
Circadian Regulation of Synaptic Plasticity

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Circadian Rhythms in Synaptic Plasticity: Electrophysiological Measures
3. Circadian Rhythms in Synaptic Plasticity: Morphological Measures
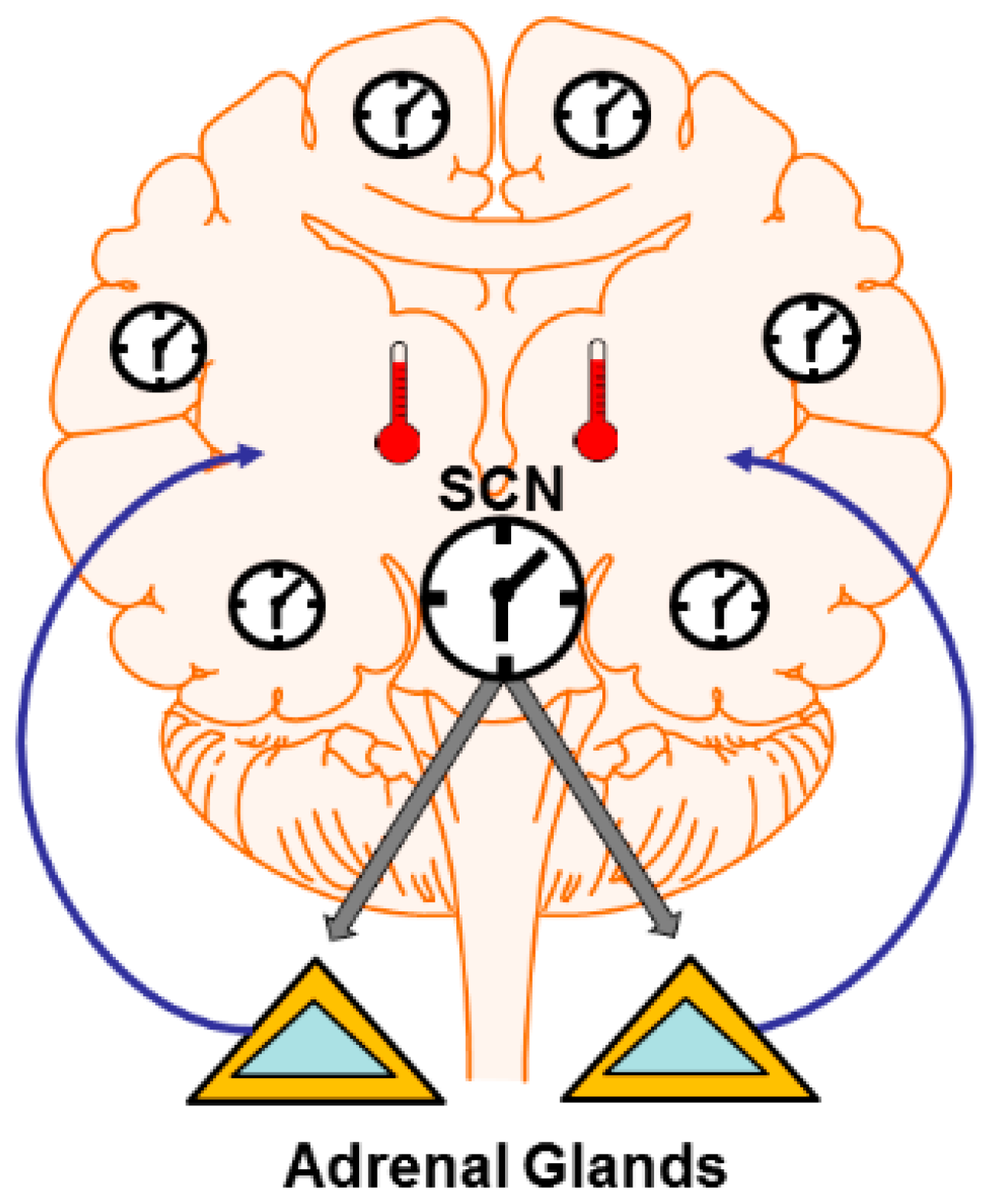
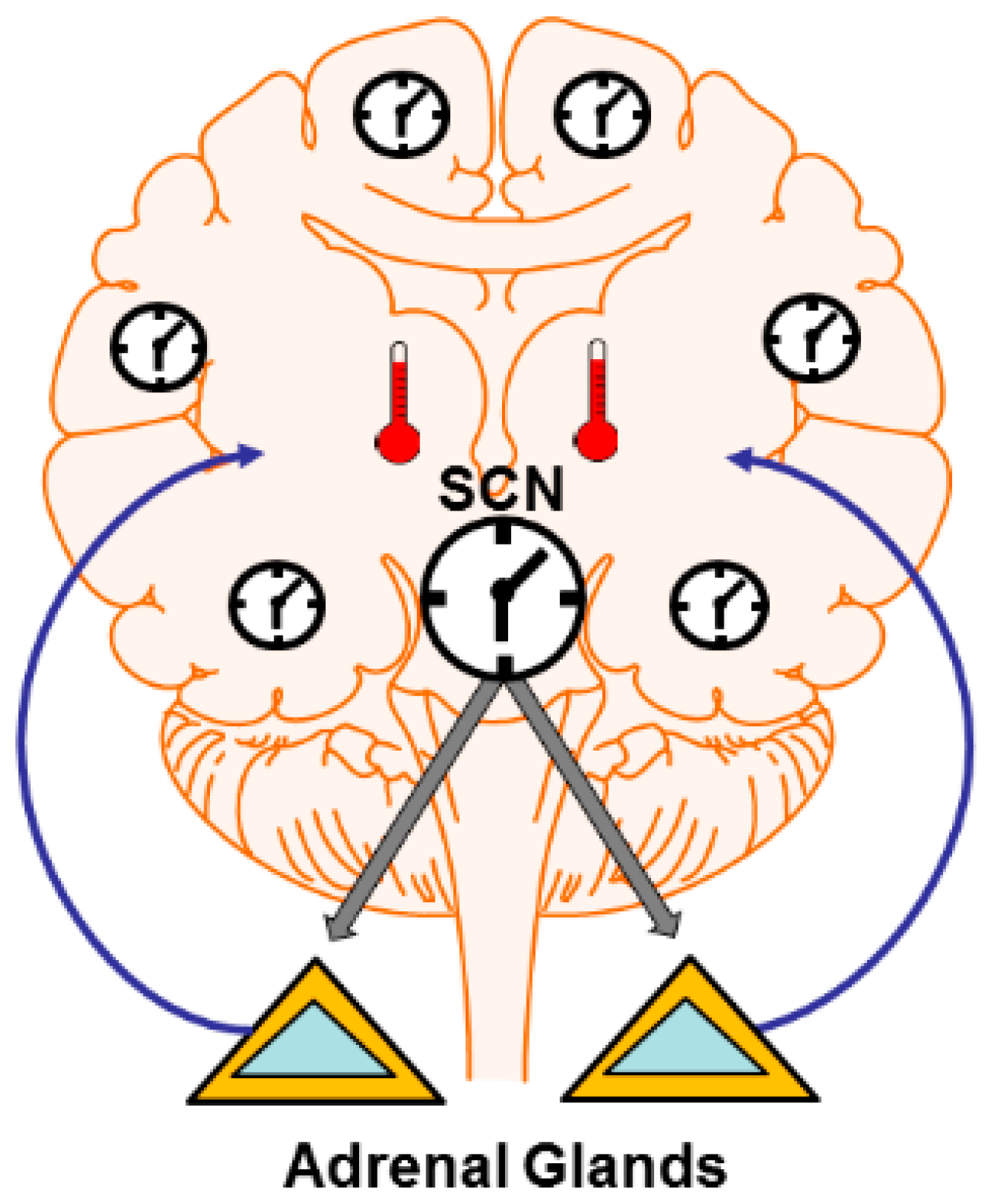
4. Mechanisms: Central and Peripheral Clocks
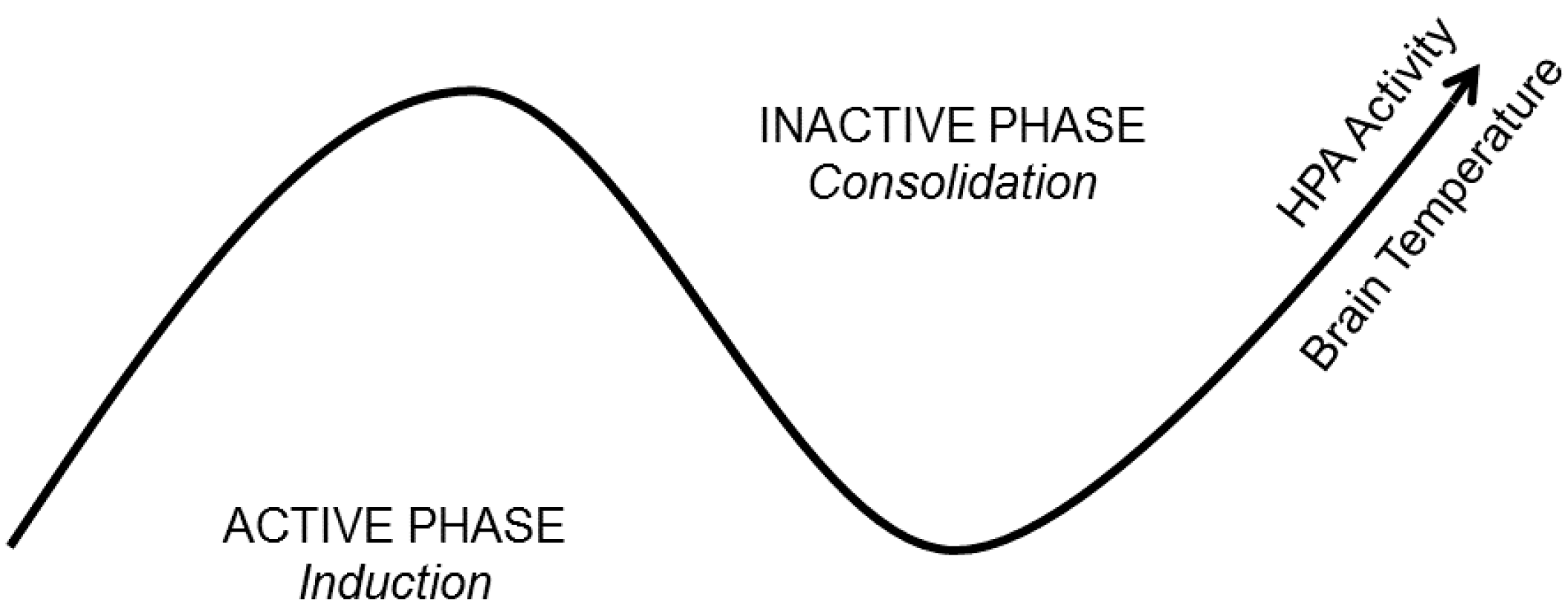
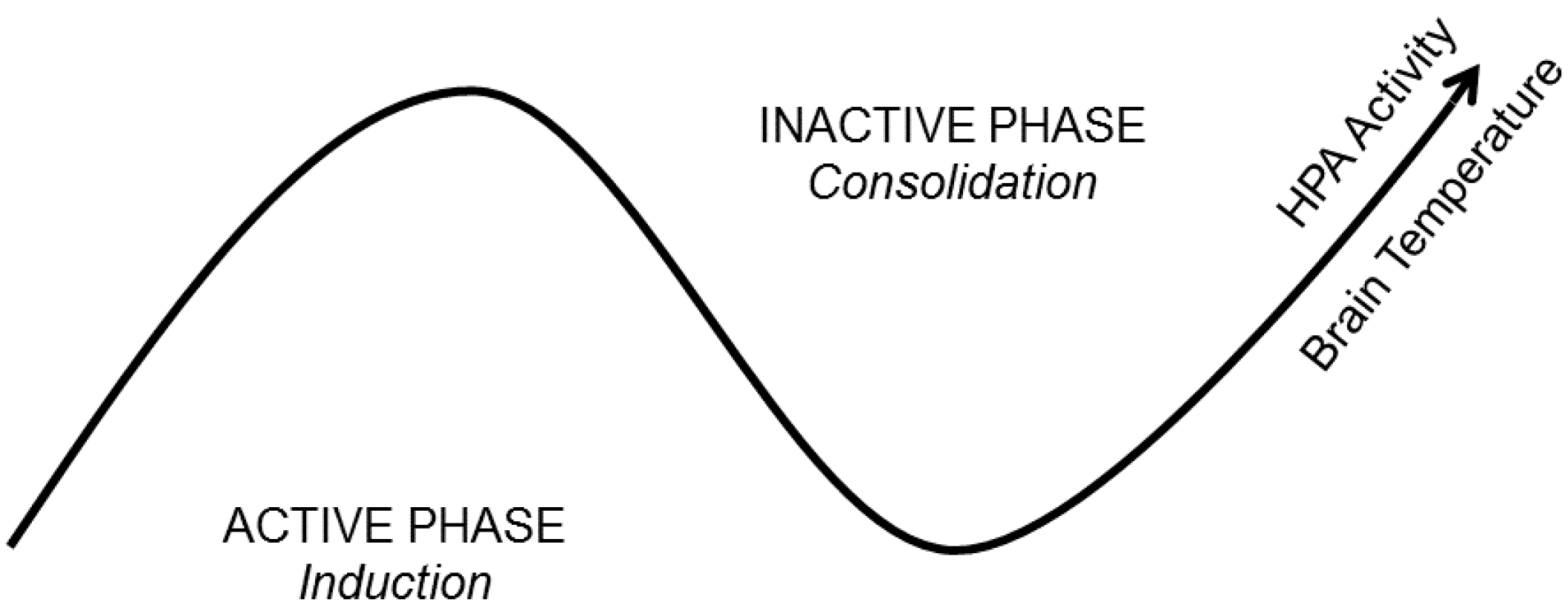
4.1. Brain Temperature
4.2. Hormone and Neuromodulator Release
4.3. Rhythms in GABAergic Inhibition
4.4. Peripheral Clocks
5. Discussion
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| Cam | calmodulin |
| LTP | Long-term potentiation |
| LTD | Long-term depression |
| SCN | Suprachiasmatic Nucleus |
| SCM | State-Clock Model |
| LNv | Lateral-Ventral Neuron |
| LD | Light-Dark |
| DD | Dark-Dark |
| TRP | transient receptor potential |
References
- Feldman, D.E. Synaptic mechanisms for plasticity in neocortex. Annu. Rev. Neurosci. 2009, 32, 33–55. [Google Scholar] [CrossRef] [PubMed]
- Steidl, S.; Rose, J.K.; Rankin, C.H. Stages of memory in the nematode Caenorhabditis elegans. Behav. Cogn. Neurosci. Rev. 2003, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.O.L. Retinal waves and visual system development. Annu. Rev. Neurosci. 1999, 22, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Cantera, R. Sleep, clocks, and synaptic plasticity. Trends Neurosci. 2014, 37, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G. Erasing synapses in sleep: Is it time to be shy? Neural Plast. 2012. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G. Why I am not shy: A reply to Tononi and Cirelli. Neural Plast. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hengen, K.B.; Torrado Pacheco, A.; McGregor, J.N.; van Hooser, S.D.; Turrigiano, G.G. Neuronal firing rate homeostasis is inhibited by sleep and promoted by wake. Cell 2016, 165, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Cirelli, C.; Tononi, G. Sleep and synaptic homeostasis. Sleep 2015, 38, 161–162. [Google Scholar] [CrossRef] [PubMed]
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012, 35, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.A.; McNaughton, B.L.; Goddard, G.V.; Douglas, R.M.; Adamec, R. Circadian rhythm of synaptic excitability in rat and monkey central nervous system. Science 1977, 197, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.M.; Teyler, T.J. Age differences in a circadian influence on hippocamapl LTP. Brain Res. 1983, 261, 69–73. [Google Scholar] [CrossRef]
