
The Enzymology of 2-Hydroxyglutarate, 2-Hydroxyglutaramate and 2-Hydroxysuccinamate and Their Relationship to Oncometabolites


Abstract
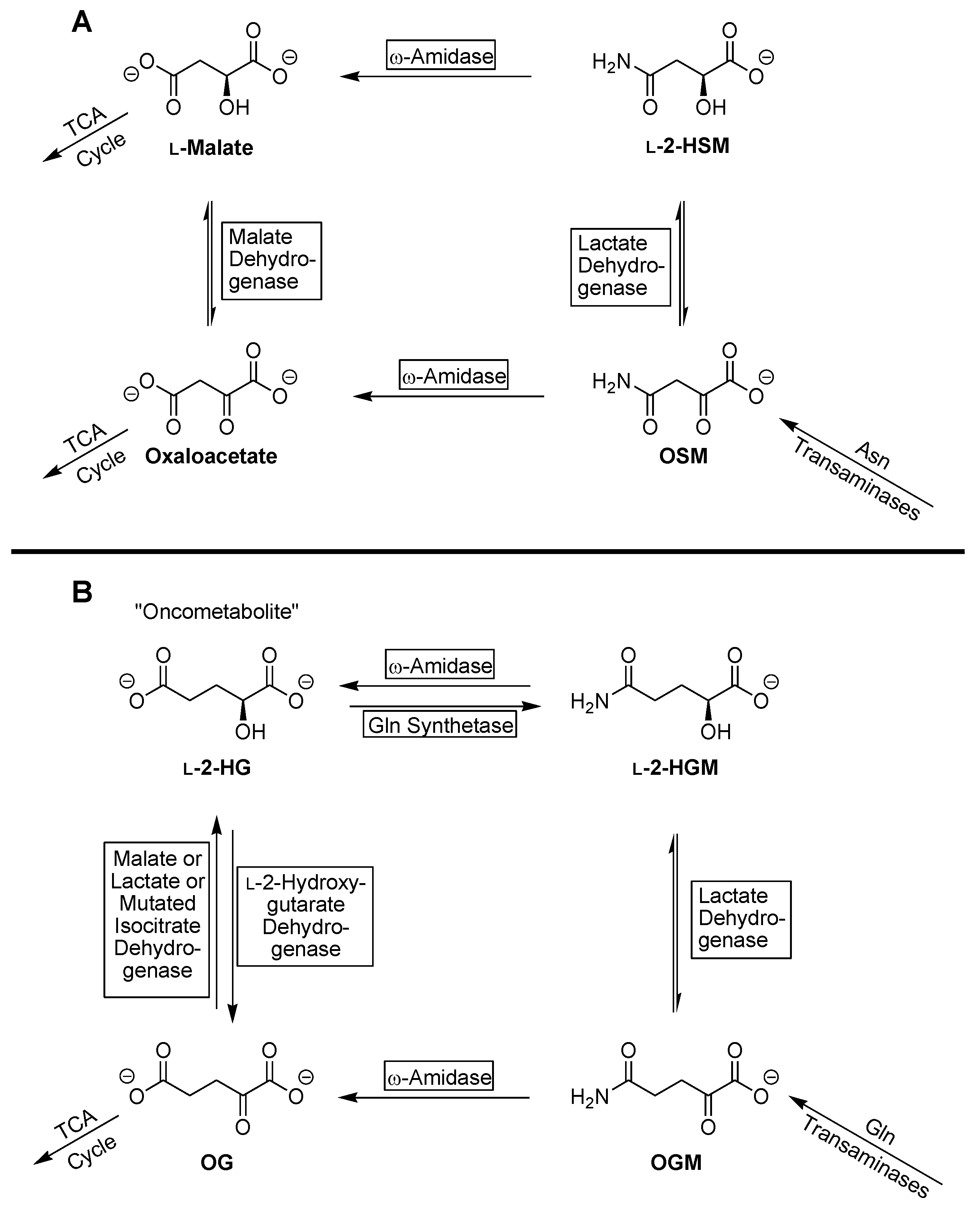
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Enzymes
2.3. Preparation of Rat Liver Cytosol
2.4. Enzyme Assays
2.4.1. ω-Amidase
2.4.2. Glutamine Synthetase (GS)
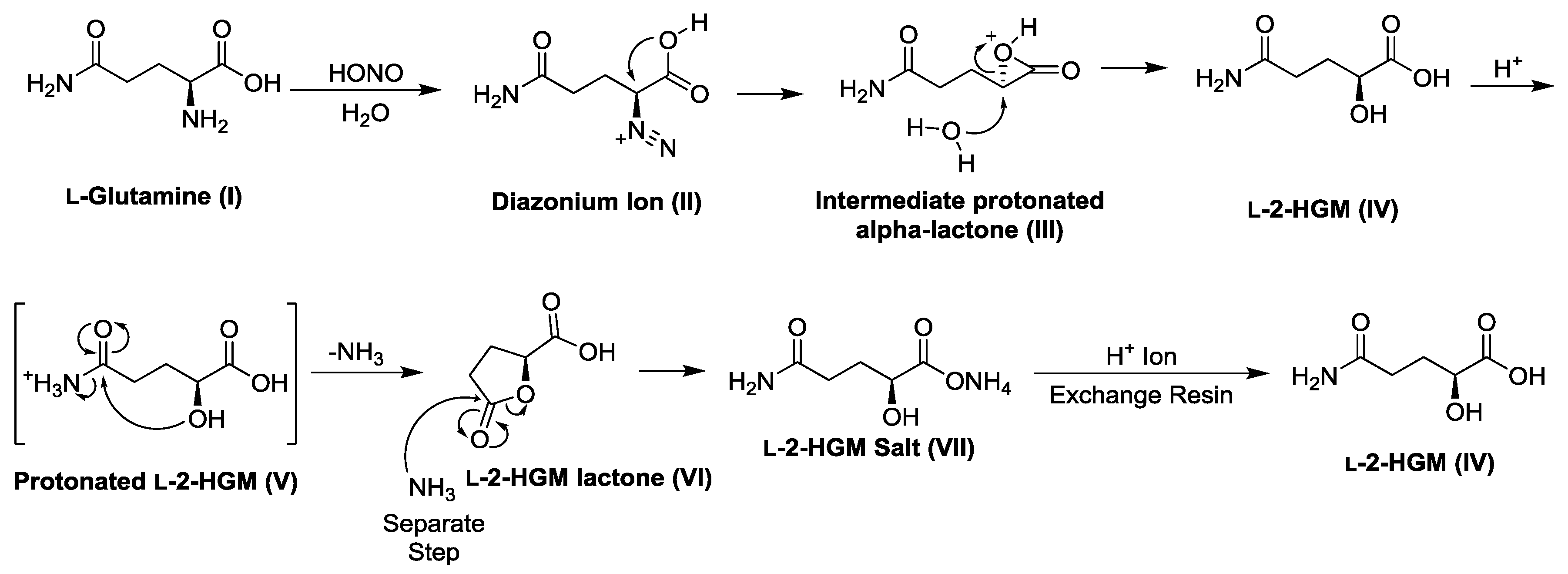
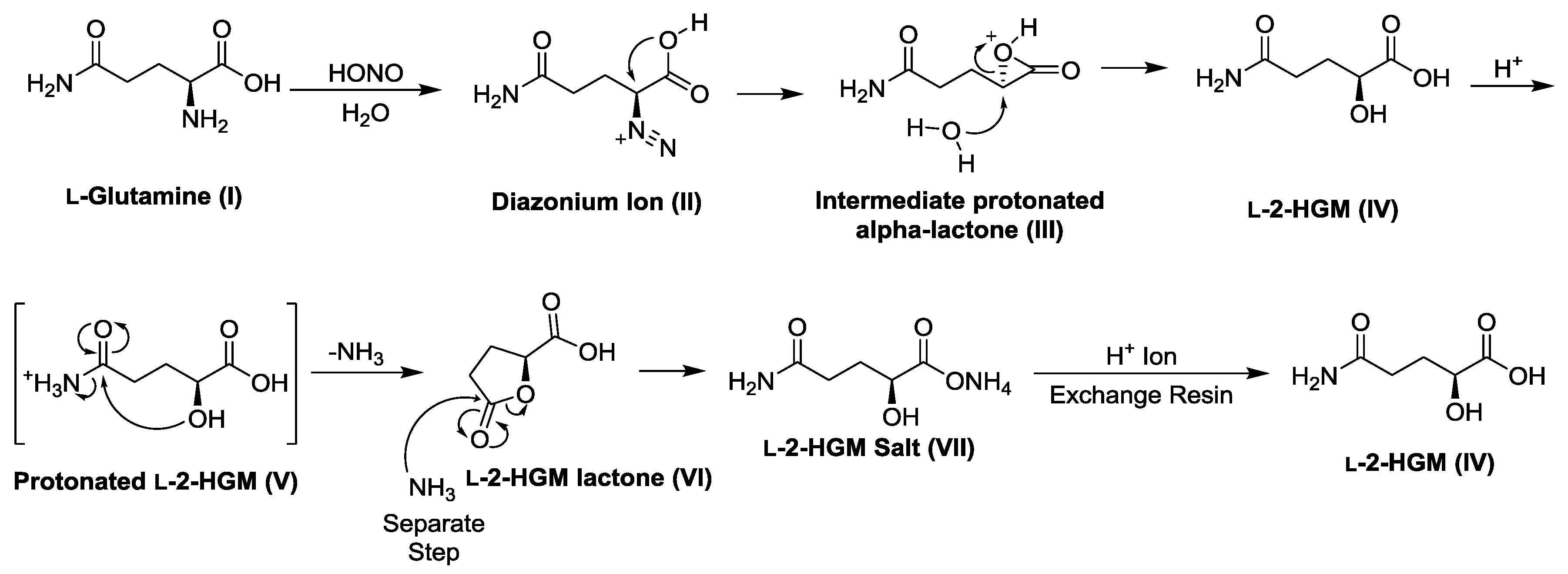
2.5. Preparation of l-2-Hydroxyglutaramic Acid (l-2-HGM; S-5-Amino-2-Hydroxy-5-Oxopentanoic Acid)
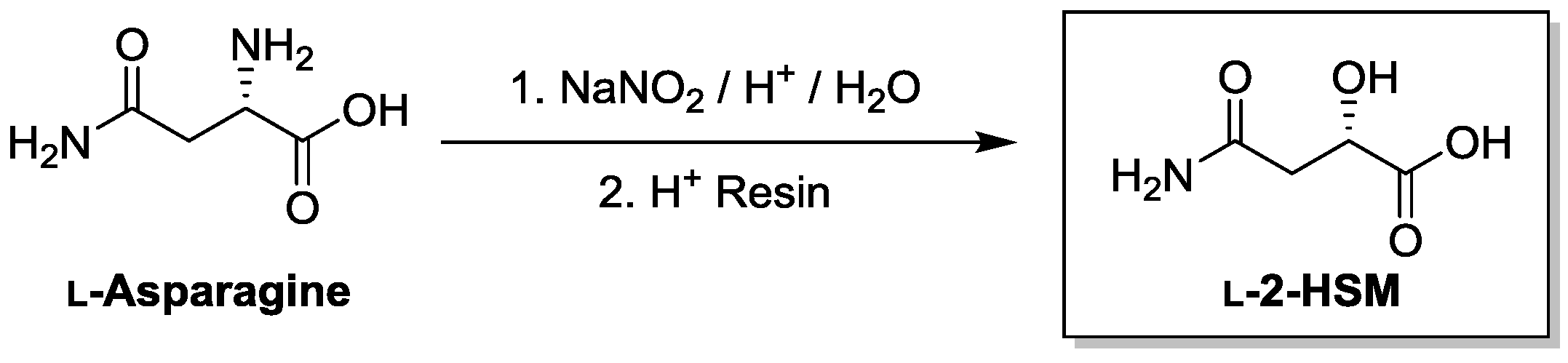
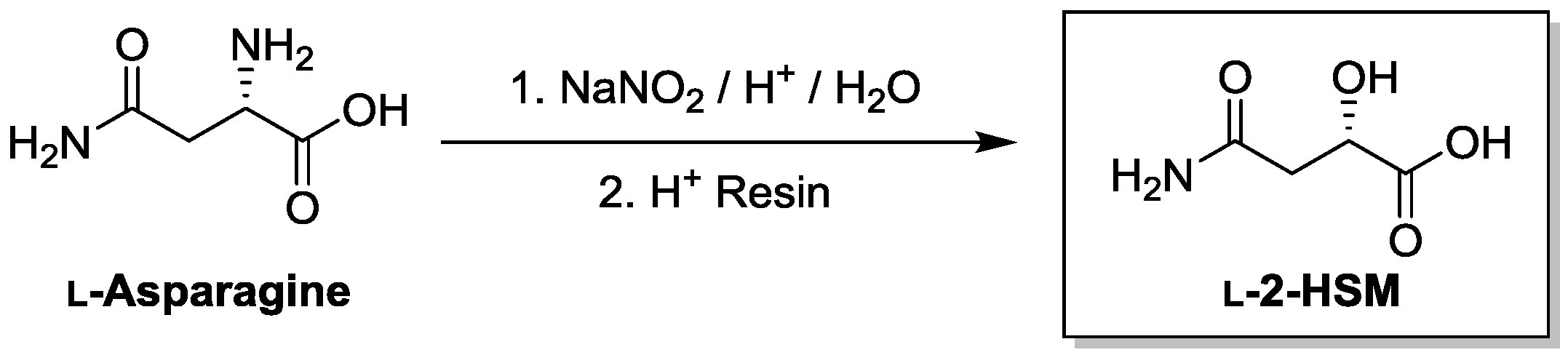
2.6. Preparation of l-2-Hydroxysuccinamic Acid (l-2-HSM; S-4-Amino-2-Hydroxy-4-Oxobutanoic Acid)
2.7. Preparation of the l- and d-Enantiomers of 2-Hydroxyglutaric Acid
2.8. Spectrophotometry
2.9. Statistics
3. Results
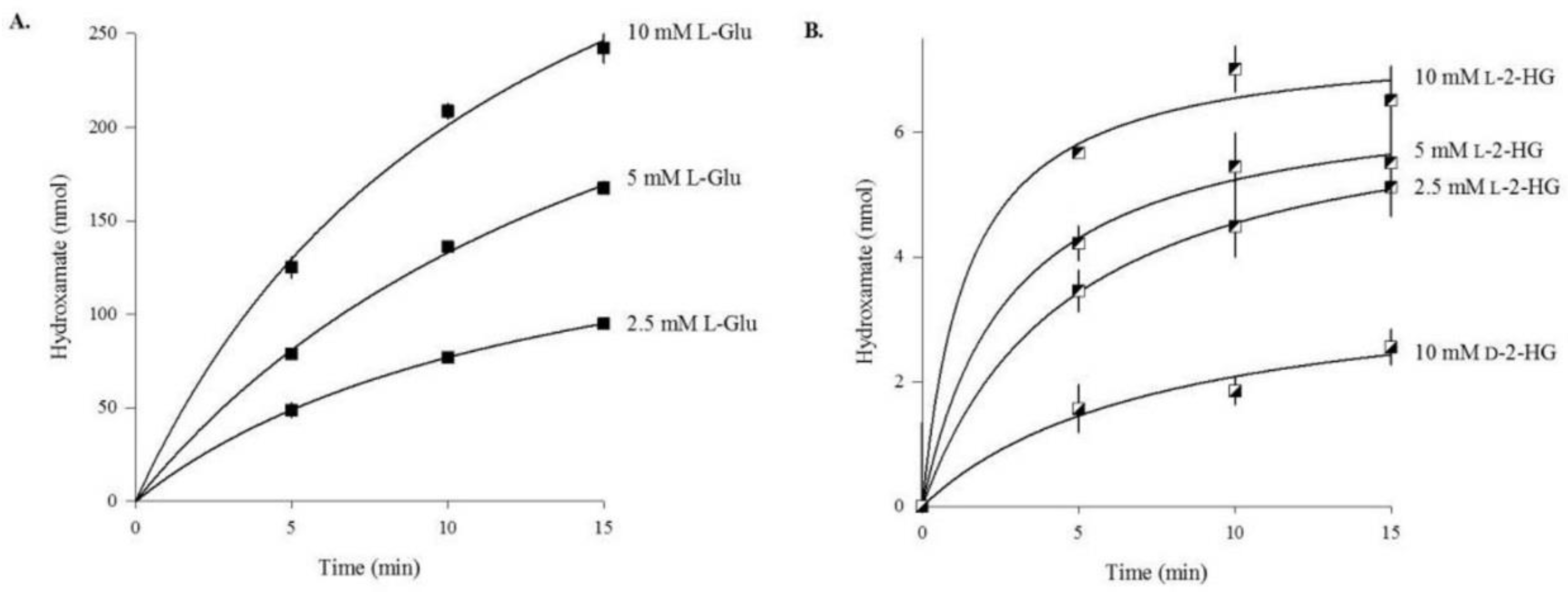
3.1. Human ω-Amidase Utilizes l-2-Hydroxyglutaramate (l-2-HGM) and l-2-Hydroxysuccinamate (l-2-HSM) as Substrates
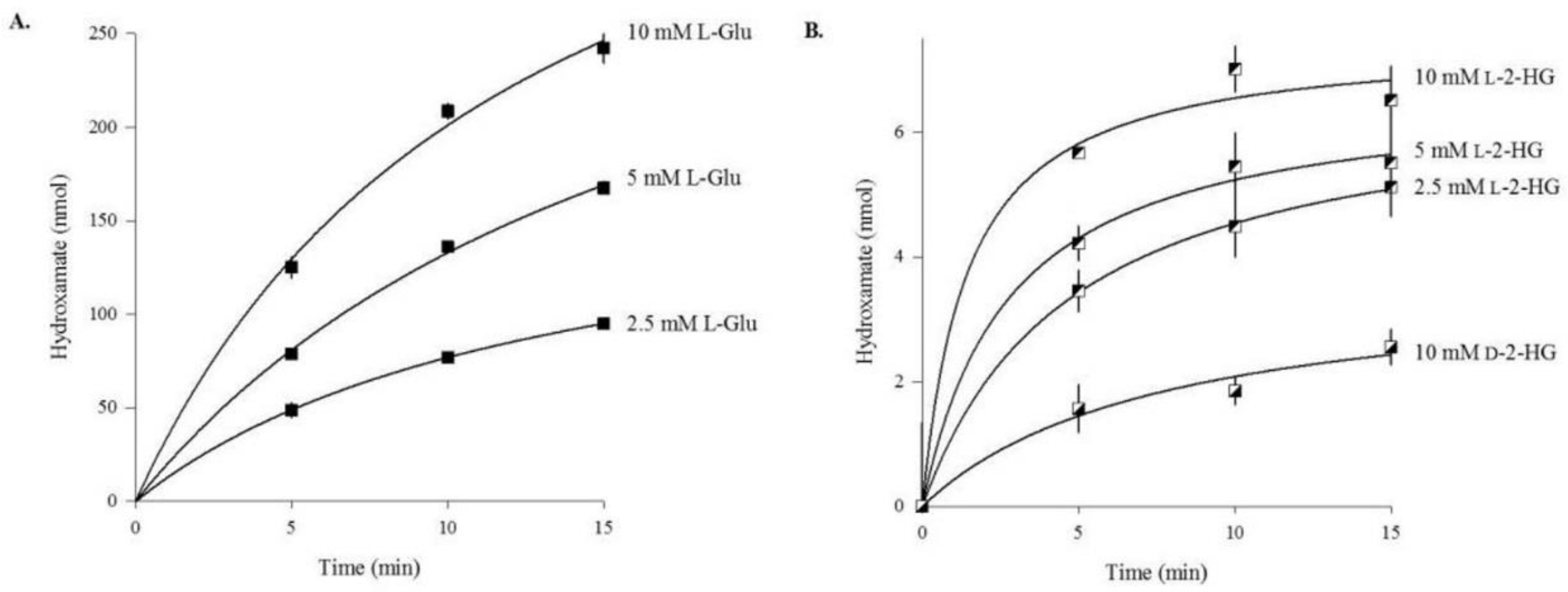
3.2. Rat Liver Cytosol Catalyzes the Hydroxaminolysis of l-2-Hydroxyglutaramate (l-2-HGM)
3.3. l- and d-2-Hydroxyglutarate Are Weak Substrates of Human Glutamine Synthetase (GS)
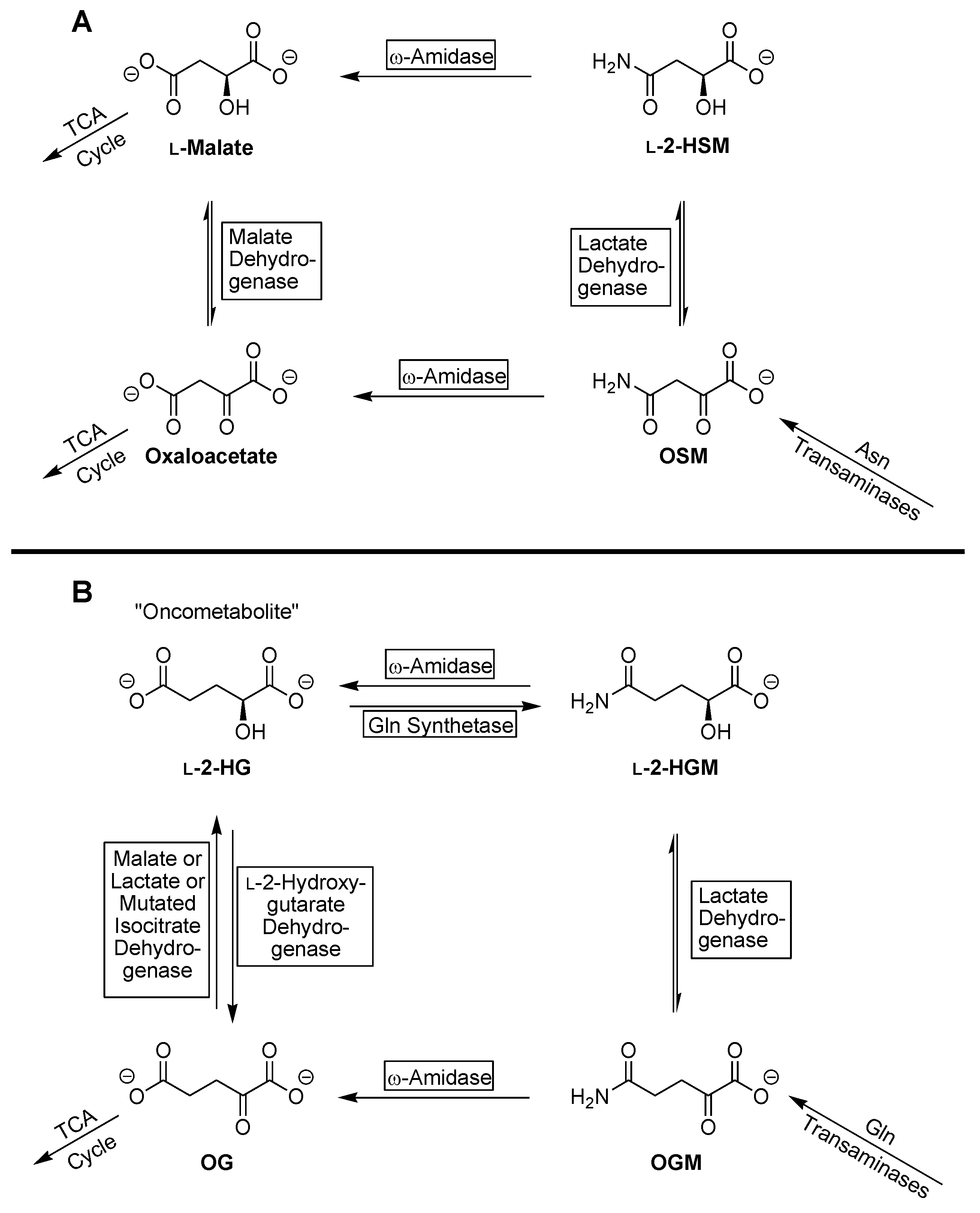
