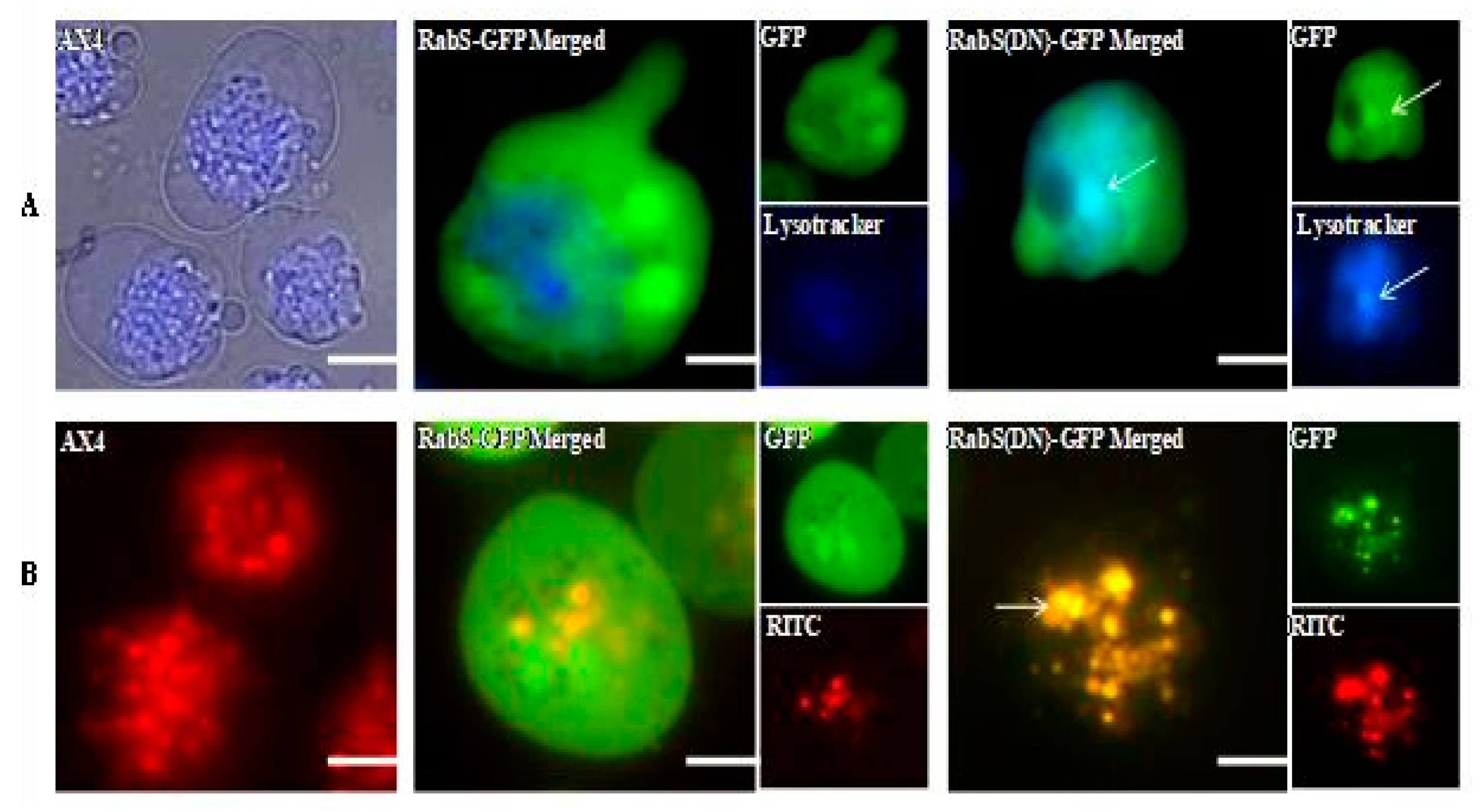
Rhodamine B isothiocynate-dextran (RITC-dextran, Sigma Aldrich, St. Louis, MO, USA) is a fluid that is internalized but not degraded by endosomes [
17]. Cells were harvested, placed in a 35 × 10 mm Petri dish containing 2 mL of HL5 medium, and supplemented with 40 µL of 100 mg/mL RITC-dextran; the cells were incubated for 60 min, washed with fresh media, and allowed to settle on a glass coverslip. All cell lines were photographed using the BrightLine
® TXRED filter set to visualize endosomes and the BrightLine
® GFP filter set was used to visualize GFP fluorescence on a Nikon 2000SE microscope with IPLab 3.7 software at 100× magnification. Quantification was performed using Fiji by ImageJ. The Pearson’s Correlation Coefficient (PCC) was reported.
2.5. Growth Rate
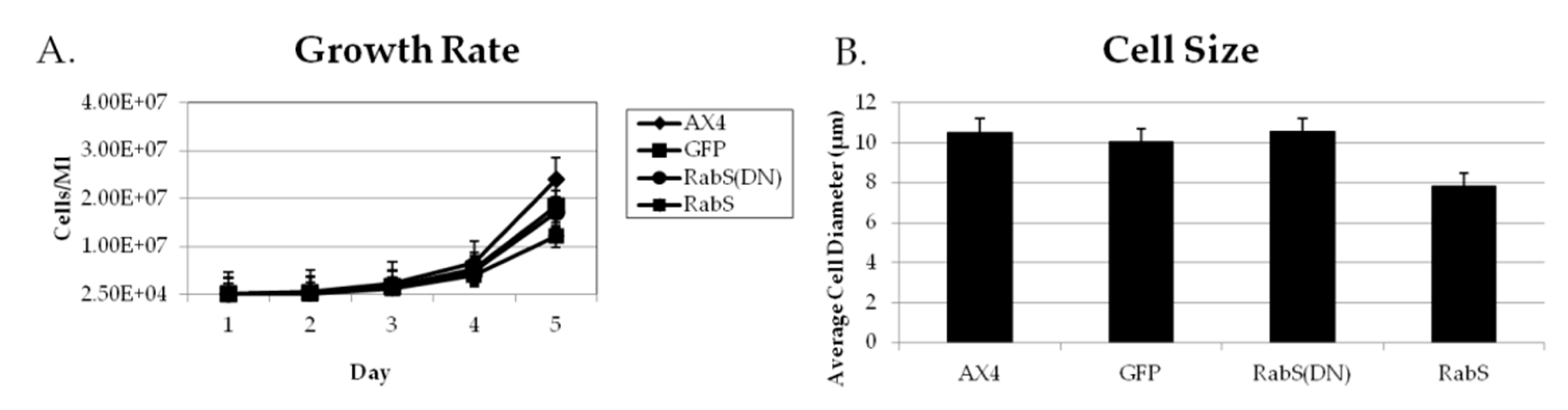
All cell lines were grown in HL5 medium until a density of 1–3 × 104 cells/ mL was obtained. Cellular growth was determined by counting the cells every 24 h over five days using a hemacytometer (Hausser Scientific, Horshman, PA, USA). Experiments were performed in triplicate.
2.6. Cell Size and Diameter
Cell size was determined by taking cells in exponential growth phase, washed in SB, and then resuspended in equal volumes of SB. Then, they were placed on a microscope slide. 50 cells were photographed and measured using a Nikon 2000SE microscope with IPLab 3.7 software (Scanalytics, Inc., Fairfax, VA, USA) with 100× magnification. Experiments were performed in triplicate.
2.7. Lysosome Visualization: LysoTracker® Blue Staining
LysoTracker
® Blue (Molecular Probes) was used to mark acidic organelles like lysosomes [
18]. Cells were harvested and allowed to adhere to coverslips; the medium was aspirated, replaced with fresh medium containing 100 nM LysoTracker
® Blue, and incubated for 30 min at room temperature. The cells were washed twice with fresh media, and fluorescence was photographed with a Nikon 2000SE microscope with IPLab 3.7 software (Scanalytics, Inc., Fairfax, VA, USA) with 100× magnification using the BrightLine
® DAPI filter set, causing lysosomes to fluoresce blue, and the BrightLine
® GFP filter set was used for comparison with GFP localization. Quantification was performed as previously described.
2.8. Secretion of Lysosomal Enzymes under Starvation Conditions
The standard secretion assay was performed as described by [
19]. Log phase cells were harvested by centrifugation, resuspended at a concentration of 5 × 10
7 cells/mL in starvation buffer (10 mM phosphate buffer, pH 6.0), and incubated at 21 °C; samples were removed at the indicated time points (0, 15, 30, 60, 90, 120, 180, 280 min), and enzyme assays were performed on the supernatant (extracellular) and cell pellets (intracellular). The α-mannosidase, N-acetylglucosaminidase, β-galactosidase, acid phosphatase, and β-glucosidase activity was measured as previously described [
19,
20]. The 0 min time point for each cell line was assigned as the 100% relative amount, and the rest of the time points were calculated relative to them. Experiments were performed in triplicate.
