
StAR Protein Stability in Y1 and Kin-8 Mouse Adrenocortical Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Source of Reagents
2.2. Radiolabeling Studies and STAR Immunoprecipitation
2.3. Immunoblot Analysis
2.4. Progesterone Radioimmunoassay
2.5. Statistical Analysis
3. Results
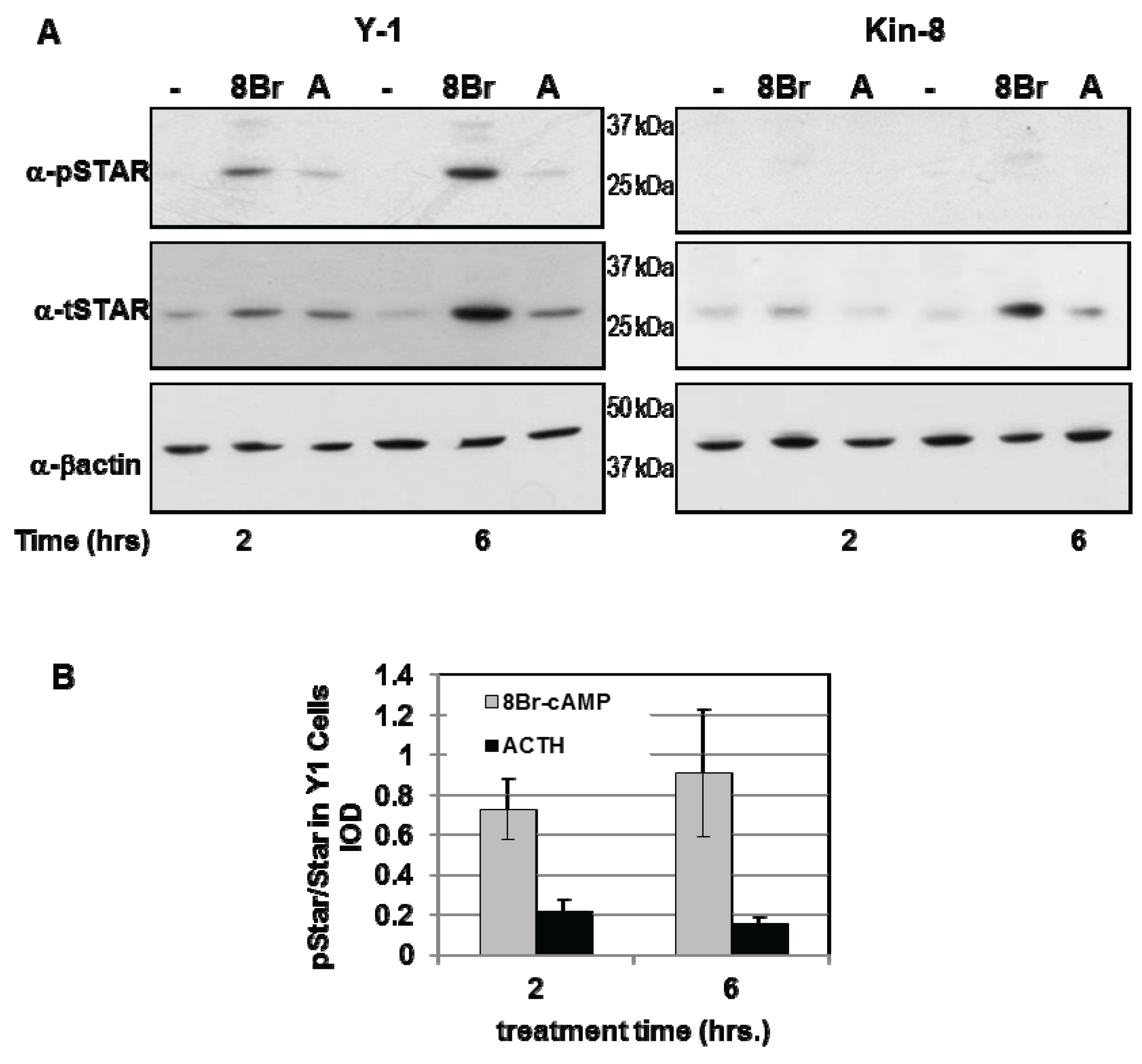
