Inpactor, Integrated and Parallel Analyzer and Classifier of LTR Retrotransposons and Its Application for Pineapple LTR Retrotransposons Diversity and Dynamics

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
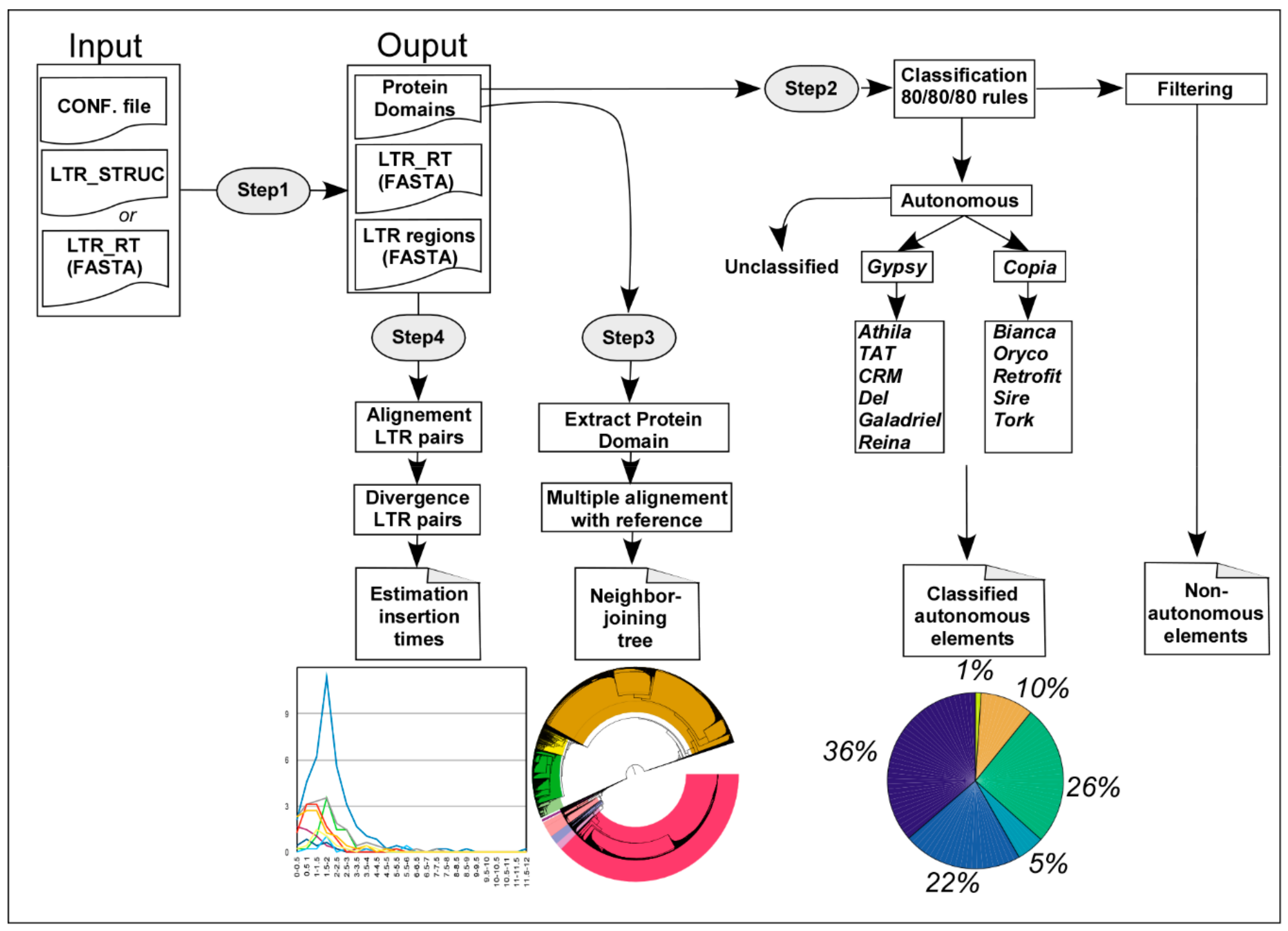
2.1. Implementation of Inpactor
2.2. Availability of Inpactor
2.3. Computational Resources
2.4. Sequence Data Sources
2.5. Identification of Repeated Elements
2.6. Annotation, Phylogenetic Analysis and Insertion Time Analysis of LTR Retrotransposons
3. Results and Discussion
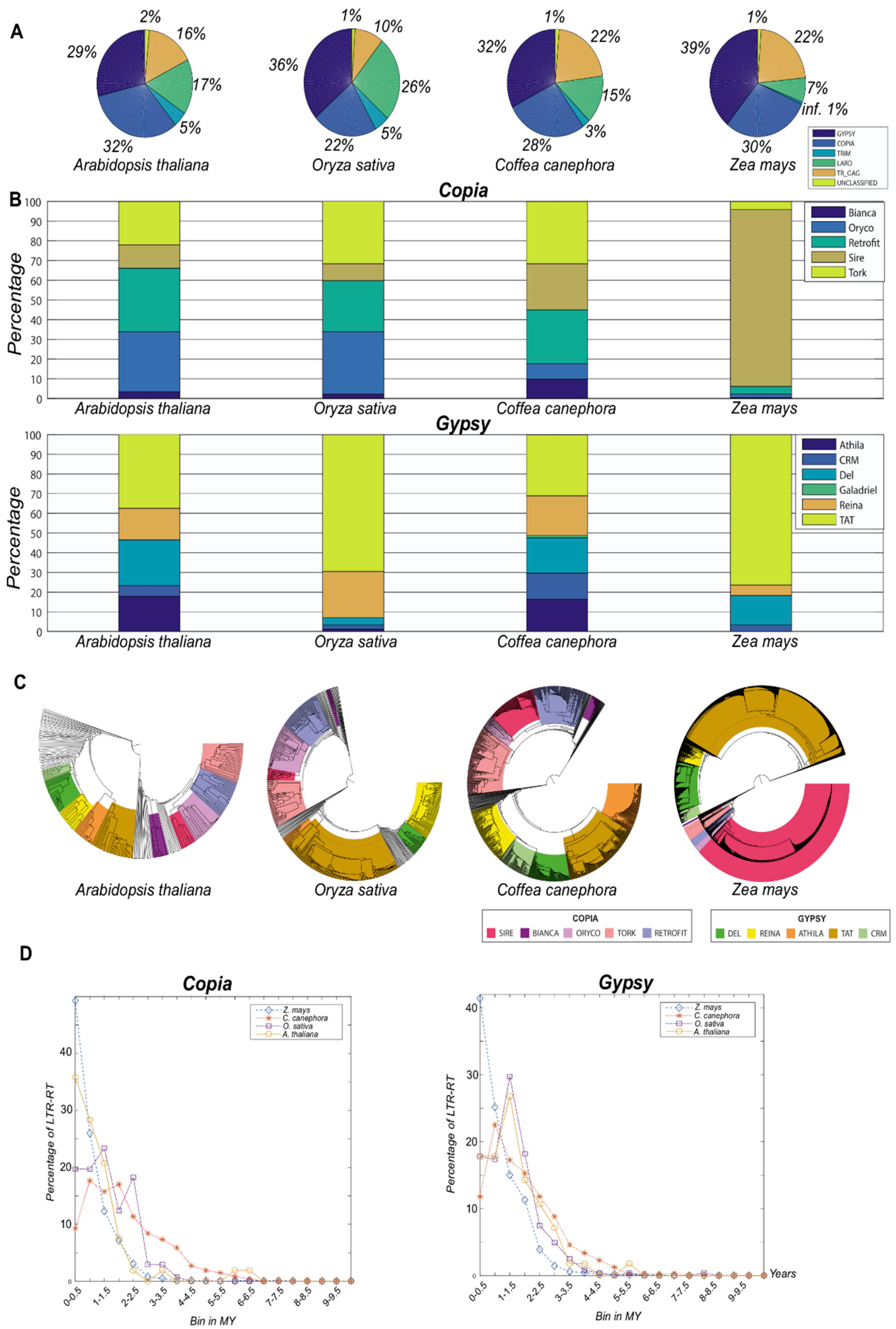
3.1. Testing Inpactor on Reference Plant Genomes
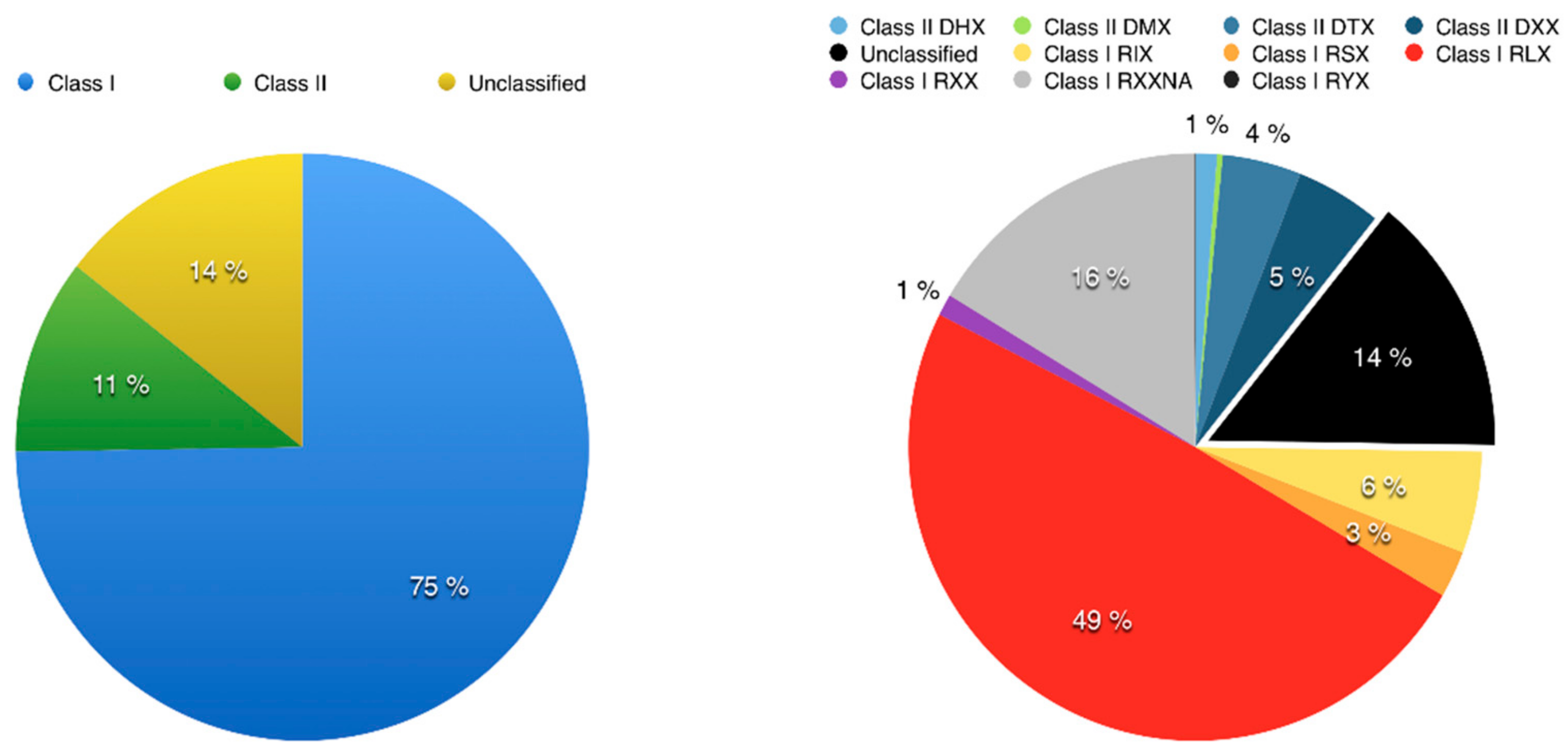
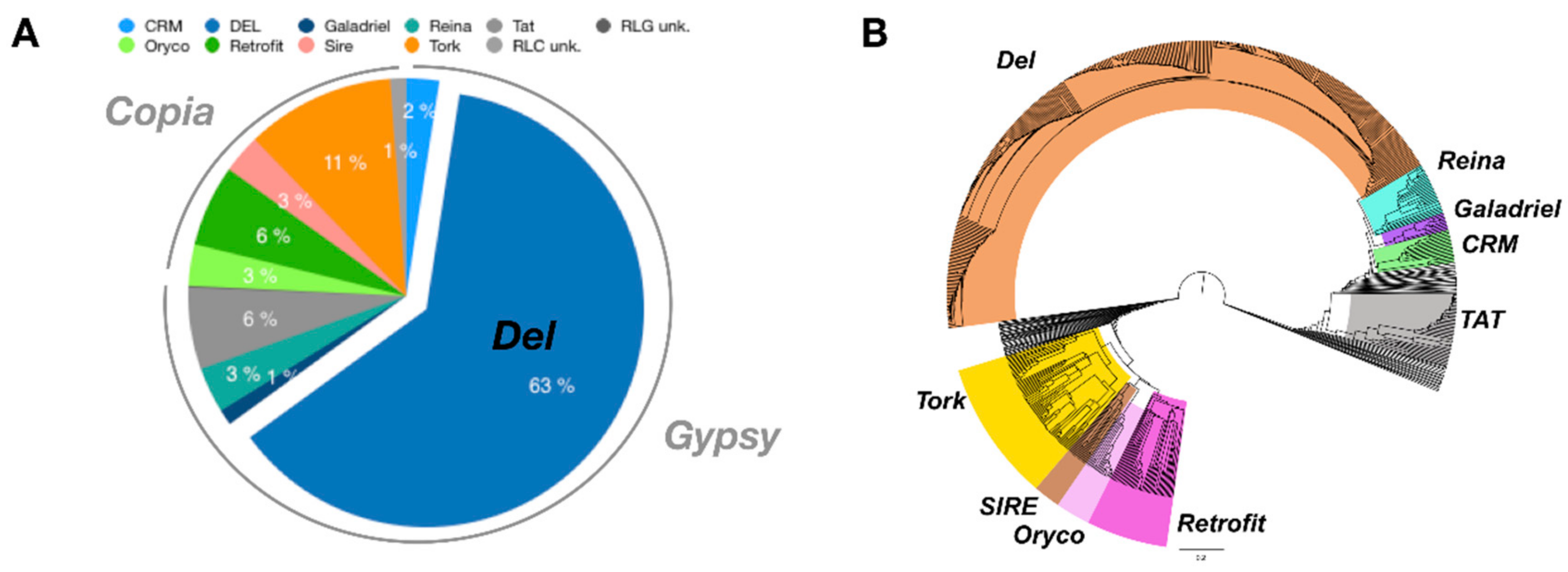
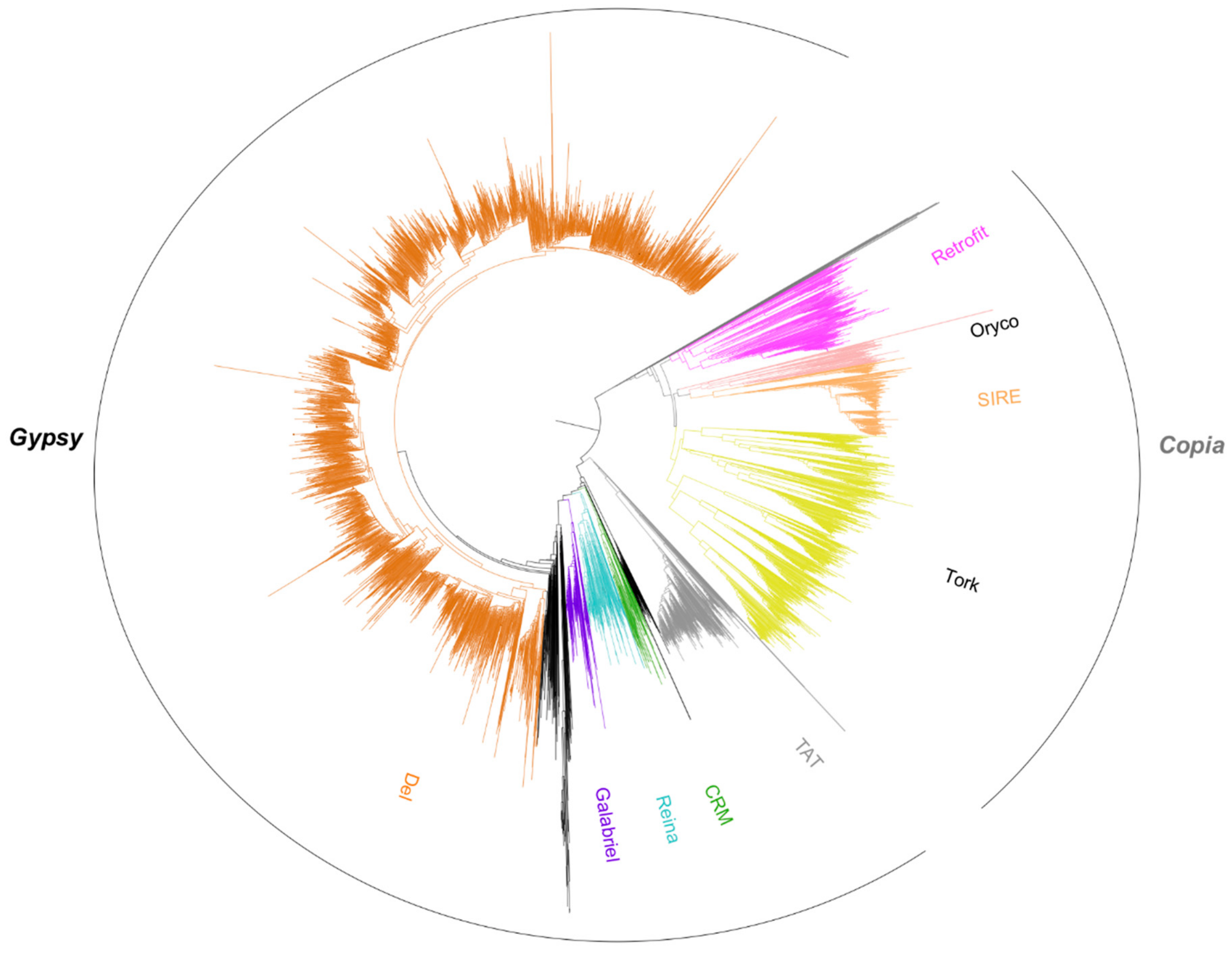
3.2. Using Inpactor on the Pineapple Genome
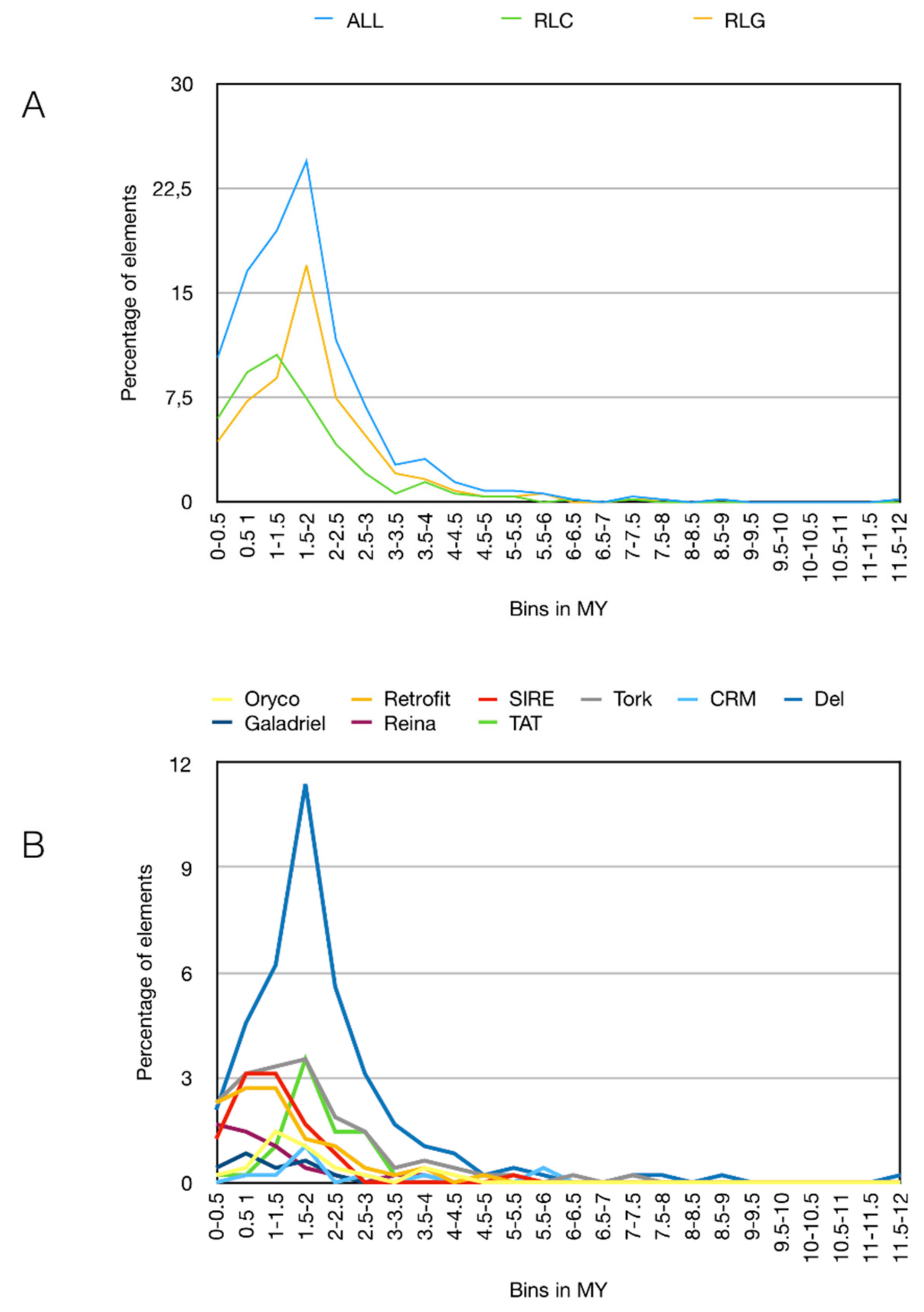
3.3. Pineapple LTR Retrotransposons Abundance and Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Choulet, F.; Alberti, A.; Theil, S.; Glover, N.; Barbe, V.; Daron, J.; Pingault, L.; Sourdille, P.; Couloux, A.; Paux, E.; et al. Structural and functional partitioning of bread wheat chromosome 3B. Science 2014, 345, 1249721. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Laclette, E.; Lyons, E. Architecture and evolution of a minute plant genome. Nature 2013, 498, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, M.I.; Hollister, J.D.; Gaut, B.S. A triptych of the evolution of plant transposable elements. Trends Plant Sci. 2010, 15, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Saniyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Makarevitch, I.; Waters, A.J.; West, P.T.; Stitzer, M.; Hirsch, C.N.; Ross-Ibarra, J.; Springer, N.M. Transposable Elements Contribute to Activation of Maize Genes in Response to Abiotic Stress. PLoS Genet. 2015, 11, e1004915. [Google Scholar]
- Todorovska, E. Retrotransposons and their Role in Plant—Genome Evolution Retrotransposons and Their Role in Plant—Genome. Biotechnol. Biotechnol. Equip. 2017, 2818, 294–305. [Google Scholar]
- Casacuberta, E.; González, J. The impact of transposable elements in environmental adaptation. Mol. Ecol. 2013, 22, 1503–1517. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Paux, E.; Roger, D.; Badaeva, E.; Gay, G.; Bernard, M.; Sourdille, P.; Feuillet, C. Characterizing the composition and evolution of homoeologous genomes in hexaploid wheat through BAC-end sequencing on chromosome 3B. Plant J. 2006, 48, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Denoeud, F.; Carretero-Paulet, L.; Dereeper, A.; Droc, G.; Guyot, R.; Pietrella, M.; Zheng, C.; Alberti, A.; Anthony, F.; Aprea, G.; et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 2014, 345, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Llorens, C.; Muñoz-Pomer, A.; Bernad, L.; Botella, H.; Moya, A. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol. Direct 2009, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Keller, B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007, 17, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Llorens, C.; Futami, R.; Covelli, L.; Domínguez-Escribá, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodríguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The Gypsy Database (GyDB) of Mobile Genetic Elements: Release 2.0. Nucleic Acids Res. 2011, 39, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Witte, C.-P.; Le, Q.H.; Bureau, T.; Kumar, A. Terminal-repeat retrotransposons in miniature (TRIM) are involved in restructuring plant genomes. Proc. Natl. Acad. Sci. USA 2001, 98, 13778–13783. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Vicient, C.M.; Peleg, O.; Anamthawat-Jonsson, K.; Bolshoy, A.; Schulman, A.H. Large retrotransposon derivatives: Abundant, conserved but nonautonomous retroelements of barley and related genomes. Genetics 2004, 166, 1437–1450. [Google Scholar] [CrossRef] [PubMed]
- Tanskanen, J.A.; Sabot, F.; Vicient, C.; Schulman, A.H. Life without GAG: The BARE-2 retrotransposon as a parasite’s parasite. Gene 2007, 390, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Chaparro, C.; Gayraud, T.; de Souza, R.F.; Domingues, D.S.; Akaffou, S.; Vanzela, A.L.L.; de Kochko, A.; Rigoreau, M.; Crouzillat, D.; Hamon, S.; et al. Terminal-repeat retrotransposons with gAG domain in plant genomes: A new testimony on the complex world of transposable elements. Genome Biol. Evol. 2015, 7, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Bergman, C.M.; Quesneville, H. Discovering and detecting transposable elements in genome sequences. Brief. Bioinform. 