2.9. Developmental Assay
Cells were grow axenically in HL5 medium on a rotary shaker at 160 r.p.m. and 1 × 10
9 cells/ mL were harvested by centrifugation. After washing three times with developing buffer (5 mM Na
2HPO
4, 5 mM Na
2HPO
4, 5 mM KH
2PO
4, 1 mM CaCl
2, 2 mM MgCl
2), cells were resuspended at a density of 2 × 10
8 cells/m; 200 µL of the cell suspension was spread evenly on a 100 mm KK
2 plate using a sterile glass spreader. The plates were wrapped with a wet paper towel, sealed in plastic wrap, inverted, and incubated at 22 °C. After 6 h, pictures were taken every 2 h to monitor the different developmental stages; this method was adapted from
www.dictybase.org. Photographs of multicellular development were taken using a model AM 4201, 25× microscope (Lutron Instruments, Tamil Nadu, India).
2.10. Cell Cohesion Assay
Cell cohesion assays were performed as described by [
21,
22,
23]. To determine developmental cell cohesion, vegetative cells were centrifuged at 700
g for 4 min, washed in KK
2, resuspended in KK
2 at 2 × 10
7 cells/mL, and starved in rotary-agitated suspension (175 r.p.m.) at 22 °C for 4 h. The cell suspension was diluted to a density of approximately 2.5 × 10
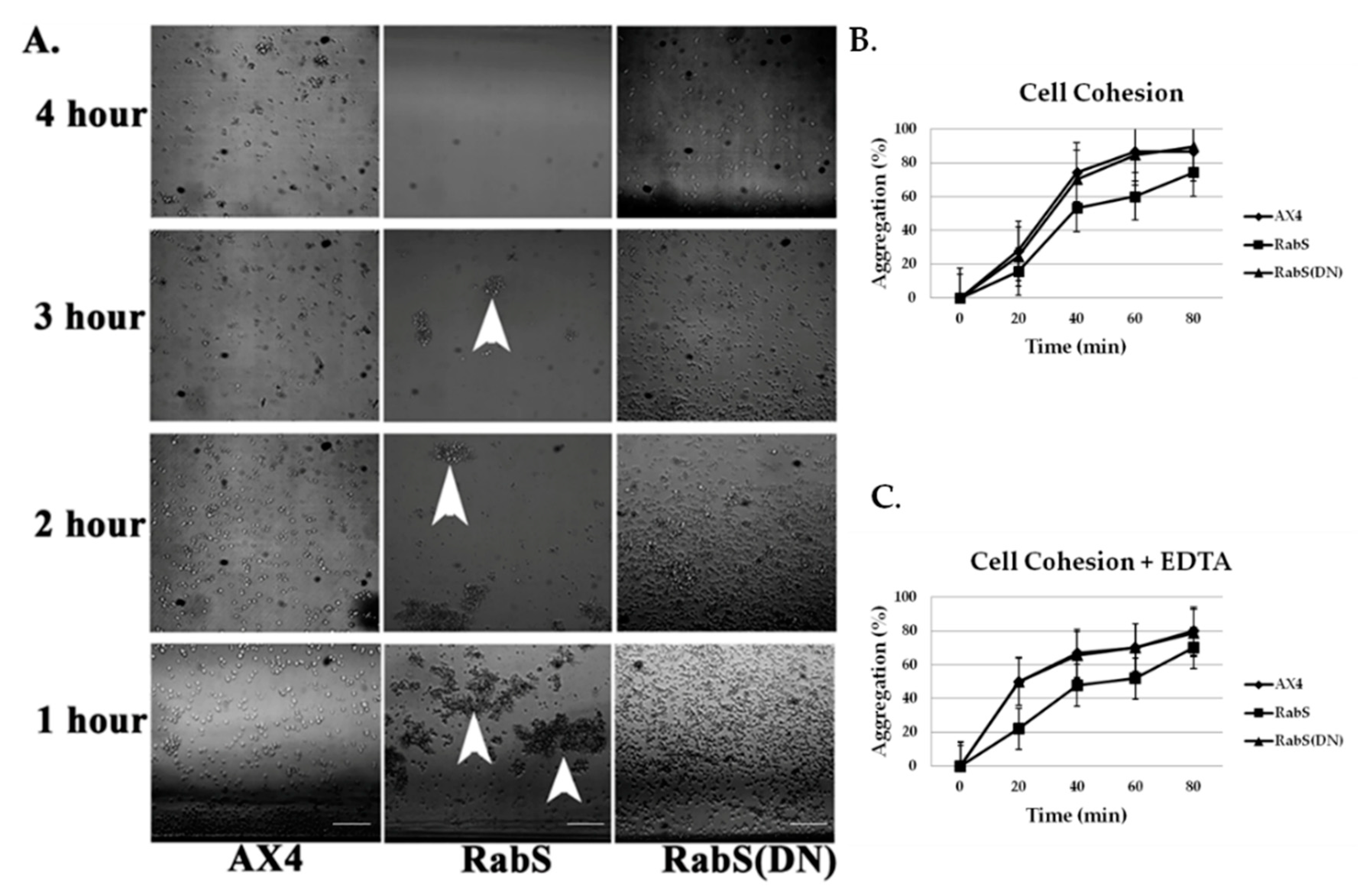
6 cells/mL, and aggregates were dispersed by vigorously vortexing for 15 s. Aggregates were allowed to reform while rotating on a platform shaker at 180 rpm at room temperature. At the indicated times (the number of non-aggregated cells, including singlets and doublets, were scored using a hemocytometer; the number of aggregating cells was determined by subtracting this number from the total number of cells and was expressed as a percentage of the total.
To assess EDTA-resistant cell-cell adhesion, cells were harvested and resuspended in KK2, starved in a rotary shaker at 22 °C for 4 h, centrifuged, and resuspended in KK2 + 10 mM EDTA; this buffer inhibits EDTA-sensitive cohesion. Aggregates were dissociated by brief vortexing, and samples (0.2 mL) were placed in plastic tubes and rotated vertically at 180 rpm at room temperature for cell re-association. The percentage of cell aggregation was monitored at regular intervals for 60 min. Experiments were performed in triplicate.
2.11. Chemotaxis in Response to cAMP
Cell lines were grown axenically in HL5 medium and 1 × 107 cells were harvested by centrifugation. Cells were washed twice with developing buffer and resuspended at a density of 5 × 106 cells/ mL. After shaking for 8 h to allow allowing for the development of the cAMP receptor, 100 µL of the cell suspension was added to an outer 2 mm trough in a 60 mm agarose plate. At the same time, 100 µL of 5 µM cAMP was added to the center trough. Images were taken every h for 4 h using a Nikon 2000SE microscope with NIB Image software at 10× magnification. The edge of the trough was included in the image as a reference point.
2.12. Flow Cytometry Assay
To induce cell death, vegetative cells in late exponential growth phase were harvested and washed twice with SB. Approximately 5 × 10
5 cell/ mL were suspended in 1 mL of SB containing 3 mM cAMP (Sigma) in a flask and incubated for 8 h at 22 °C. Cells were washed with 1 mL of SB which was replaced afterward with either 1 mL of SB or 1 mL of SB containing 0.1 mM differentiation inducing factor DIF-1 [1-(3,5-dichloro-2,6-dihydroxy-4-methoxyphenyl)-hexanone-1-one], (Sigma) and incubated for 30 h at 22 °C. The cells were resuspended at a density of 1 × 10
6 cell/ mL in SB and divided into 300 µL aliquots. The aliquots were placed into test tubes containing 1 µg/mL Propidium Iodide (PI) stain, and they were incubated for 10 min at 22 °C without washing. Cytometry analysis was then performed on a FACSCalibur cytometer from Becton Dickinson using CellQuest software [
16]. Experiments were performed in triplicate.
2.13. Determining Cell Viability Due to ATP Levels
The number of viable cells in culture was determined based on measuring the quantity of ATP, an indication of the presence of active cells [
24]. The CellTiter-Glo
® Luminescent Cell Viability Assay kit (Promega) was used for this test. Approximately 5 × 10
5 vegetative cells were harvested, resuspended in 100 µL of 10 mM MES (Sigma), and then transferred into opaque-walled 96-well plates [
25]. Cells were lysed by adding 100 µL of the CellTiter-Glo reagent, mixed on a shaker for 2 min, and incubated at room temperature for 10 min. Microtiter
® Plate Illuminometer B36580 with Revelation MLX software was used to measure the ATP levels [
25]. Experiments were performed in triplicate.
2.14. Cellular Re-Growth Assay
Vegetative cells in the late exponential growth phase were harvested and washed twice with SB. Approximately 5 × 10
5 cells were added to 1 mL of SB containing 3 mM cAMP in a two Lab-Tek chamber and incubated for 8 h at 22 °C. The cells were washed with 1 mL of SB that was later replaced with either 1 mL of SB or 1 mL of SB containing 0.1 mM DIF-1 and incubated for 30 h at 22 °C. From each chamber, 0.5 mL of SB was removed and replaced with 1 mL of HL5 medium and incubated for 72 h at 22 °C. Cells were detached and counted using a hemocytometer and phase contrast optics. Finally, the ratio of the number of cells re-growing in the DIF-1 chamber to the number of cells in the control chamber was calculated [
16]. Experiments were performed in triplicate.