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Yizhak, K.; Chaneton, B.; Gottlieb, E.; Ruppin, E. Modeling cancer metabolism on a genome scale. Mol. Syst. Biol. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, C.; Yang, Z.; Zhang, D.; Ma, X.; Mills, G.; Liu, Z. Homeostasis of redox status derived from glucose metabolic pathway could be the key to understanding the Warburg effect. Am. J. Cancer Res. 2015, 5, 1265–1280. [Google Scholar] [PubMed]
- Tran, Q.; Lee, H.; Park, J.; Kim, S.H.; Park, J. Targeting cancer metabolism—Revisiting the Warburg effects. Toxicol. Res. 2016, 32, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.S.; Li, L.Y.; Guan, Y.D.; Yang, J.M.; Cheng, Y. Anticancer strategies based on the metabolic profile of tumor cells: therapeutic targeting of the Warburg effect. Acta. Pharmacol. Sin. 2016, 37, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Gaude, E.; Hilvo, M.; Frezza, C. The metabolic alterations of cancer cells, Methods Enzymol. 2014, 542, 1–23. Methods Enzymol. 2014, 542, 1–23. [Google Scholar] [PubMed]
- Morin, A.; Letouzé, E.; Gimenez-Roqueplo, A.P.; Favier, J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int. J. Cancer 2014, 135, 2237–2248. [Google Scholar] [CrossRef] [PubMed]