2007, 8, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Lerat, E. Identifying repeats and transposable elements in sequenced genomes: How to find your way through the dense forest of programs. Heredity 2010, 104, 520–533. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.; Scossa, F.; Bolger, M.E.; Lanz, C.; Maumus, F.; Tohge, T.; Quesneville, H.; Alseekh, S.; Sørensen, I.; Lichtenstein, G.; et al. The genome of the stress-tolerant wild tomato species Solanum pennellii. Nat. Genet. 2014, 46, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Slotte, T.; Hazzouri, K.M.; Ågren, J.A.; Koenig, D.; Maumus, F.; Guo, Y.-L.; Steige, K.; Platts, A.E.; Escobar, J.S.; Newman, L.K.; et al. The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat. Genet. 2013, 45, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Abrusán, G.; Grundmann, N.; Demester, L.; Makalowski, W. Teclass—A tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 2009, 25, 1329–1330. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Keswani, U.; Ranganathan, N.; Guibotsy, M.L.; Levine, D. Exploring repetitive DNA landscapes using REPCLASS, a tool that automates the classification of transposable elements in eukaryotic genomes. Genome Biol. Evol. 2009, 1, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Hoede, C.; Arnoux, S.; Moisset, M.; Chaumier, T.; Inizan, O.; Jamilloux, V.; Quesneville, H. PASTEC: An automatic transposable element classification tool. PLoS ONE 2014, 9, e91929. [Google Scholar] [CrossRef] [PubMed]