2.15. Statistical Analysis
Statistical analysis for the endocytosis, exocytosis, phagocytosis, and membrane recycling assays was performed using by running the data through a 2-way ANOVA to test for significance at p < 0.01. Statistical analysis for the growth, cell size, and volume assays was performed by running the data through a student’s t-test with p ≤ 0.05. Statistical analysis for the secretion of lysosomal enzyme assays was performed using by running the data through a 2-way ANOVA to test for significance at p < 0.05. Statistical analysis for the chemotaxis assay was per formed by running the data through a student’s t-test with p ≤ 0.05. Statistical analysis for regrowth, cell death, and metabolic activity assays was performed by running data run through 2-way ANOVAs to test for significance at p < 0.05.
4. Discussion
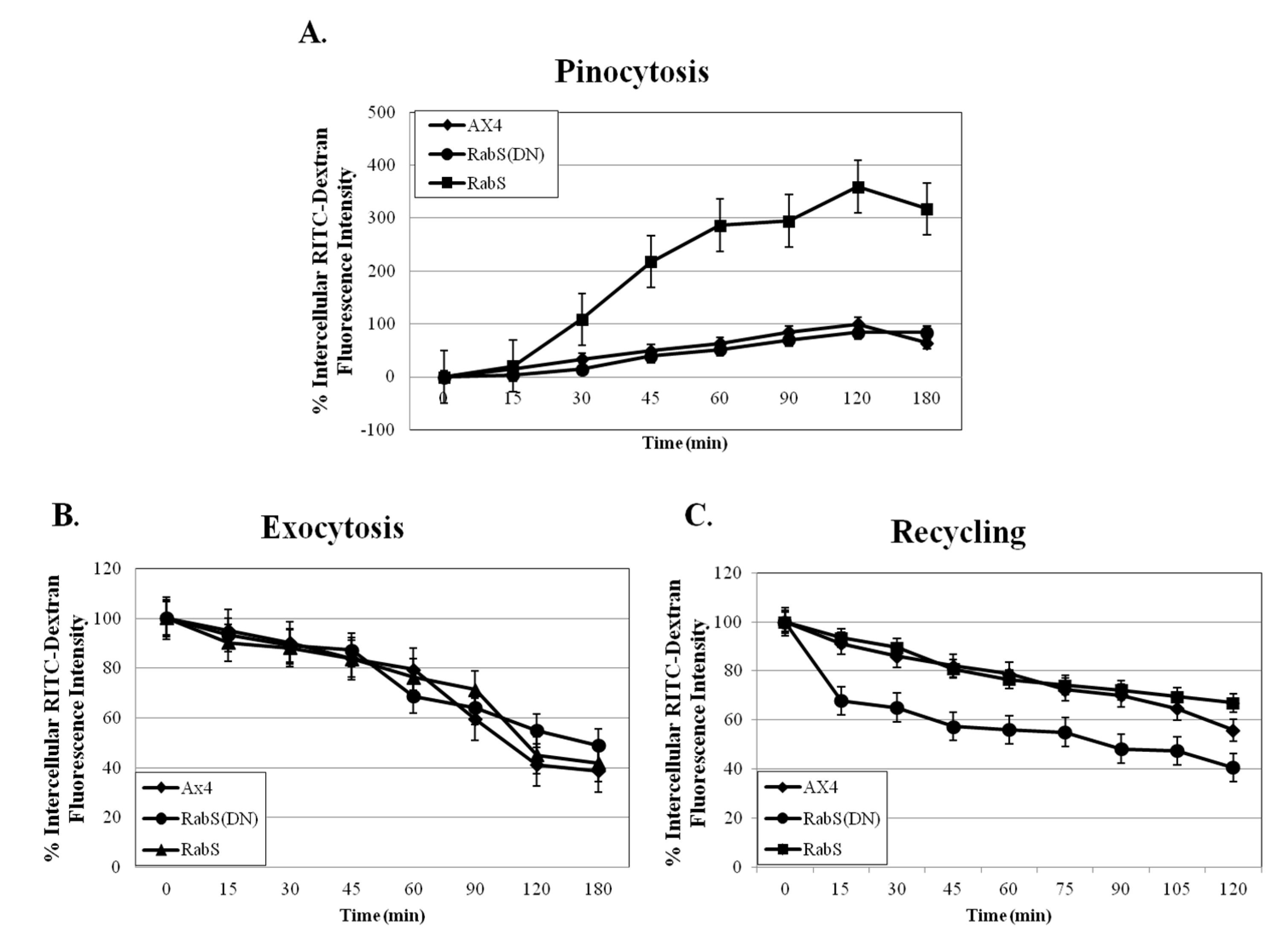
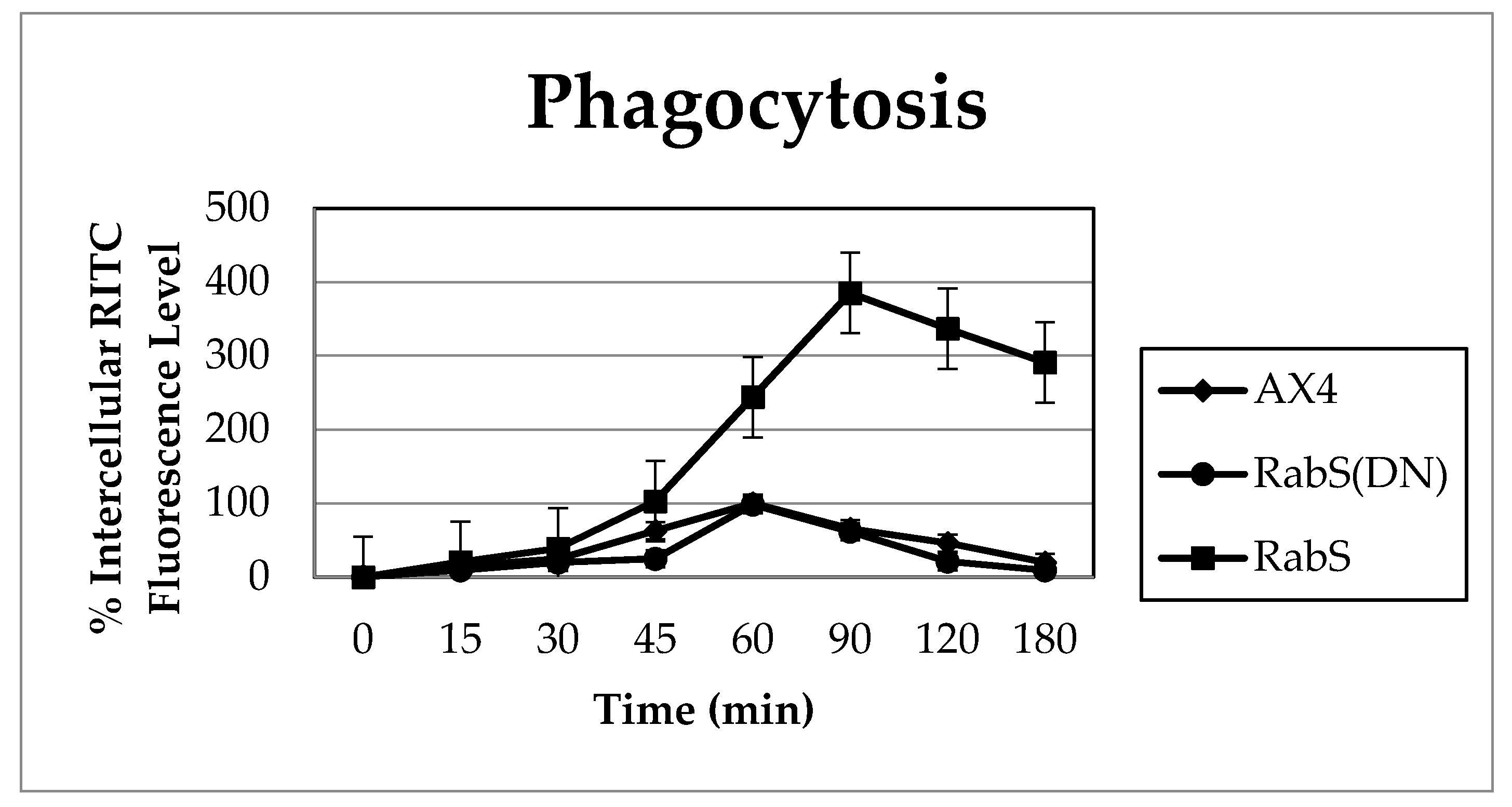
Upon analysis, it was found that the rates of both phagocytosis and pinocytosis were significantly increased in cells overexpressing DdRabS. Furthermore, cells expressing the DdRabS(DN) protein showed a significantly increased rate of recycling. The RabS(DN) protein may be down-regulating DdRabS signaling pathways accounting for the increased rate of recycling observed in the GDP-bound protein. Another possibility is that DdRabS may normally function to negatively regulate the process of endocytic recycling in
Dictyostelium; this is similar to the findings [
30] that DdRab11 acts to negatively regulate the rate of phagocytosis as expression of DdRab11(DN) resulted in an increase in this process. The correlation in the rates of phagocytosis, pinocytosis, and growth rate observed in DdRabS overexpressing cells has also been previously observed in the overexpression of
Dictyostelium GTPase RacH [
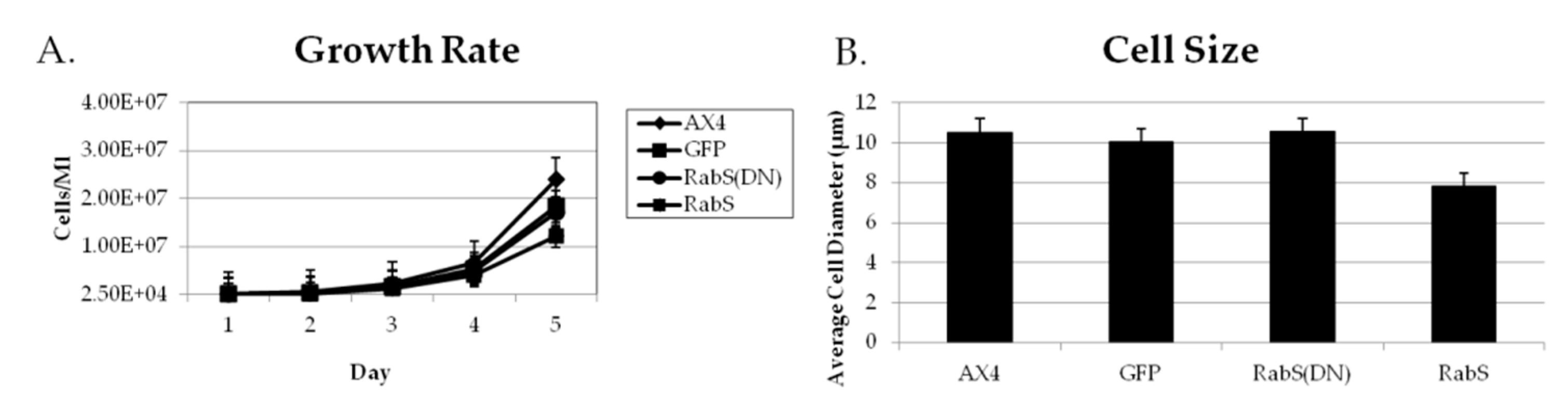
31], which plays a role in endocytic vesicular trafficking. The smaller size of the cells in the RabS overexpressing cell line could also contribute to the decreased rates of phagocytosis, pinocytosis, and growth.