- Shim, E.H.; Livi, C.B.; Rakheja, D.; Tan, J.; Benson, D.; Parekh, V.; Kho, E.Y.; Ghosh, A.P.; Kirkman, R.; Velu, S.; et al. l-2-Hydroxyglutarate: An epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 2014, 4, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Van Schaftingen, E.; Rzem, R.; Veiga-Da-Cunha, M. l-2-Hydroxyglutaric aciduria, a disorder of metabolite repair. J. Inherit. Metab. Dis. 2009, 32, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Patay, Z.; Orr, B.A.; Shulkin, B.L.; Hwang, S.N.; Ying, Y.; Broniscer, A.; Boop, F.A.; Elliso, D.W. Successive distinct high-grade gliomas in l-2-hydroxyglutaric aciduria. J. Inherit. Metab. Dis. 2015, 38, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Shi, E.H.; Sudarshan, S. Another small molecule in the oncometabolite mix: l-2-Hydroxyglutarate in kidney cancer. Oncoscience 2015, 2, 483–486. [Google Scholar]
- Patay, Z.; Mills, J.C.; Löbel, U.; Lambert, A.; Sablauer, A.; Ellison, D.W. Cerebral neoplasms in l-2 hydroxyglutaric aciduria: 3 new cases and meta-analysis of literature data. Am. J. Neuroradiol. 2012, 33, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia induces production of l-2-hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Nadtochiy, S.M.; Schafer, X.; Fu, D.; Nehrke, K.; Munger, J.; Brookes, P.S. Acidic pH is a metabolic switch for 2-hydroxyglutarate generation and signaling. J. Biol. Chem. 2016, 291, 20188–20197. [Google Scholar] [CrossRef] [PubMed]