- Bowden, J.B.; Abraham, W.C.; Harris, K.M. Differential effects of strain, circadian cycle, and stimulation pattern on LTP and concurrent LTD in the dentate gyrus of freely moving rats. Hippocampus 2012, 22, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, D.; Wang, L.M.; Colwell, C.S. Circadian regulation of hippocampal long-term potentiation. J. Biol. Rhythms. 2005, 20, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, A.V.; Horowitz, J.M.; Fuller, C.A. Diurnal modulation of long-term potentiation in the hamster hippocampal slice. Brain Res. 1999, 833, 311–314. [Google Scholar] [CrossRef]
- Rawashdeh, O.; Jilg, A.; Jedlicka, P.; Slawska, J.; Thomas, L.; Saade, A.; Schwarzacher, S.W.; Stehle, J.H. Period1 coordinates hippocampal rhythms and memory processing with daytime. Hippocampus 2014, 24, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Kondratova, A.A.; Dubrovsky, Y.V.; Antoch, M.P.; Kondratov, R.V. Circadian clock proteins control adaptation to novel environment and memory formation. Aging 2010, 2, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, S.M.; Phan, T.X.; Saraf, A.; Chen, X.; Storm, D.R. Genetic disruption of the core circadian clock impairs hippocampus-dependent memory. Learn. Mem. 2014, 21, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Mahan, K.L. Circadian oscillations within the hippocampus support hippocampus-dependent memory processing. Front. Mol. Neurosci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Mahan, K.L.; Phan, T.; Han, S.; Wang, H.; Chan, G.C.K.; Scheiner, Z.S.; Storm, D.R. Circadian oscillation of hippocampal mapk activity and camp: Implications for memory persistence. Nat. Neurosci. 2008, 11, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Hanada, Y.; Kawamura, H. Circadian rhythms in synaptic excitability of the dorsal lateral geniculate nucleus in the rat. Int. J. Neurosci. 1984, 22, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Parekh, P.K.; McClung, C.A. Circadian mechanisms underlying reward-related neurophysiology and synaptic plasticity. Front. Psychiatry 2016. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, B.; Dryer, S.E.; Hardin, P.E. Circadian rhythms in olfactory responses of drosophila melanogaster. Nature 1999, 400, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Page, T.L.; Koelling, E. Circadian rhythm in olfactory response in the antennae controlled by the optic lobe in the cockroach. J. Insect Physiol. 2003, 49, 697–707. [Google Scholar] [CrossRef]