It is possible that by overexpressing DdRabS, downstream signals triggering phagocytosis are being up-regulated, or that it may play a role in regulating the formation of the phagocytic cup or phagosome maturation. It has been proposed that formation of the phagocytic cup in many cells involves recruitment of endo-lysosomal vesicles [
32,
33,
34]. If DdRabS is involved either directly or indirectly in the formation or maturation of the phagosomes, overexpressing it may in turn be causing more phagosomes to form resulting in the increased rate of phagocytosis observed. The fact that the DdRabS(DN) mutant cells displayed phagocytic rates similar to that of AX4 cells suggests an indirect role for DdRabS in phagocytosis. DdRabS may also be responsible for trafficking key players in the formation or maturation of phagosomes. Mutant DdRabS(DN) cells would still be able to traffic these proteins; however, they would not be able to leave the membrane after docking due to their GDP-locked form.
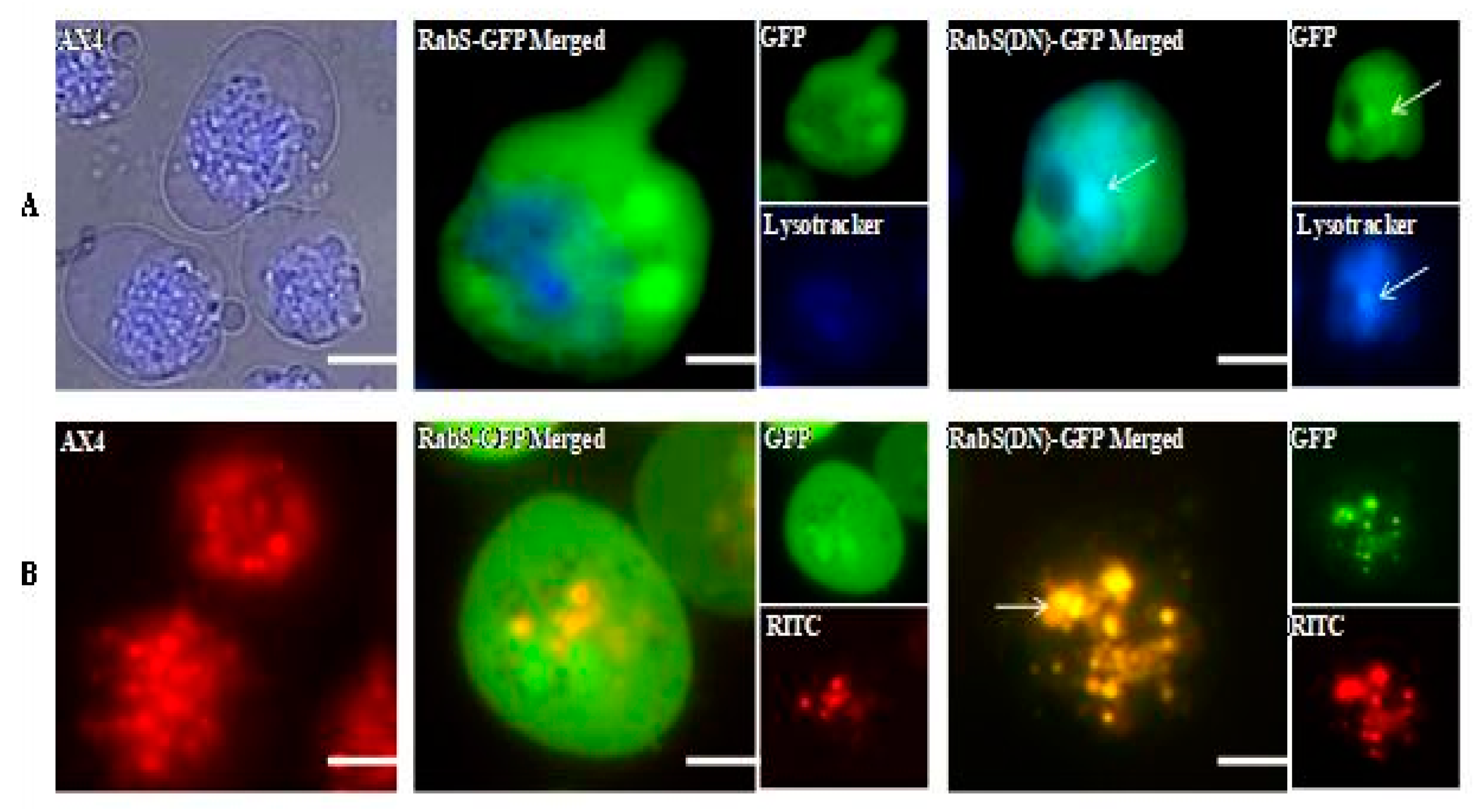
DdRabS has been shown to localize between the ER-Golgi membranes and the CV system [
15], and it also partially colocalizes with endosomes. DdRabS(DN) cells colocalize to endosomes and lysosomes, and it has been hypothesized that DdRabS may also be involved in trafficking along the endo-lysosomal pathway as well. In this study, it was demonstrated that overexpressing DdRabS causes a significant increase in the rates of growth, phagocytosis, and pinocytosis, and that cells expressing DdRabS(DN) are defective in endocytic recycling. It has been demonstrated that DdRab2 plays a role in vesicular trafficking, and given the similar locations of DdRab2 and DdRabS, this prompted us to investigate a possible role for DdRabS along the endo-lysosomal pathway [
35].