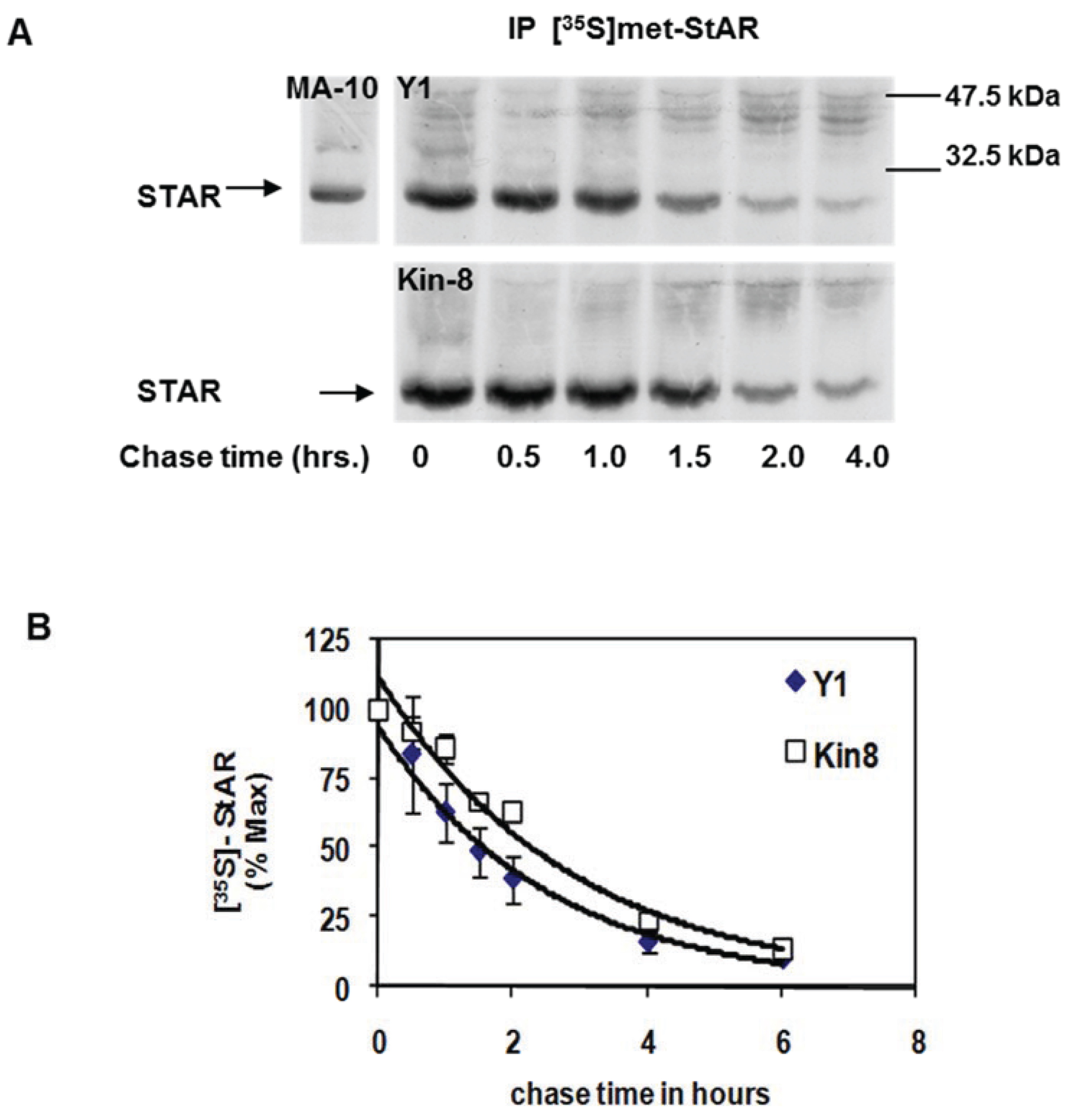
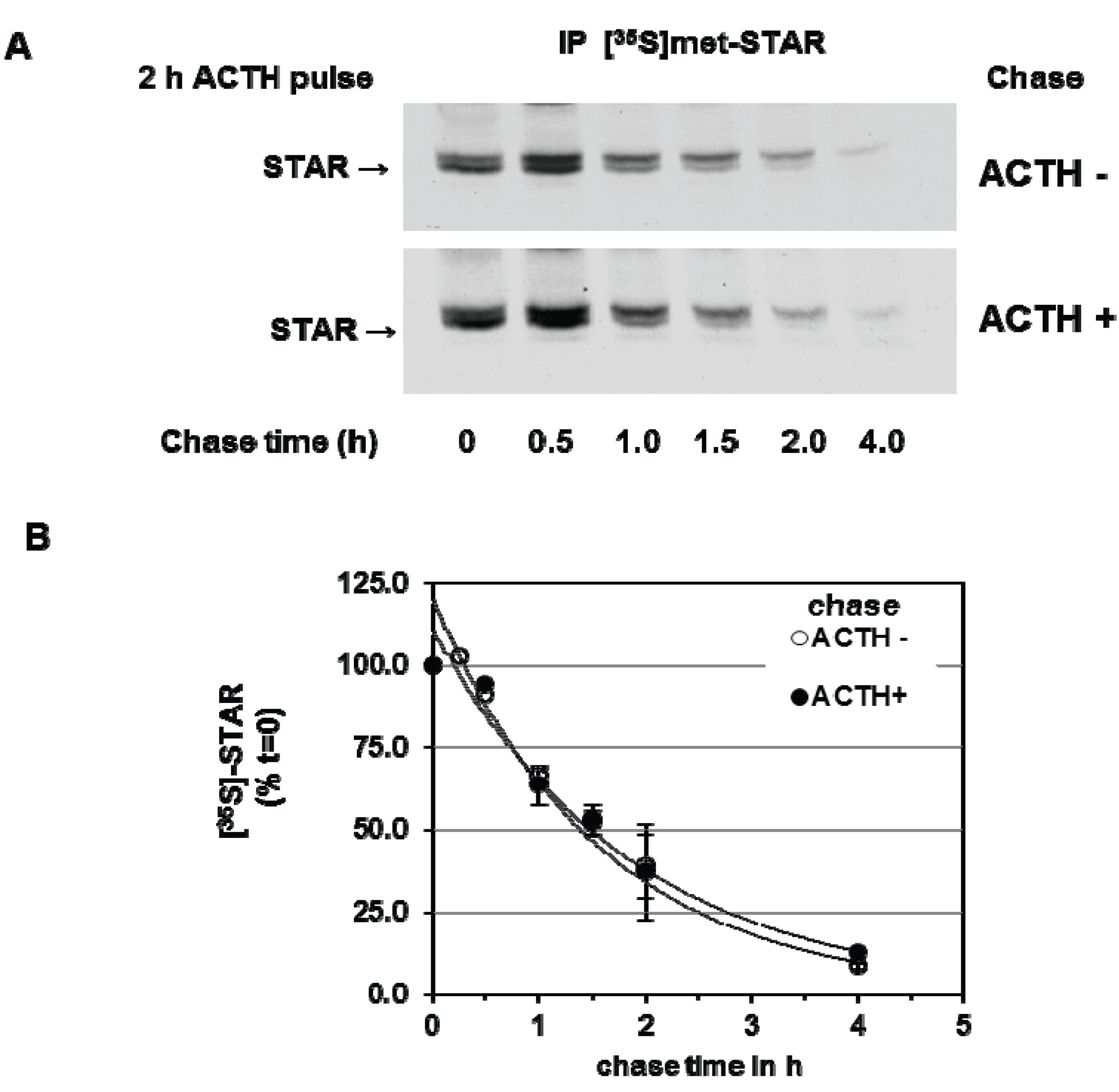
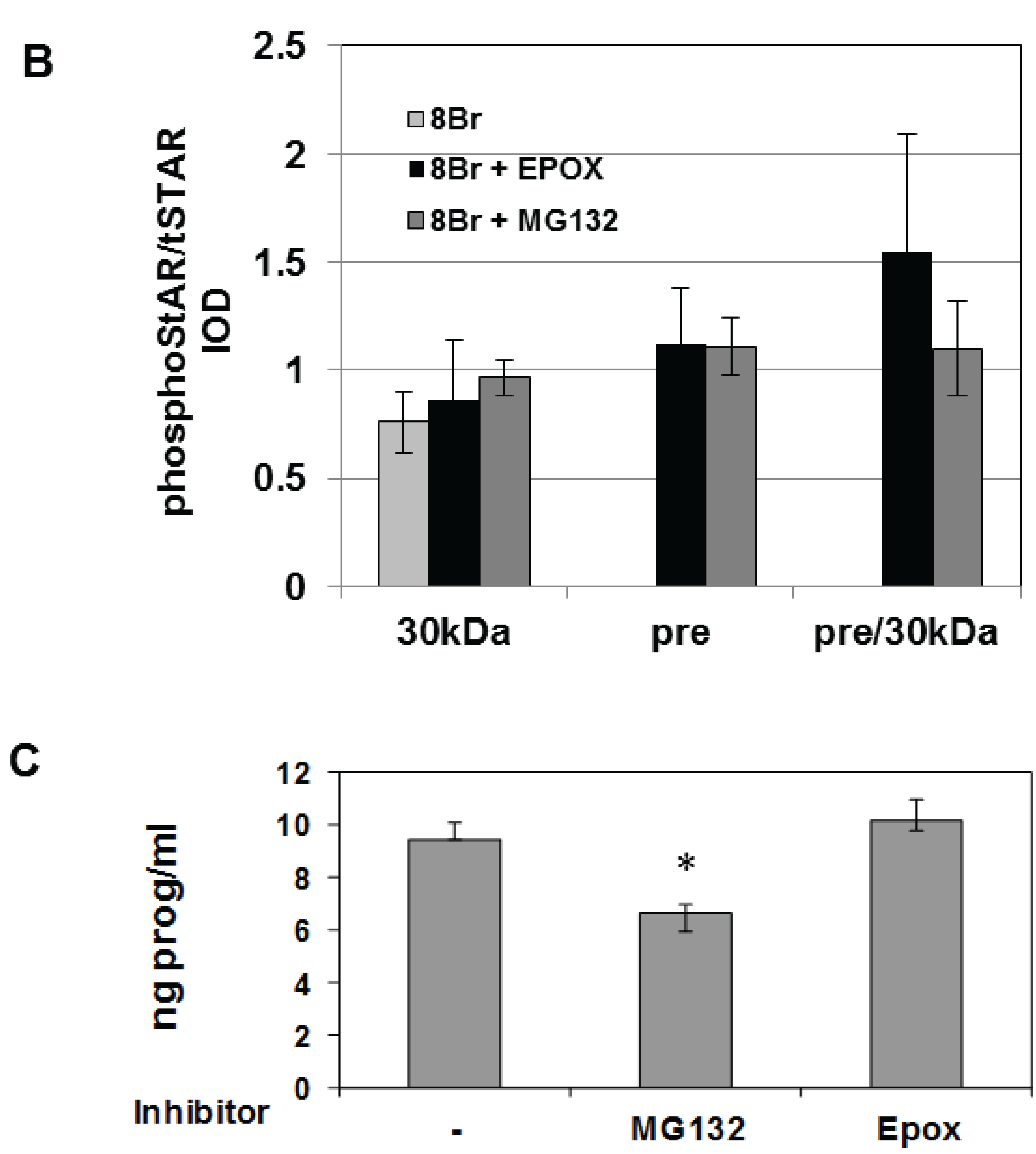