- Steinbiss, S.; Kastens, S.; Kurtz, S. LTRsift: A graphical user interface for semi-automatic classification and postprocessing of de novo detected LTR retrotransposons. Mob. DNA 2012, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Monat, C.; Tando, N.; Tranchant-Dubreuil, C.; Sabot, F. LTRclassifier: A website for fast structural LTR retrotransposons classification in plants. Mob. Genet. Elem. 2016, 6, e1241050. [Google Scholar] [CrossRef] [PubMed]
- Orozco, S.; Jeferson, A. Aplicación de la inteligencia artificial en la bioinformática, avances, definiciones y herramientas. UGCiencia 2016, 22, 159–171. [Google Scholar] [CrossRef]
- Arango-López, J.; Orozco-Arias, S.; Salazar, J.A.; Guyot, R. Application of Data Mining Algorithms to Classify Biological Data: The Coffea canephora Genome Case. Adv. Comput. 2017, 735, 156–170. [Google Scholar]
- Maizel, J.R. Supercomputing in molecular biology: Applications to sequence analysis. IEEE Eng. Med. Biol. Mag. Q. Mag. Eng. Med. Biol. Soc. 1988, 7, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Arias, S.; Tabares-Soto, R.; Ceballos, D.; Guyot, R. Parallel Programming in Biological Sciences, Taking Advantage of Supercomputing in Genomics. Adv. Comput. 2017, 735, 627–643. [Google Scholar]
- Gropp, W.; Lusk, E.; Doss, N.; Skjellum, A. A high-performance, portable implementation of the MPI message passing interface standard. Parallel Comput. 1996, 22, 789–828. [Google Scholar] [CrossRef]
- Tabares Soto, R. Programación Paralela Sobre Arquitecturas Heterogéneas. Master’s Thesis, Universidad Nacional de Colombia, Manizales, Colombia, 2016. [Google Scholar]