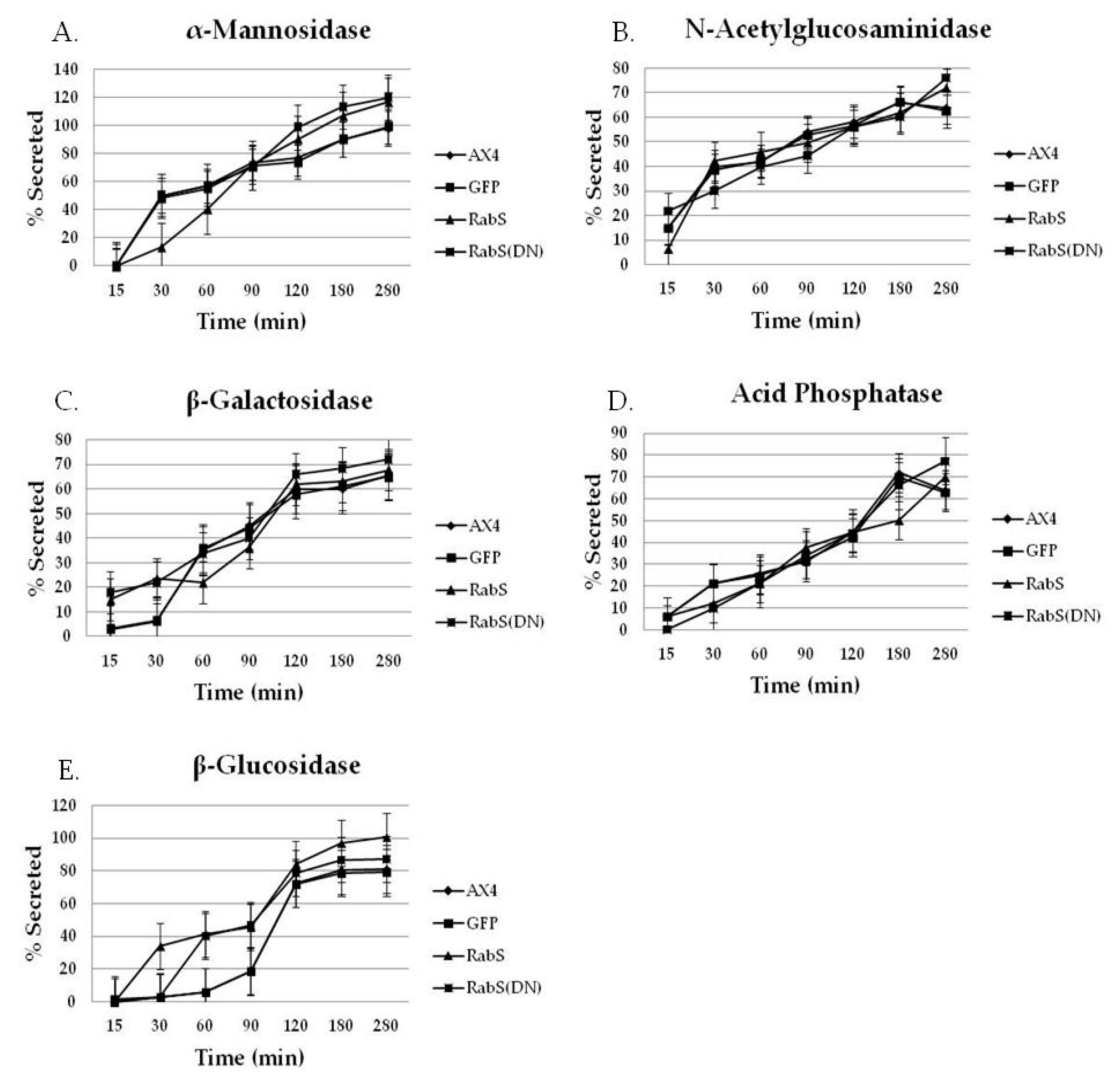
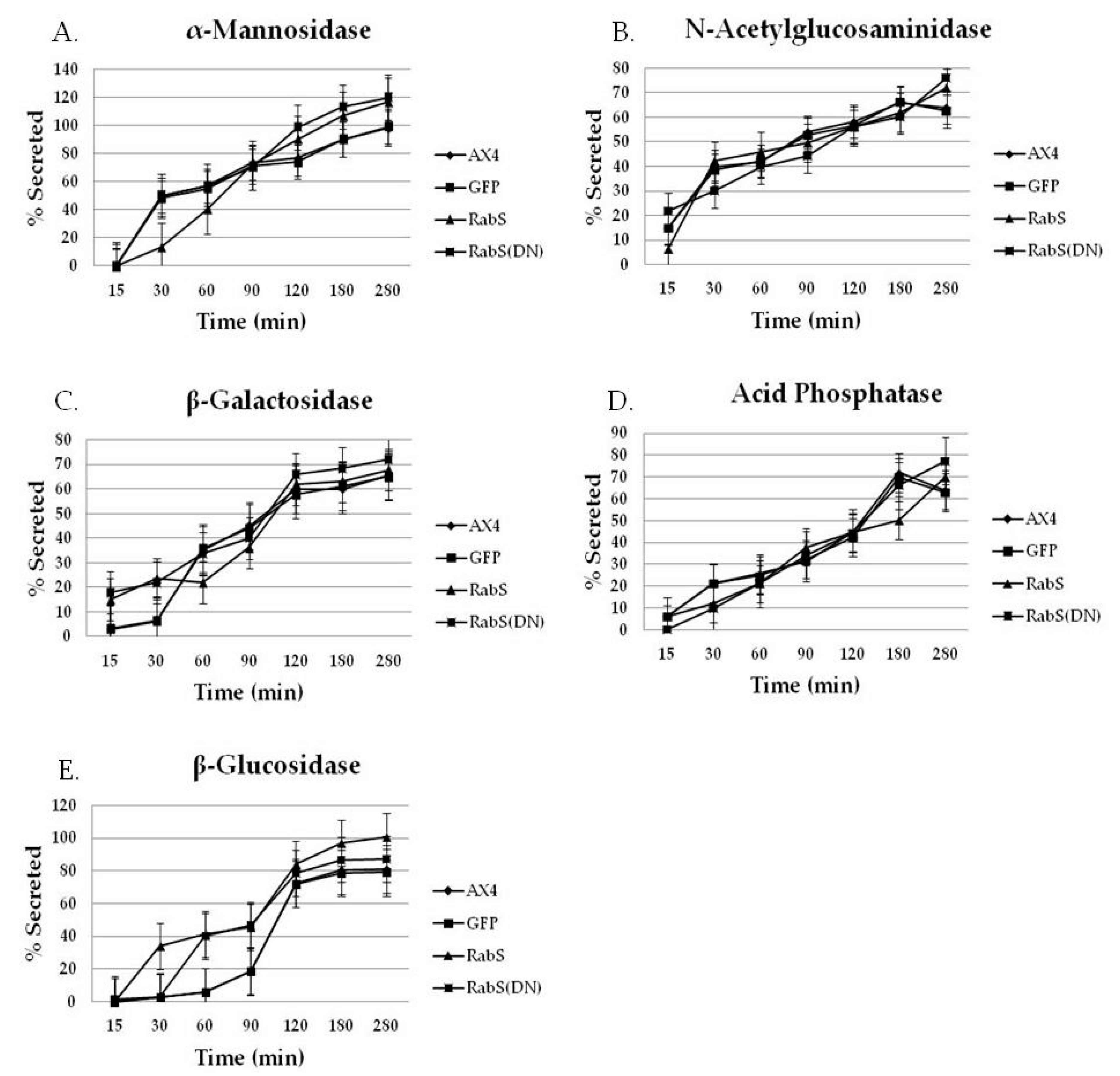
Both DdRabS overexpressing and DdRabS(DN) cells exhibited an up-regulation in the secretion of lysosomal enzymes, with the greatest change being observed in the oversecretion of β-glucosidase. DdRabS is deficient in development, cell-cell adhesion, chemotaxis, regrowth, and viability, which could be related to the increased release of lysosomal enzymes. However, it shows an increase in cell death in response to DIF-1. This data suggests that the DdRabS protein functions in the transport of proteins from the ER-Golgi-CV as well as along the endo-lysosomal pathway, and that after docking with the membrane, the GDP-locked DdRabS(DN) is not able to leave the membrane accounting for the differential localization observed. Under normal conditions, DdRabS may only be dropping its cargo to the endosomes and lysosomes and then continuing on. This would explain why DdRabS-GFP does not show localization with these organelles in the overexpressing cell line. It is not able to accumulate on the membranes as in the DN form. This microscopic evidence coupled with the graphical evidence suggests a role for DdRabS along the endo-lysosomal pathway [
35].
Normally, Rab proteins are activated by the exchange of GDP for GTP, which is triggered by guanine nucleotide exchange factors (GEFs). Once an individual transport step is completed, GTPase-activating proteins (GAPs) accelerate Rab GTP hydrolysis, allowing recognition by a GDP dissociation inhibitor (GDI), which sequesters the Rab in the cytosol until it is recruited to a membrane and begins the transport cycle again [
6,
36]. We propose that DdRabS(DN) is unable to leave the endosomal and lysosomal membranes because it is locked in its GDP-bound state preventing it from converting to the GTP-bound form after delivering its cargo; thus, it accumulates in these organelles accounting for its differential localization.
Statistically significant differences were found in the secretion of lysosomal enzymes, both between the cell lines and the time points samples were taken at. After following standard secretion conditions [
19], more than 50% of the total cellular activity of several lysosomal glycosidases was secreted within a few hours; these conditions are particularly useful because the lysosomal enzymes are secreted in the absence of any appreciable enzyme synthesis or degradation. Thus, by monitoring the release of lysosomal enzymes, the functioning of the lysosomal vesicles involved in secretion can be observed [
19]. As compared to all tested cell lines, we observed that DdRabS mutants slightly oversecrete α-mannosidase, N-acetylglucosaminidase, and β-galactosidase and significantly oversecrete β-glucosidase. The secretion of acid phosphatase appeared unaffected.