- Tanoue, S.; Krishnan, P.; Krishnan, B.; Dryer, S.E.; Hardin, P.E. Circadian clocks in antennal neurons are necessary and sufficient for olfaction rhythms in Drosophila. Curr. Biol. 2004, 14, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Nitabach, M.N. Circadian control of membrane excitability in Drosophila melanogaster lateral ventral clock neurons. J. Neurosci. 2008, 28, 6493–6501. [Google Scholar] [CrossRef] [PubMed]
- Sheeba, V.; Gu, H.; Sharma, V.K.; O’Dowd, D.K.; Holmes, T.C. Circadian- and light-dependent regulation of resting membrane potential and spontaneous action potential firing of Drosophila circadian pacemaker neurons. J. Neurophysiol. 2008, 99, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Pyza, E.; Meinertzhagen, I.A. Daily and circadian rhythms of synaptic frequency in the first visual neuropile of the housefly’s (Musca domestica L.) optic lobe. Proc. Biol. Sci. 1993, 254, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Pyza, E.; Meinertzhagen, I.A. Daily rhythmic changes of cell size and shape in the first optic neuropil in Drosophila melanogaster. J. Neurobiol. 1999, 40, 77–88. [Google Scholar] [CrossRef]
- Woznicka, O.; Gorlich, A.; Sigrist, S.J.; Pyza, E.M. BRP-170 and BRP190 isoforms of bruchpilot protein differentially contribute to the frequency of synapses and synaptic circadian plasticity in the visual system of Drosophila. Front. Cell. Neurosci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Barth, M.; Schultze, M.; Schuster, C.M.; Strauss, R. Circadian plasticity in photoreceptor cells controls visual coding efficiency in Drosophila melanogaster. PLoS ONE 2010, 5, e9217. [Google Scholar] [CrossRef] [PubMed]
- Weber, P.; Kula-Eversole, E.; Pyza, E. Circadian control of dendrite morphology in the visual system of Drosophila melanogaster. PLoS ONE 2009, 4, e4290. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.P.; Berni, J.; Ceriani, M.F. Circadian remodeling of neuronal circuits involved in rhythmic behavior. PLoS Biol. 2008, 6, e69. [Google Scholar] [CrossRef] [PubMed]