- Castro, J.L.A.; Leiss, E. Introducción a la Computación Paralela; Editorial Venezolana, Universidad de Los Andes: Mérida, Venezuela, 2004; ISBN 980-12-0752-3. [Google Scholar]
- Zhang, J.; Liu, J.; Ming, R. Genomic analyses of the CAM plant pineapple. J. Exp. Bot. 2014, 65, 3395–3404. [Google Scholar] [CrossRef] [PubMed]
- Carlier, J.D.; Sousa, N.H.; Santo, T.E.; d’Eeckenbrugge, G.C.; Leitão, J.M. A genetic map of pineapple (Ananas comosus (L.) Merr.) including SCAR, CAPS, SSR and EST-SSR markers. Mol. Breed. 2012, 29, 245–260. [Google Scholar] [CrossRef]
- Ong, W.D.; Voo, C.L.Y.; Kumar, S.V. Development of ESTs and data mining of pineapple EST-SSRs. Mol. Biol. Rep. 2012, 39, 5889–5896. [Google Scholar] [CrossRef] [PubMed]
- Thomson, K.G.; Thomas, J.E.; Dietzgen, R.G. Retrotransposon-like sequences integrated into the genome of pineapple, Ananas comosus. Plant Mol. Biol. 1998, 38, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Ming, R.; VanBuren, R.; Wai, C.M.; Tang, H.; Schatz, M.C.; Bowers, J.E.; Lyons, E.; Wang, M.-L.; Chen, J.; Biggers, E.; et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 2015, 47, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.M.; McDonald, J.F. LTR STRUC: A novel search and identification program for LTR retrotransposons. Bioinformatics 2003, 19, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Jurka, J.; Klonowski, P.; Dagman, V.; Pelton, P. CENSOR—A program for identification and elimination of repetitive elements from DNA sequences. Comput. Chem. 1996, 20, 119–121. [Google Scholar] [CrossRef]