It has been proposed that there are three functional classes of lysosomal vesicles in
Dictyostelium; two of these classes are secretory, and the other is not. While acid phosphatase is believed to belong to one of the secretory classes, α-mannosidase, N-acetylglucosaminidase, β-galactosidase, and β-glucosidase are believed to belong to the other [
19,
37,
38]. Another possibility presented by the Cardelli group [
37] is that acid phosphatase secretion is differentially regulated compared to the glycosidases. Our results indicate that DdRabS is involved with the class of lysosomal vesicles containing glycosidases as the secretion of acid phosphatase appeared unaltered when compared to the control cell lines. The secretion of β-glucosidase was highest in DdRabS overexpressing cells and while this phenotype was rescued slightly by DdRabS(DN) cells, the cells still oversecreted the enzyme. These results are consistent with a role for DdRabS in the secretion of lysosomal enzymes. The oversecretion observed may be related to improper acidification of endosomes. It was indicated in previous reports on
Acanthamoeba that hydrolase transport and secretion is pH dependent [
31,
39]. Starvation initiates the developmental cycle in
Dictyostelium, which is, in part, characterized by induced secretion of lysosomal hydrolases relative to secretion during growth [
10].
Regulated secretion is also characteristic of
Dictyostelium development; when cells reach high density and during early development, they secrete lysosomal hydrolases [
3,
10,
13], cell-density sensing factor (CMF) [
3,
40,
41,
42] and cell-counting factor (CF) [
3,
43,
44,
45,
46,
47], which can regulate aggregate size during development. Countin, a subunit of CF, negatively regulates adhesion by regulating the expression of CAMs [
44]. In mutants that over-secrete countin, there is decreased adhesion, and in mutants where countin is disrupted (countin 2), there is increased adhesion [
44]. It is possible that overexpressing DdRabS also causes an oversecretion of the lysosomal hydrolase countin accounting for the decrease in adhesion; it is also feasible that this oversecretion could be related to the elevated rate of death the RabS cell line.
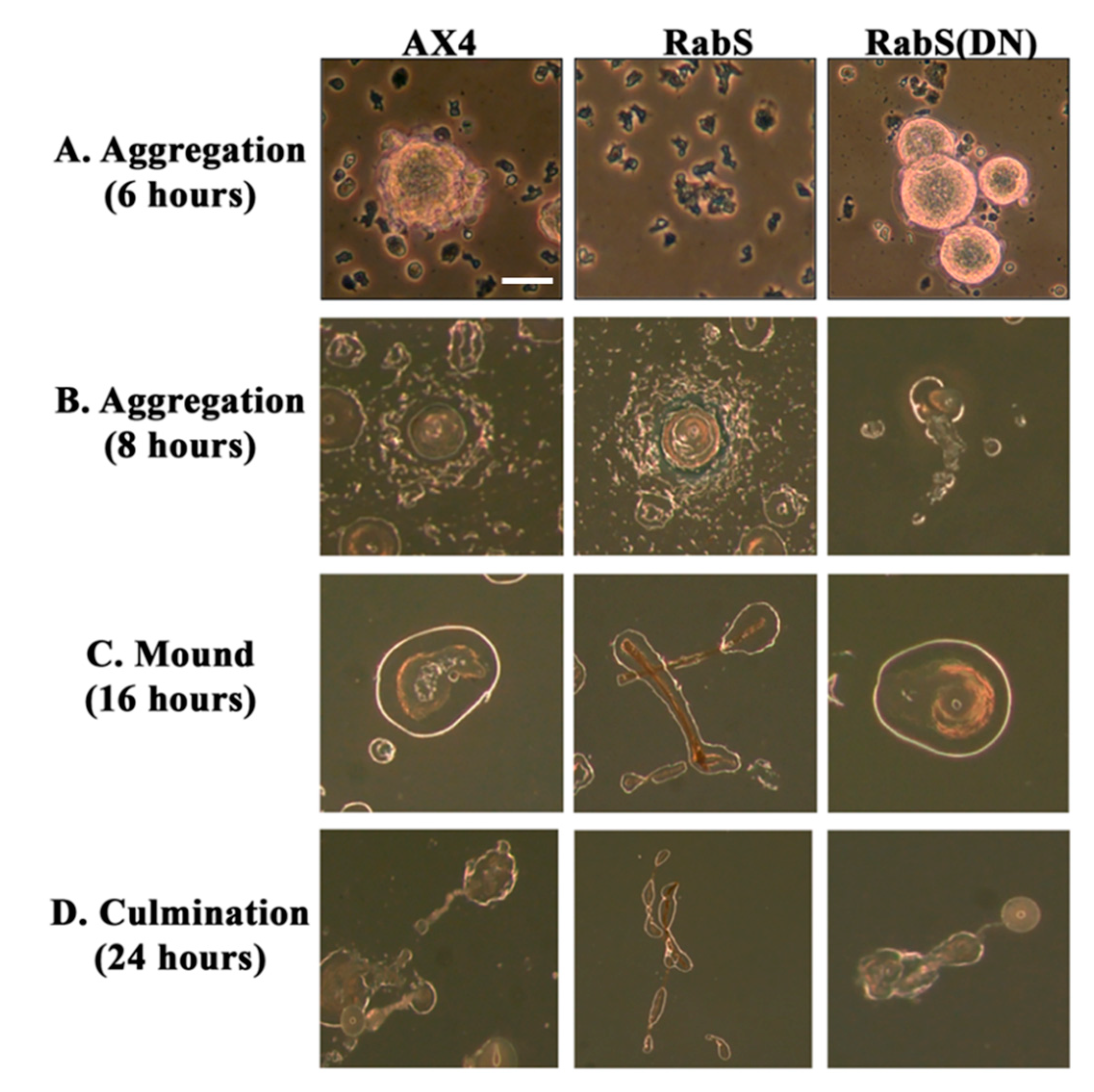
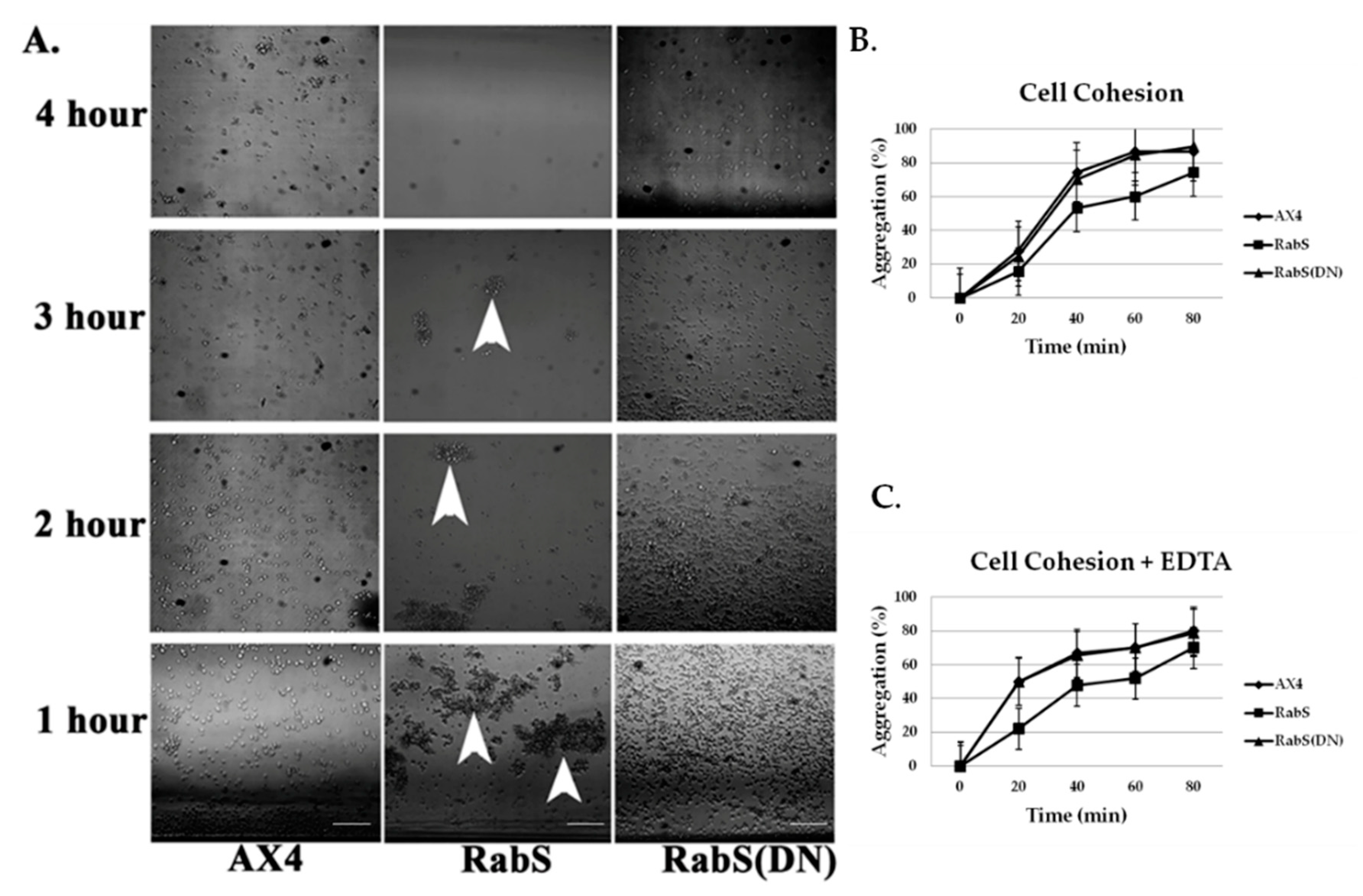
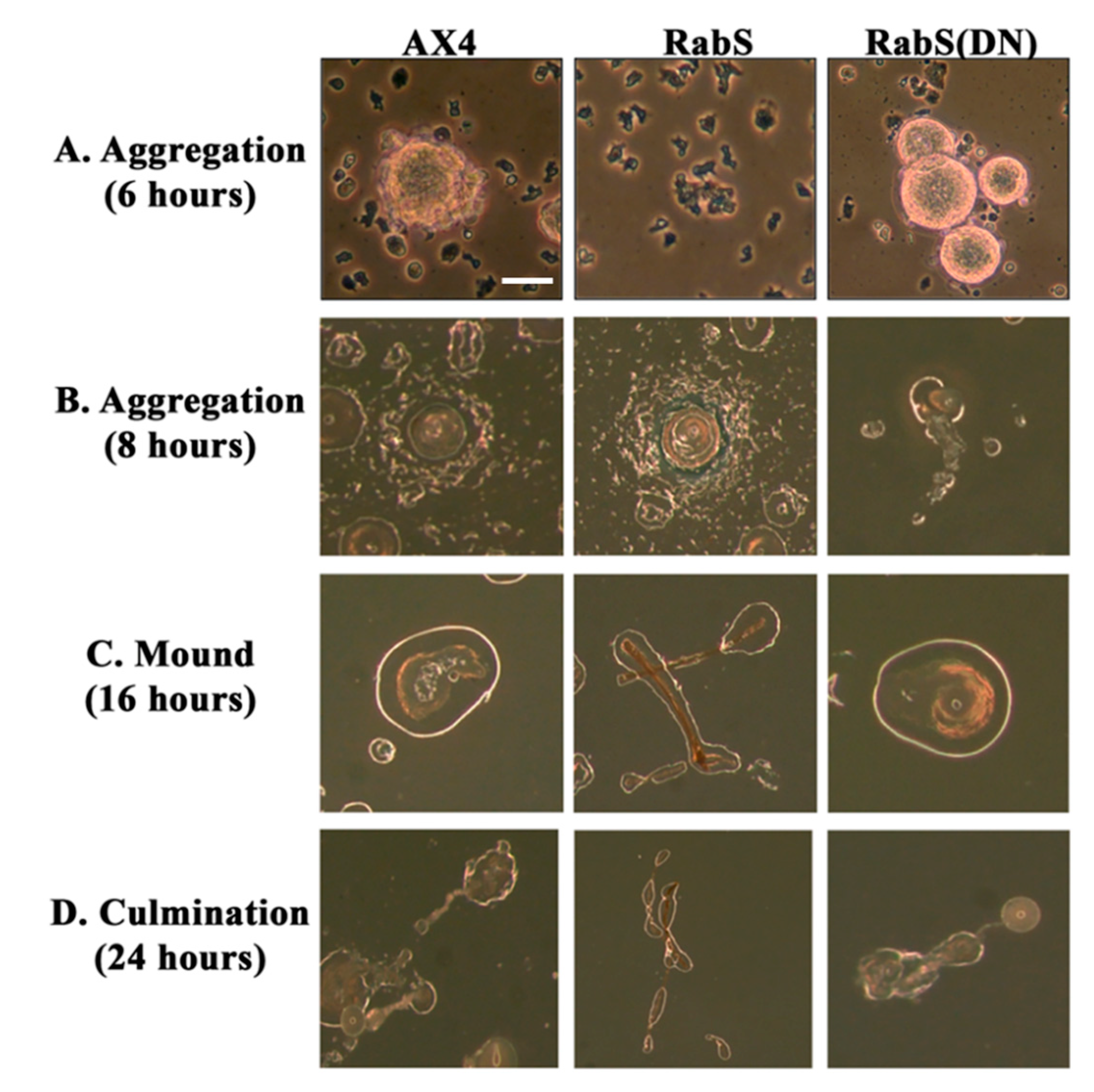
In the developmental assay, DdRabS overexpressing cells failed to complete development; the cells were unable to aggregate normally and did not develop fruiting body structures. DdRabS overexpressing cells were also less cohesive than AX4 cells at both the EDTA-sensitive and EDTA-insensitive contact sites. DdRabS overexpressing cells also displayed a decreased sensitivity and migratory response to the chemoattractant cAMP, and they showed an increased sensitivity to DIF-1 causing an increase in induced cell death. In DdRabS(DN) cells, all of these phenotypes were rescued. We propose that the overexpression of DdRabS interferes with the function of other small GTPase proteins crucial for these processes to proceed normally.