- Petsakou, A.; Sapsis, T.P.; Blau, J. Circadian rhythms in rho1 activity regulate neuronal plasticity and network hierarchy. Cell 2015, 162, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Ferreiro, M.J.; Menhert, K.I.; Casanova, G.; Olivera, A.; Cantera, R. Rhythmic changes in synapse numbers in Drosophila melanogaster motor terminals. PLoS ONE 2013, 8, e67161. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, K.I.; Beramendi, A.; Elghazali, F.; Negro, P.; Kyriacou, C.P.; Cantera, R. Circadian changes in Drosophila motor terminals. Dev. Neurobiol. 2007, 67, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, K.I.; Cantera, R. A peripheral pacemaker drives the circadian rhythm of synaptic boutons in Drosophila independently of synaptic activity. Cell Tissue Res. 2008, 334, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Ferreiro, M.J.; Casanova, G.; Olivera, A.; Cantera, R. Synaptic vesicles in motor synapses change size and distribution during the day. Synapse 2010, 64, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Vollrath, L.; Spiwoks-Becker, I. Plasticity of retinal ribbon synapses. Microsc. Res. Tech. 1996, 35, 472–487. [Google Scholar] [CrossRef]
- Sterling, P.; Matthews, G. Structure and function of ribbon synapses. Trends Neurosci. 2005, 28, 20–29. [Google Scholar] [CrossRef] [PubMed]
- McNulty, J.A. Synaptic ribbons in the pineal organ of the goldfish: Circadian rhythmicity and the effects of constant light and constant darkness. Cell Tissue Res. 1981, 215, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Emran, F.; Rihel, J.; Adolph, A.R.; Dowling, J.E. Zebrafish larvae lose vision at night. Proc. Natl. Acad. Sci. USA 2010, 107, 6034–6039. [Google Scholar] [CrossRef] [PubMed]
- Appelbaum, L.; Wang, G.; Yokogawa, T.; Skariah, G.M.; Smith, S.J.; Mourrain, P.; Mignot, E. Circadian and homeostatic regulation of structural synaptic plasticity in hypocretin neurons. Neuron 2010, 68, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Jasinska, M.; Grzegorczyk, A.; Woznicka, O.; Jasek, E.; Kossut, M.; Barbacka-Surowiak, G.; Litwin, J.A.; Pyza, E. Circadian rhythmicity of synapses in mouse somatosensory cortex. Eur. J. Neurosci. 2015, 42, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Liston, C.; Cichon, J.M.; Jeanneteau, F.; Jia, Z.; Chao, M.V.; Gan, W.-B. Circadian glucocorticoid oscillations promote learning-dependent synapse formation and maintenance. Nat. Neurosci. 2013, 16, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Hojo, Y.; Komatsuzaki, Y.; Okamoto, M.; Kato, A.; Takeda, T.; Kawato, S. Hippocampal spine changes across the sleep-wake cycle: Corticosterone and kinases. J. Endocrinol. 2015, 226, M13–M27. [Google Scholar] [CrossRef] [PubMed]