- Birney, E.; Durbin, R. Using GeneWise in the. Genome Res. 2000, 10, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- SanMiguel, P.; Gaut, B.S.; Tikhonov, A.; Nakajima, Y.; Bennetzen, J.L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 1998, 20, 43. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Bennetzen, J.L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 12404–12410. [Google Scholar] [CrossRef] [PubMed]
- Jette, M.; Grondona, M. SLURM: Simple Linux Utility for Resource Management. In Workshop on Job Scheduling Strategies for Parallel Processing; Springer: Berlin/Heidelberg, Germany, 2003; pp. 44–60. [Google Scholar]
- Furlani, J.L.; Osel, P.W. Abstract Yourself with Modules. In Proceedings of the 10th USENIX Conference on System Administrationm, Chicago, IL, USA, 29 September–4 October 1996; pp. 193–204. [Google Scholar]
- Tello-ruiz, M.K.; Stein, J.; Wei, S.; Preece, J.; Olson, A.; Naithani, S.; Amarasinghe, V.; Dharmawardhana, P.; Jiao, Y.; Mulvaney, J.; et al. Gramene 2016: Comparative plant genomics and pathway resources. Nucleic Acids Res. 2017, 44, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Bocs, S.; Rouard, M.; Guignon, V.; Ravel, S.; Tranchant-Dubreuil, C.; Poncet, V.; Garsmeur, O.; Lashermes, P.; Droc, G. The coffee genome hub: A resource for coffee genomes. Nucleic Acids Res. 2015, 43, D1028–D1035. [Google Scholar] [CrossRef] [PubMed]
- Duprat, E.; Feuillet, C.; Quesneville, H. Considering Transposable Element Diversification in De Novo Annotation Approaches. Genome Res. 2011, 6, e16526. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 23 May 2018).