Overexpression of DdRabS caused very abnormal chemotaxis. Chemotaxis was not completely abolished as cells did migrate slightly and cluster together forming a loose type of aggregate. However, their migration towards the cAMP was minimal. This could be due to the cells being unable to detect cAMP or to the cells being unable to migrate towards the source of cAMP. Binding of cAMP causes dissociation of the heterotrimeric G protein. This in turn causes activation of small GTP-binding proteins of the Ras family [
48,
49]. It is possible that by overexpressing DdRabS, GTP-binding proteins of the Ras family are unable to be activated. As stated previously, some GDP/GTP exchange proteins (GEP), GAPs, and GDIs, which are crucial for GTPase activation, show wide substrate specificities [
49,
50,
51]. It is possible that these GTP-activating proteins are binding with DdRabS, leaving them unavailable to activate the Ras family GTPases that are crucial for the signaling cascade of chemotaxis. During our investigation, we observed a possible role for DdRabS in phagocytosis in the formation of the phagocytic cup. F-actin is essential for the formation of the phagocytic cup and is also an integral member of the actin cytoskeleton [
34]. It is possible that DdRabS may play a role in the regulation of F-actin, which would explain the phenotypes observed in both phagocytosis and motility. This is supported by the suggestion of [
34] that there is a connection between the actin cytoskeleton and membrane trafficking.
Cell-cell contact, as well as cAMP, plays a key role in cellular differentiation and gene expression in
Dictyostelium. Several studies have suggested that cell-cell contact has a major influence on the expression of many developmentally regulated genes [
21]. In cells overexpressing DdRabS, cell–cell adhesion is reduced in both EDTA-sensitive, and EDTA-insensitive contacts. Previous reports have implicated Ras family proteins as being crucial for downstream signaling to adhesion molecules DdCAD-1 and gp80 [
21,
23]; some GEP, GAP, and GDI that are critical for Ras GTPase activation show a wide substrate specificity [
49,
50,
51] and will bind to several Rab isoforms [
52]. It is possible that through overexpressing DdRabS, binding proteins required for Ras activation are instead bound to DdRabS causing Ras to remain inactive resulting in neither DdCAD-1 nor gp80 being signaled. These results indicate that overexpressing DdRabS may have an effect on both DdCAD-1 (EDTA-sensitive contacts) and gp80 (EDTA-insensitive contacts), possibly by affecting GTPase proteins in charge of signaling these adhesion molecules. This would explain the difficulty DdRabS overexpressing cells have with the aggregation process. The fact that the DN mutant does not show decreased cohesion supports this as it is locked in its GDP-bound form leaving the GEP’s available for Ras activation.