- Gamble, K.L.; Berry, R.; Frank, S.J.; Young, M.E. Circadian clock control of endocrine factors. Nature reviews. Endocrinology 2014, 10, 466–475. [Google Scholar] [PubMed]
- Ito, C.; Tomioka, K. Heterogeneity of the peripheral circadian systems in Drosophila melanogaster: A review. Front. Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Heller, H.C. Temperature, thermoregulation and sleep. In Principles and Practice of Sleep Medcine, 4th ed.; Kryger, M.H., Roth, T., Dement, W.C., Eds.; Elsevier: Philadelphia, PA, USA, 2005; pp. 292–304. [Google Scholar]
- Stevenson, R.D. Body size and limits to the daily range of body temperature in terrestrial ectotherms. Am. Nat. 1985, 125, 102–117. [Google Scholar] [CrossRef]
- Moser, E.; Mathiesen, I.; Andersen, P. Association between brain temperature and dentate field potentials in exploring and swimming rats. Science 1993, 259, 1324–1326. [Google Scholar] [CrossRef] [PubMed]
- Franken, P.; Dijk, D.J.; Tobler, I.; Borbely, A.A. Sleep deprivation in rats: Effects on EEG power spectra, vigilance states, and cortical temperature. Am. J. Physiol. 1991, 261, R198–R208. [Google Scholar] [PubMed]
- Franken, P.; Tobler, I.; Borbely, A.A. Effects of 12-h sleep deprivation and of 12-h cold exposure on sleep regulation and cortical temperature in the rat. Physiol. Behav. 1993, 54, 885–894. [Google Scholar] [CrossRef]
- Zhong, Y.; Wu, C.-F. Neuronal activity and adenylyl cyclase in environment-dependent plasticity of axonal outgrowth in Drosophila. J. Neurosci. 2004, 24, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Peng, I.F.; Berke, B.A.; Zhu, Y.; Lee, W.-H.; Chen, W.; Wu, C.-F. Temperature-dependent developmental plasticity of drosophila neurons: Cell-autonomous roles of membrane excitability, Ca2+ influx, and camp signaling. J. Neurosci. 2007, 27, 12611–12622. [Google Scholar] [CrossRef] [PubMed]
- Vanin, S.; Bhutani, S.; Montelli, S.; Menegazzi, P.; Green, E.W.; Pegoraro, M.; Sandrelli, F.; Costa, R.; Kyriacou, C.P. Unexpected features of drosophila circadian behavioural rhythms under natural conditions. Nature 2012, 484, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, B.; Normoyle, K.; Jackson, K.; Spitler, K.; Sharrock, M.F.; Miller, C.; Best, C.; Llano, D.; Du, R. Brain temperature and its fundamental properties: A review for clinical neuroscientists. Front. Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.M.; Xu, H.; Clapham, D.E. Trp ion channels in the nervous system. Curr. Opin. Neurobiol. 2004, 14, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Talavera, K.; Nilius, B.; Voets, T. Neuronal trp channels: Thermometers, pathfinders and life-savers. Trends Neurosci. 2008, 31, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, K.; Suzuki, M.; Mizuno, A.; Tominaga, M. Effects of body temperature on neural activity in the hippocampus: Regulation of resting membrane potentials by transient receptor potential vanilloid 4. J. Neurosci. 2007, 27, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Martins, D.; Tavares, I.; Morgado, C. “Hotheaded”: The role OF TRPV1 in brain functions. Neuropharmacology 2014, 85, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Van Cauter, E. Endocrine physiology. In Principles and Practice of Sleep Medicine, 5th ed.; Kryger, M., Roth, T., Dement, W.C., Eds.; Elsevier: Philadelphia, PA, USA, 2005; pp. 266–282. [Google Scholar]
- Wang, L.M.; Suthana, N.A.; Chaudhury, D.; Weaver, D.R.; Colwell, C.S. Melatonin inhibits hippocampal long-term potentiation. Eur. J. Neurosci. 2005, 22, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- O’Neal-Moffitt, G.; Pilli, J.; Kumar, S.S.; Olcese, J. Genetic deletion of MT1/MT2 melatonin receptors enhances murine cognitive and motor performance. Neuroscience 2014, 277, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Cardinali, D.P.; Srinivasan, V.; Spence, D.W.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin—A pleiotropic, orchestrating regulator molecule. Prog. Neurobiol. 2011, 93, 350–384. [Google Scholar] [CrossRef] [PubMed]
- Komatsuzaki, Y.; Hatanaka, Y.; Murakami, G.; Mukai, H.; Hojo, Y.; Saito, M.; Kimoto, T.; Kawato, S. Corticosterone induces rapid spinogenesis via synaptic glucocorticoid receptors and kinase networks in hippocampus. PLoS ONE 2012, 7, e34124. [Google Scholar] [CrossRef] [PubMed]
- Tse, Y.C.; Bagot, R.C.; Wong, T.P. Dynamic regulation of nmdar function in the adult brain by the stress hormone corticosterone. Front. Cell. Neurosci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Joels, M.; Krugers, H.; Karst, H. Stress-induced changes in hippocampal function. Prog. Brain Res. 2008, 167, 3–15. [Google Scholar] [PubMed]