- Du, J.; Tian, Z.; Hans, C.S.; Laten, H.M.; Cannon, S.B.; Jackson, S.A.; Shoemaker, R.C.; Ma, J. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: Insights from genome-wide analysis and multi-specific comparison. Plant J. 2010, 63, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Dupeyron, M.; de Souza, R.F.; Hamon, P.; de Kochko, A.; Crouzillat, D.; Couturon, E.; Domingues, D.S.; Guyot, R. Distribution of Divo in Coffea genomes, a poorly described family of angiosperm LTR-Retrotransposons. Mol. Genet. Genom. 2017, 292, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. LTR-FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal-repeat retrotransposons. Plant Physiol. 2017, 176, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef] [PubMed]
- Marco, A.; Marín, I. How Athila retrotransposons survive in the Arabidopsis genome. BMC Genom. 2008, 9, 219. [Google Scholar] [CrossRef] [PubMed]
- Pélissier, T.; Tutois, S.; Deragon, J.M.; Tourmente, S.; Genestier, S.; Picard, G. Athila, a new retroelement from Arabidopsis thaliana. Plant Mol. Biol. 1995, 29, 441–452. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Total Average Sequential Runtime in Seconds | Sequential Standard Deviation in Seconds | Total Average Parallel Runtime in Seconds | Parallel Standard Deviation in Seconds | Number of Cores | Speed-Up |
|---|---|---|---|---|---|---|
| Arabidopsis thaliana | 995.3 | 14.42 | 361.28 | 5.82 | 4 | 2.8 |
| 201.16 | 12.65 | 8 | 4.9 | |||
| 134.5 | 9.24 | 16 | 7.4 | |||
| 158.47 | 11.28 | 32 | 6.3 | |||
| Oryza sativa | 3228.7 | 94.07 | 1099.67 | 42.48 | 4 | 2.9 |
| 677.25 | 41.65 | 8 | 4.8 | |||
| 428.02 | 24.64 | 16 | 7.5 | |||
| 412.75 | 18.61 | 32 | 7.8 | |||
| Coffea canephora | 9569.48 | 11.91 | 3292.15 | 155.64 | 4 | 2.9 |
| 2029.39 | 108.92 | 8 | 4.7 | |||
| 1143.97 | 23.64 | 16 | 8.4 | |||
| 1015.44 | 31.71 | 32 | 9.4 | |||
| Zea mays | 65,031.07 | 1143.79 | 22,186.47 | 306.43 | 4 | 2.9 |
| 11,657.74 | 582.24 | 8 | 5.6 | |||
| 8452.74 | 394.94 | 16 | 7.7 | |||
| 7907.58 | 495.85 | 32 | 8.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orozco-Arias, S.; Liu, J.; Tabares-Soto, R.; Ceballos, D.; Silva Domingues, D.; Garavito, A.; Ming, R.; Guyot, R. Inpactor, Integrated and Parallel Analyzer and Classifier of LTR Retrotransposons and Its Application for Pineapple LTR Retrotransposons Diversity and Dynamics. Biology 2018, 7, 32. https://doi.org/10.3390/biology7020032
Orozco-Arias S, Liu J, Tabares-Soto R, Ceballos D, Silva Domingues D, Garavito A, Ming R, Guyot R. Inpactor, Integrated and Parallel Analyzer and Classifier of LTR Retrotransposons and Its Application for Pineapple LTR Retrotransposons Diversity and Dynamics. Biology. 2018; 7(2):32. https://doi.org/10.3390/biology7020032
Chicago/Turabian StyleOrozco-Arias, Simon, Juan Liu, Reinel Tabares-Soto, Diego Ceballos, Douglas Silva Domingues, Andréa Garavito, Ray Ming, and Romain Guyot. 2018. "Inpactor, Integrated and Parallel Analyzer and Classifier of LTR Retrotransposons and Its Application for Pineapple LTR Retrotransposons Diversity and Dynamics" Biology 7, no. 2: 32. https://doi.org/10.3390/biology7020032
APA StyleOrozco-Arias, S., Liu, J., Tabares-Soto, R., Ceballos, D., Silva Domingues, D., Garavito, A., Ming, R., & Guyot, R. (2018). Inpactor, Integrated and Parallel Analyzer and Classifier of LTR Retrotransposons and Its Application for Pineapple LTR Retrotransposons Diversity and Dynamics. Biology, 7(2), 32. https://doi.org/10.3390/biology7020032