- Mekhail, K.; Gunaratnam, L.; Bonicalzi, M.E.; Lee, S. HIF activation by pH-dependent nucleolar sequestration of VHL. Nat. Cell Biol. 2004, 6, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Oldham, W.M.; Clish, C.B.; Yang, Y.; Loscalzo, J. Hypoxia-mediated increases in l-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 2015, 22, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Struys, E.A.; Verhoeven, N.M.; Ten Brink, H.J.; Wickenhagen, W.V.; Gibson, K.M.; Jakobs, C. Kinetic characterization of human hydroxyacid-oxoacid transhydrogenase: relevance to d-2-hydroxyglutaric and γ-hydroxybutyric acidurias. J. Inherit. Metab. Dis. 2005, 28, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, G.; Lindstedt, G.; Lindstedt, S. Metabolism of 2-amino-5-hydroxyadipic acid in the rat. Arch. Biochem. Biophys. 1967, 119, 347–352. [Google Scholar] [CrossRef]
- Chalmers, R.A.; Lawson, A.M.; Watts, R.W.; Tavill, A.S.; Kamerling, J.P.; Hey, E.; Ogilvie, D. d-2-Hydroxyglutaric aciduria: case report and biochemical studies. J. Inherit. Metab. Dis. 1980, 3, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Achouri, Y.; Noël, G.; Vertommen, D.; Rider, M.H.; Veiga-Da-Cunha, M.; Van Schaftingen, E. Identification of a dehydrogenase acting on d-2-hydroxyglutarate. Biochem. J. 2004, 381, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Struys, E. d-2-Hydroxyglutaric aciduria: unravelling the biochemical pathway and the genetic defect. J. Inherit. Metab. Dis. 2006, 29, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kranendijk, M.; Struys, E.A.; Van Schaftingen, E.; Gibson, K.M.; Kanhai, W.A.; Van Der Knaap, M.S.; Amiel, J.; Buist, N.R.; Das, A.M.; De Klerk, J.B.; et al. IDH 2 mutations in patients with d-2-hydroxyglutaric aciduria. Science 2010, 330. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Radivoyevitch, T.; Maciejewski, J.P.; Van Noorden, C.J.; Bleeker, F.E. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim. Biophys. Acta. 2014, 1846, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro. Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Sotgia, F. Oncogenes induce the cancer-associated fibroblast phenotype: metabolic symbiosis and “fibroblast addiction” are new therapeutic targets for drug discovery. Cell Cycle 2013, 12, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Katt, W.P.; Cerione, R.A. Glutaminase regulation in cancer cells: A druggable chain of events. Drug Discov. Today 2014, 19, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; L2-OSMan, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Gerken, T.; Girard, C.A.; Tung, Y.C.; Webby, C.J.; Saudek, V.; Hewitson, K.S.; Yeo, G.S.; McDonough, M.A.; Cunliffe, S.; McNeill, L.A.; et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 2007, 318, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Carbonneau, M.; Gagné, L.M.; Lalonde, M.E.; Germain, M.A.; Motorina, A.; Guiot, M.C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Mühlhausen, C.; Salomons, G.S.; Lukacs, Z.; Struys, E.A.; Van Der Knaap, M.S.; Ullrich, K.; Santer, R. Combined D2-/L2-hydroxyglutaric aciduria (SLC25A1 deficiency): Clinical course and effects of citrate treatment. J. Inherit. Metab. Dis. 2014, 37, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Nota, B.; Struys, E.A.; Pop, A.; Jansen, E.E.; Fernandez Ojeda, M.R.; Kanhai, W.A.; Kranendijk, M.; Van Dooren, S.J.; Bevova, M.R.; Sistermans, E.A.; et al. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined d-2- and l-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2013, 92, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Boufersaoui, A.; Yang, C.; Ko, B.; Rakheja, D.; Guevara, G.; Hu, Z.; DeBerardinis, R.J. Quantitative metabolic flux analysis reveals an unconventional pathway of fatty acid synthesis in cancer cells deficient for the mitochondrial citrate transport protein. Available online: http://www.sciencedirect.com/science/article/pii/S1096717616302166 (accessed on 14 November 2016).