- Krugers, H.J.; Hoogenraad, C.C.; Groc, L. Stress hormones and AMPA receptor trafficking in synaptic plasticity and memory. Nat. Rev. Neurosci. 2010, 11, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, F.; Lu, D.; Ha, P.; Costacurta, P.; Chavez, R.; Heller, H.C.; Ruby, N.F. Dysrhythmia in the suprachiasmatic nucleus inhibits memory processing. Science 2014, 346, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Ruby, N.F.; Fernandez, F.; Garrett, A.; Klima, J.; Zhang, P.; Sapolsky, R.; Heller, H.C. Spatial memory and long-term object recognition are impaired by circadian arrhythmia and restored by the GABAAAntagonist pentylenetetrazole. PLoS ONE 2013, 8, e72433. [Google Scholar]
- Ruby, N.F.; Hwang, C.E.; Wessells, C.; Fernandez, F.; Zhang, P.; Sapolsky, R.; Heller, H.C. Hippocampal-dependent learning requires a functional circadian system. Proc. Natl. Acad. Sci. USA 2008, 105, 15593–15598. [Google Scholar] [CrossRef] [PubMed]
- Henny, P.; Jones, B.E. Projections from basal forebrain to prefrontal cortex comprise cholinergic, gabaergic and glutamatergic inputs to pyramidal cells or interneurons. Eur. J. Neurosci. 2008, 27, 654–670. [Google Scholar] [CrossRef] [PubMed]
- Unal, G.; Joshi, A.; Viney, T.J.; Kis, V.; Somogyi, P. Synaptic targets of medial septal projections in the hippocampus and extrahippocampal cortices of the mouse. J. Neurosci. 2015, 35, 15812–15826. [Google Scholar] [CrossRef] [PubMed]
- Frank, M. Sleep and synaptic plasticity in the developing and adult brain. Sleep Neuronal Plast. Brain Funct. 2014, 25, 123–149. [Google Scholar]
- Stickgold, R. Sleep-dependent memory consolidation. Nature 2005, 437, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Turrigiano, G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu. Rev. Neurosci. 2011, 34, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Tononi, G.; Cirelli, C. Sleep and synaptic homeostasis: A hypothesis. Brain Res. Bull. 2003, 62, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Tononi, G.; Cirelli, C. Sleep and the price of plasticity: From synaptic and cellular homeostasis to memory consolidation and integration. Neuron 2014, 81, 12–34. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G. The mystery of sleep function: Current perspectives and future directions. Rev. Neurosci. 2006, 17, 375–392. [Google Scholar] [CrossRef] [PubMed]