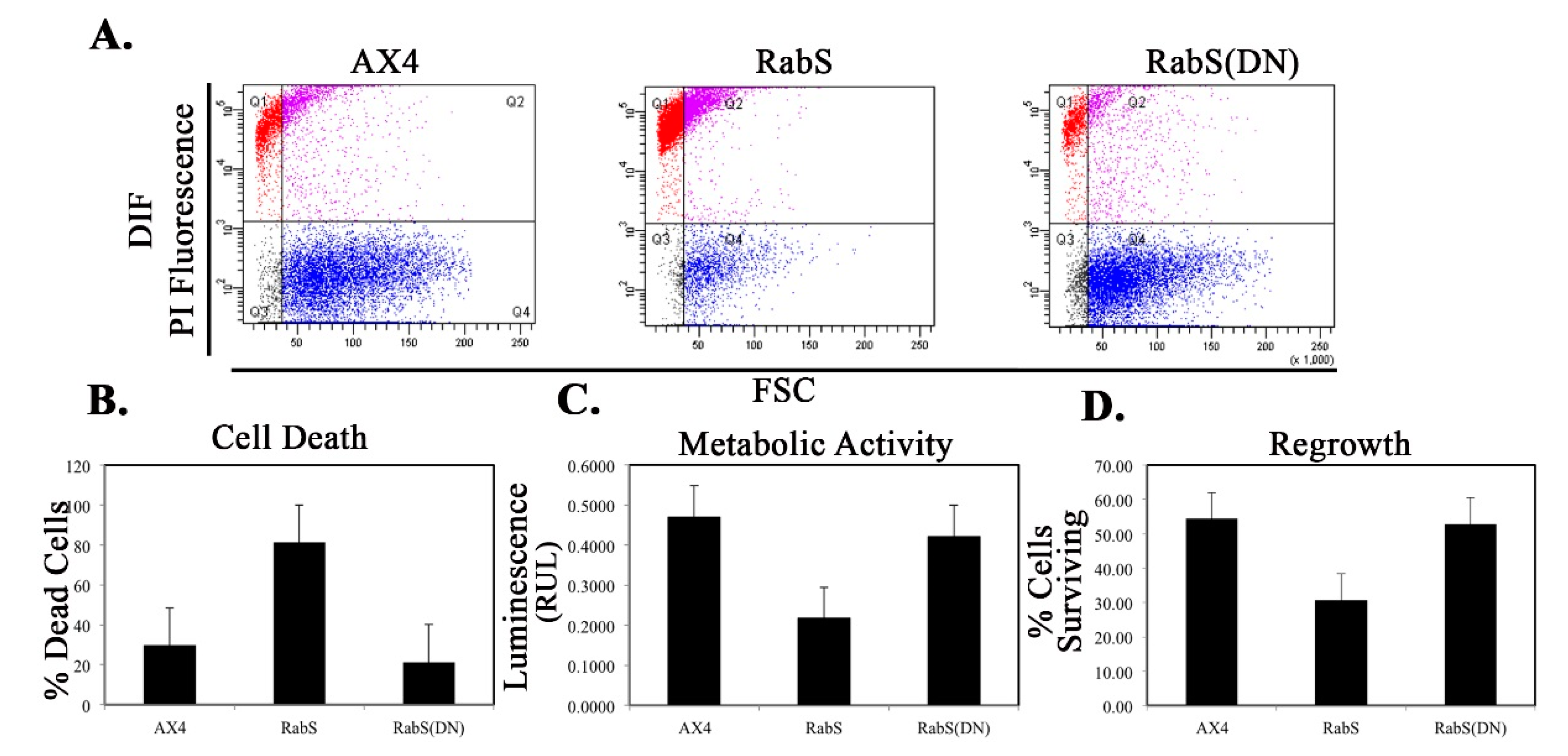
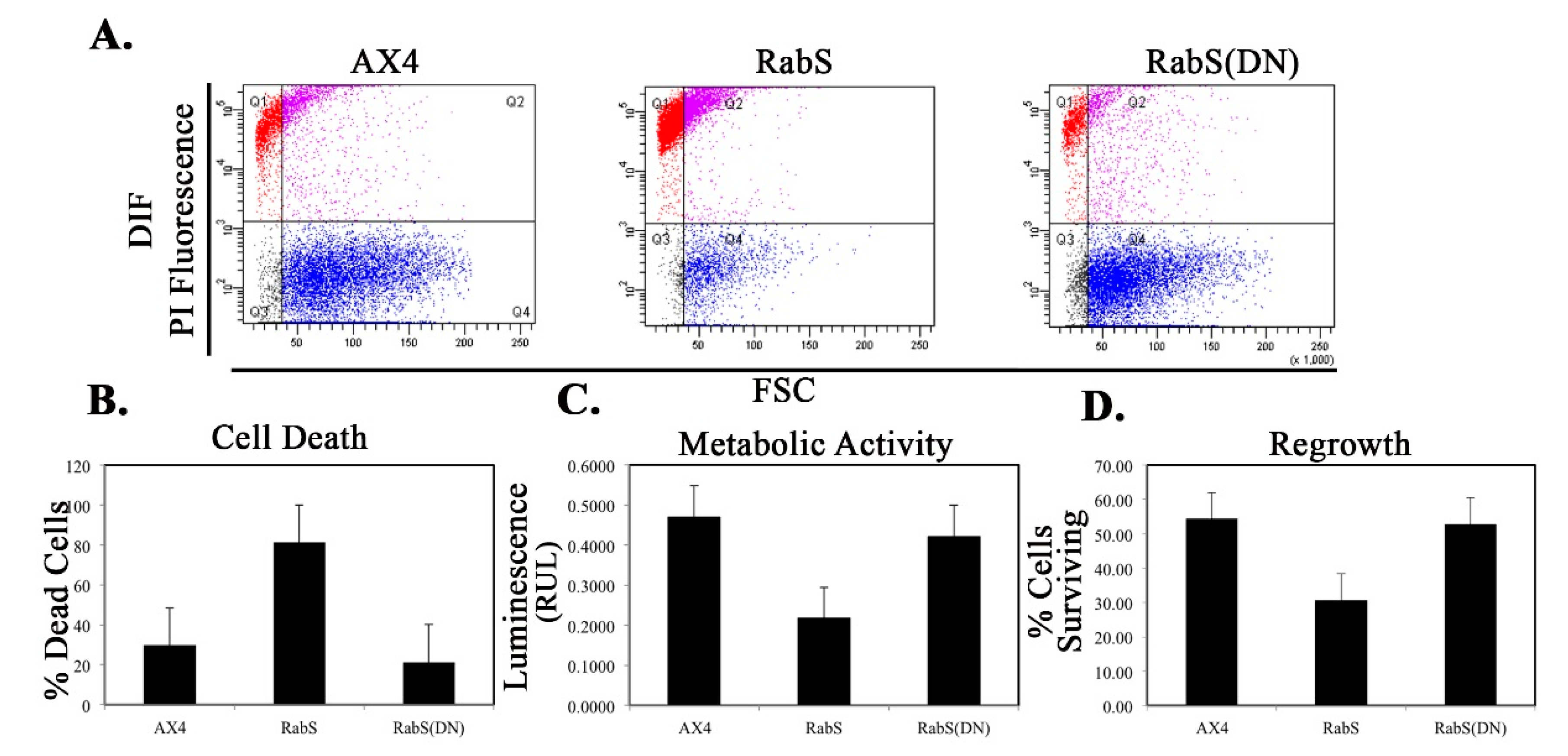
We tested the effects of DdRabS overexpression on induced cell death by incubating cells first with cAMP then DIF-1 as described above. DdRabS overexpressing cells showed a significant increase in autophagic induced cell death as compared to the controls. These cells also had decreased ATP levels and were unable to re-grow after DIF-1 treatment; this confirmed that these cells have died. Our data showed that although DdRabS overexpressing cells showed great difficulty aggregating, they did eventually form small loose aggregates that later gave rise to abnormal, elongated, small mound structures. The two types of prestalk cells, prestalk A (pstA) and prestalk B (pstB), are first detectable in the
Dictyostelium mound. pstB cells are found at the base and are sensitive to cAMP, and pstA cells are in the apex and form the tip of the stalk. In the presence of cAMP, pstB cells are more slowly induced by DIF-1 [
53]. It is possible that DdRabS overexpressing cells, which may have a decreased sensitivity to cAMP, possess a greater number of pstB cells being induced by DIF-1. The base of the stalk would then form. However, it would not actually form the stalk or spore head. This theory supports our data finding that DdRabS overexpressing cells do not form fruiting body structures and are very sensitive to induced autophagic cell death by DIF-1.

 ) DdRabS(DN). DdRabS overexpressing and DdRabS(DN) cells show abnormal secretion of all enzymes assayed. Secretion of the lysosomal enzyme β-glucosidase is increased in DdRabS overexpressing and DdRabS(DN) cell lines. Cells overexpressing the DdRabS protein had a higher rate of secretion of β-glucosidase compared to the WT AX4 cells. DdRabS overexpressing cells reached 100% secreted enzyme after 280 min whereas the AX4 cells peaked at 80% secretion at 280 min. The DdRabS(DN) cell line has increased secretion of β-glucosidase early on, but by 120 min, secretion is only slightly more than the WT AX4 cell line. Data represent the mean of three independent experiments. Data was run through a 2-way ANOVA to test for significance at p < 0.05.
) DdRabS(DN). DdRabS overexpressing and DdRabS(DN) cells show abnormal secretion of all enzymes assayed. Secretion of the lysosomal enzyme β-glucosidase is increased in DdRabS overexpressing and DdRabS(DN) cell lines. Cells overexpressing the DdRabS protein had a higher rate of secretion of β-glucosidase compared to the WT AX4 cells. DdRabS overexpressing cells reached 100% secreted enzyme after 280 min whereas the AX4 cells peaked at 80% secretion at 280 min. The DdRabS(DN) cell line has increased secretion of β-glucosidase early on, but by 120 min, secretion is only slightly more than the WT AX4 cell line. Data represent the mean of three independent experiments. Data was run through a 2-way ANOVA to test for significance at p < 0.05.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}