- Linster, C.L.; Van Schaftingen, E.; Hanson, A.D. Metabolite damage and its repair or pre-emption. Nat. Chem. Biol. 2013, 9, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Van Schaftingen, E.; Rzem, R.; Marbaix, A.; Collard, F.; Veiga-Da-Cunha, M.; Linster, C.L. Metabolite proofreading, a neglected aspect of intermediary metabolism. J. Inherit. Metab. Dis. 2103, 36, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Enzymatic preparation of α-keto acids. J. Biol. Chem. 1952, 197, 309–317. [Google Scholar] [PubMed]
- Meister, A. Reduction of α,γ-diketo and α-keto acids catalyzed by muscle preparations and by crystalline lactic dehydrogenase. J. Biol. Chem. 1950, 184, 117–129. [Google Scholar] [PubMed]
- Meister, A. Preparation of enzymatic reactions of the keto analogues of asparagine and glutamine. J. Biol. Chem. 1953, 200, 571–589. [Google Scholar] [PubMed]
- Pamiljans, V.; Krishnaswamy, P.R.; Dumville, G.; Meister, A. Studies on the mechanism of glutamine synthesis; isolation and properties of the enzyme from sheep brain. Biochemistry 1962, 1, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Vorhaben, J.E.; Smith, D.D.; Campbell, J.W. Characterization of glutamine synthetase from avian liver mitochondria. Int. J. Biochem. 1982, 14, 747–756. [Google Scholar] [CrossRef]
- Krasnikov, B.F.; Nostramo, R.; Pinto, J.T.; Cooper, A.J.L. Assay and purification of ω-amidase/Nit2, a ubiquitously expressed putative tumor suppressor, that catalyzes the deamidation of the α-keto acid analogues of glutamine and asparagine. Anal. Biochem. 2009, 391, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Listrom, C.D.; Morizono, H.; Rajagopal, B.S.; McCann, M.T.; Tuchman, M.; Allewell, N.M. Expression, purification, and characterization of recombinant human glutamine synthetase. Biochem. J. 1997, 328, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Jeitner, T.M.; Cooper, A.J.L. Inhibition of human glutamine synthetase by l-methionine-S,R-sulfoximine-relevance to the treatment of neurological diseases. Metab. Brain Dis. 2014, 29, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Jeitner, T.M.; Kristoferson, E.; Azcona, J.A.; Pinto, J.T.; Stalnecker, C.; Erickson, J.W.; Kung, H.F.; Li, J.; Ploessl, K.; Cooper, A.J.L. Fluorination at the 4 position alters the substrate behavior of l-glutamine and l-glutamate: Implications for positron emission tomography of neoplasias. J. Fluor. Chem. 2016, 192, 58–67. [Google Scholar] [CrossRef]
- Krasnikov, B.F.; Kim, S.Y.; McConoughey, S.J.; Ryu, H.; Xu, H.; Stavrovskaya, I.; Iismaa, S.E.; Mearns, B.M.; Ratan, R.R.; Blass, J.P.; et al. Transglutaminase activity is present in highly purified nonsynaptosomal mouse brain and liver mitochondria. Biochemistry 2005, 44, 7830–7843. [Google Scholar] [CrossRef] [PubMed]
- Hersh, L.B. Rat liver І-amidase. Purification and properties. Biochemistry 1971, 10, 2884–2891. [Google Scholar] [CrossRef] [PubMed]
- Hersh, L.B. Rat liver І-amidase. Kinetic evidence for an acyl-enzyme intermediate. Biochemistry 1972, 11, 2251–2256. [Google Scholar] [CrossRef] [PubMed]
- Ronzio, R.A.; Wilk, S.; Rowe, W.B.; Meister, A. Preparation and studies on the characterization of sheep brain glutamine synthetase. Biochemistry 1969, 8, 2670–2674. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, N.D.H.; Joy, K.W. 2-Hydroxysuccinamic acid: a product of asparagine metabolism in plants. Biochem. Biophys. Res. Commun. 1978, 81, 186–192. [Google Scholar] [CrossRef]
- Zhang, Q.; Marsolais, F. Identification and characterization of omega-amidase as an enzyme metabolically linked to asparagine transamination in Arabidopsis. Phytochemistry. 2014, 99, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Tice, S.V. Transamination from glutamine to α-keto acids. J. Biol. Chem. 1950, 187, 173–187. [Google Scholar] [PubMed]
- Meister, A.; Sober, H.A.; Tice, S.V.; Fraser, P.E. Transamination and associated deamidation of asparagine and glutamine. J. Biol. Chem. 1952, 197, 319–330. [Google Scholar] [PubMed]
- Meister, A.; Levintow, L.; Greenfield, R.E.; Abendschein, P.A. Hydrolysis and transfer reactions catalyzed by ω-amidase preparations. J. Biol. Chem. 1955, 215, 441–460. [Google Scholar] [PubMed]
- Otani, T.T.; Meister, A. ω-Amide and ω-amino acid derivatives of α-ketoglutaric and oxalacetic acids. J. Biol. Chem. 1957, 224, 137–148. [Google Scholar]