- Benington, J.H.; Frank, M.G. Cellular and molecular connections between sleep and synaptic plasticity. Prog. Neurobiol. 2003, 69, 77–101. [Google Scholar] [CrossRef]
- Seibt, J.; Dumoulin, M.; Aton, S.J.; Naidoo, J.; Watson, A.; Coleman, T.; Frank, M.G. Protein synthesis during sleep consolidates cortical plasticity in vivo. Curr. Biol. 2012, 22, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Vecsey, C.G.; Peixoto, L.; Choi, J.H.K.; Wimmer, M.; Jaganath, D.; Hernandez, P.J.; Blackwell, J.; Meda, K.; Park, A.J.; Hannenhalli, S.; et al. Genomic analysis of sleep deprivation reveals translational regulation in the hippocampus. Physiol. Genom. 2012, 44, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Mongrain, V.; Hernandez, S.A.; Pradervand, S.; Dorsaz, S.; Curie, T.; Hagiwara, G.; Gip, P.; Heller, H.C.; Franken, P. Separating the contribution of glucocorticoids and wakefulness to the molecular and electrophysiological correlates of sleep homeostasis. Sleep 2010, 33, 1147–1157. [Google Scholar] [PubMed]
- Dijk, D.J.; Czeisler, C.A. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J. Neurosci. 1995, 15, 3526–3538. [Google Scholar] [PubMed]
- Lazar, A.S.; Lazar, Z.I.; Dijk, D.-J. Circadian regulation of slow waves in human sleep: Topographical aspects. NeuroImage 2015, 116, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Vyazovskiy, V.V.; Riedner, B.A.; Cirelli, C.; Tononi, G. Sleep homeostasis and cortical synchronization: II. A local field potential study of sleep slow saves in the rat. Sleep 2007, 30, 1631–1642. [Google Scholar] [PubMed]
- Kobayashi, Y.; Ye, Z.; Hensch, T.K. Clock genes control cortical critical period timing. Neuron 2015, 86, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Dabbish, N.S.; Raizen, D.M. Gabaergic synaptic plasticity during a developmentally regulated sleep-like state in C. elegans. J. Neurosci. 2011, 31, 15932–15943. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frank, M.G. Circadian Regulation of Synaptic Plasticity. Biology 2016, 5, 31. https://doi.org/10.3390/biology5030031
Frank MG. Circadian Regulation of Synaptic Plasticity. Biology. 2016; 5(3):31. https://doi.org/10.3390/biology5030031
Chicago/Turabian StyleFrank, Marcos G. 2016. "Circadian Regulation of Synaptic Plasticity" Biology 5, no. 3: 31. https://doi.org/10.3390/biology5030031
APA StyleFrank, M. G. (2016). Circadian Regulation of Synaptic Plasticity. Biology, 5(3), 31. https://doi.org/10.3390/biology5030031