- Jaisson, S.; Veiga-Da-Cunha, M.; Van Schaftingen, E. Molecular identification of ω-amidase, the enzyme that is functionally coupled with glutamine transaminases, as the putative tumor suppressor Nit2. Biochimie 2009, 91, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L. Asparagine transaminase from rat liver. J. Biol. Chem. 1977, 252, 2032–2038. [Google Scholar] [PubMed]
- Cooper, A.J.L.; Shurubor, Y.I.; Dorai, T.; Pinto, J.T.; Isakova, E.P.; Deryabina, Y.I.; Denton, T.T.; Krasnikov, B.F. ω-Amidase: An underappreciated, but important enzyme in l-glutamine and l-asparagine metabolism; relevance to sulfur and nitrogen metabolism, tumor biology and hyperammonemic diseases. Amino Acids 2016, 48, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Denton, T.T. Alternative functions of the brain transsulfuration pathway represent an underappreciated aspect of brain redox biochemistry with significant potential for therapeutic engagement. Free Radic. Biol. Med. 2015, 78, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Ta, T.C.; Joy, K.W.; Ireland, R.J. Role of asparagine in the photorespiratory nitrogen metabolism of pea leaves. Plant Physiol. 1985, 78, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Moraga-Amador, D.A.; MacPhee-Quiggley, K.M.; Keefer, J.F.; Schuster, S.M. Asparagine catabolism in rat liver mitochondria. Arch. Biochem. Biophys. 1989, 268, 314–326. [Google Scholar] [CrossRef]
- Stephani, R.A.; Meister, A. Structure of the dimeric α-keto acid analogue of asparagine. J. Biol. Chem. 1971, 246, 7115–7118. [Google Scholar] [PubMed]
- Cooper, A.J.L.; Raps, S.P.; Meister, A. Fluorometric determination of α-ketosuccinamic acid in rat tissues. Anal. Biochem. 1987, 167, 312–320. [Google Scholar] [CrossRef]
- Ta, T.C.; Joy, K.W.; Ireland, R.J. Utilization of the amide groups of asparagine and 2-hydroxysuccinamic acid by young pea leaves. Plant Physiol. 1984, 75, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, W.W.; Collins, R.; Holmberg-Schiavone, L.; Jones, T.A.; Karlberg, T.; Mowbray, S.L. Crystal structures of mammalian glutamine synthetases illustrate substrate-induced conformational changes and provide opportunities for drug and herbicide design. J. Mol. Biol. 2008, 375, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Becker-Kettern, J.; Paczia, N.; Conrotte, J.F.; Kay, D.P.; Guignard, C.; Jung, P.P.; Linster, C.L. Saccharomyces cerevisiae forms d-2-hydroxyglutarate and couples its degradation to d-lactate formation via a cytosolic transhydrogenase. J. Biol. Chem. 2016, 291, 6036–6058. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Enzyme Added (ng) | Specific Activity μmol/min/mg a |
|---|---|---|
| 2-Oxoglutaramate (2-OGM) b | 72.8 | 3.0 ± 0.9 (3) |
| Succinamate c | 146 | 3.4 ± 0.3 (3) |
| l-2-Hydroxyglutaramate (l-2-HGM) c | 291 | 1.4 ± 0.1 (7) |
| l-2-Hydroxysuccinamate (l-2-HSM) d | 146 | 0.51 ± 0.18 (5) |
| Addition | Hydroxamate Formation nmol/min/mg |
|---|---|
| Succinamate (50 mM) | 17 ± 2 |
| Succinamate + glycylglycine (30 mM) | 8.9 ± 0.8 b |
| Succinamate + l-2-HSM (50 mM) | 12 ± 1 c |
| l-2-HGM | 26 ± 1 |
| l-2-HGM + glycylglycine (30 mM) | 3.6 ± 0.2 d |
| l-2-HGM + l-2-HSM (25 mM) | 18 ± 1 d |
| Addition | 2-OG Formation nmol/min/mg |
|---|---|
| 2-OGM (3 mM) | 13 ± 1 |
| 2-OGM + glycylglycine (30 mM) | 7.9 ± 0.7 b |
| 2-OGM + l-2-HSM (25 mM) | 4.5 ± 0.5 b |
| 2-OGM + succinamate (50 mM) | 2.9 ± 0.2 c |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hariharan, V.A.; Denton, T.T.; Paraszcszak, S.; McEvoy, K.; Jeitner, T.M.; Krasnikov, B.F.; Cooper, A.J.L. The Enzymology of 2-Hydroxyglutarate, 2-Hydroxyglutaramate and 2-Hydroxysuccinamate and Their Relationship to Oncometabolites. Biology 2017, 6, 24. https://doi.org/10.3390/biology6020024
Hariharan VA, Denton TT, Paraszcszak S, McEvoy K, Jeitner TM, Krasnikov BF, Cooper AJL. The Enzymology of 2-Hydroxyglutarate, 2-Hydroxyglutaramate and 2-Hydroxysuccinamate and Their Relationship to Oncometabolites. Biology. 2017; 6(2):24. https://doi.org/10.3390/biology6020024
Chicago/Turabian StyleHariharan, Vivek A., Travis T. Denton, Sarah Paraszcszak, Kyle McEvoy, Thomas M. Jeitner, Boris F. Krasnikov, and Arthur J. L. Cooper. 2017. "The Enzymology of 2-Hydroxyglutarate, 2-Hydroxyglutaramate and 2-Hydroxysuccinamate and Their Relationship to Oncometabolites" Biology 6, no. 2: 24. https://doi.org/10.3390/biology6020024
APA StyleHariharan, V. A., Denton, T. T., Paraszcszak, S., McEvoy, K., Jeitner, T. M., Krasnikov, B. F., & Cooper, A. J. L. (2017). The Enzymology of 2-Hydroxyglutarate, 2-Hydroxyglutaramate and 2-Hydroxysuccinamate and Their Relationship to Oncometabolites. Biology, 6(2), 24. https://doi.org/10.3390/biology6020024