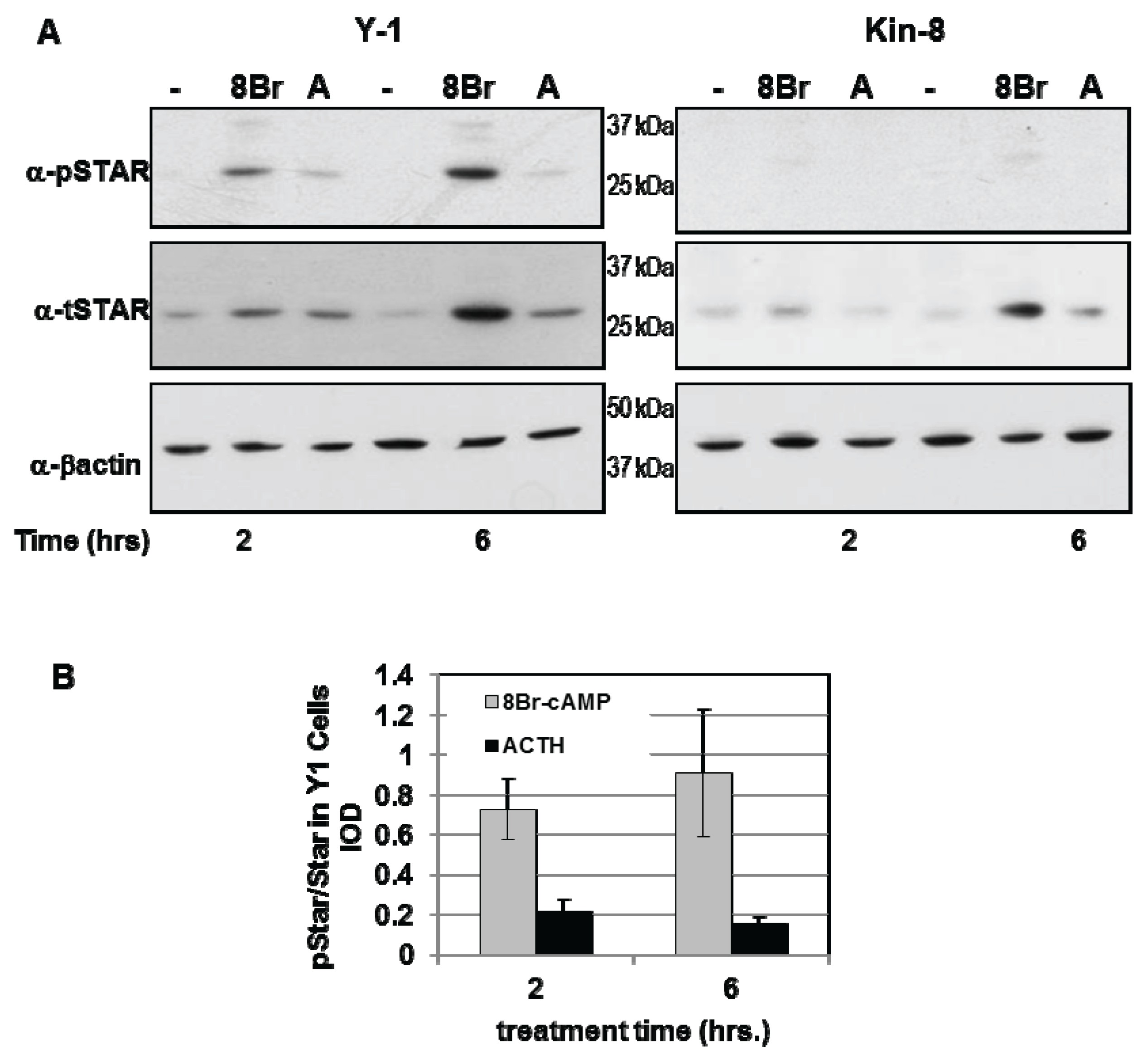
3.1. STAR Phosphorylation and Protein Half-Life in Y1 and Kin-8 Cells
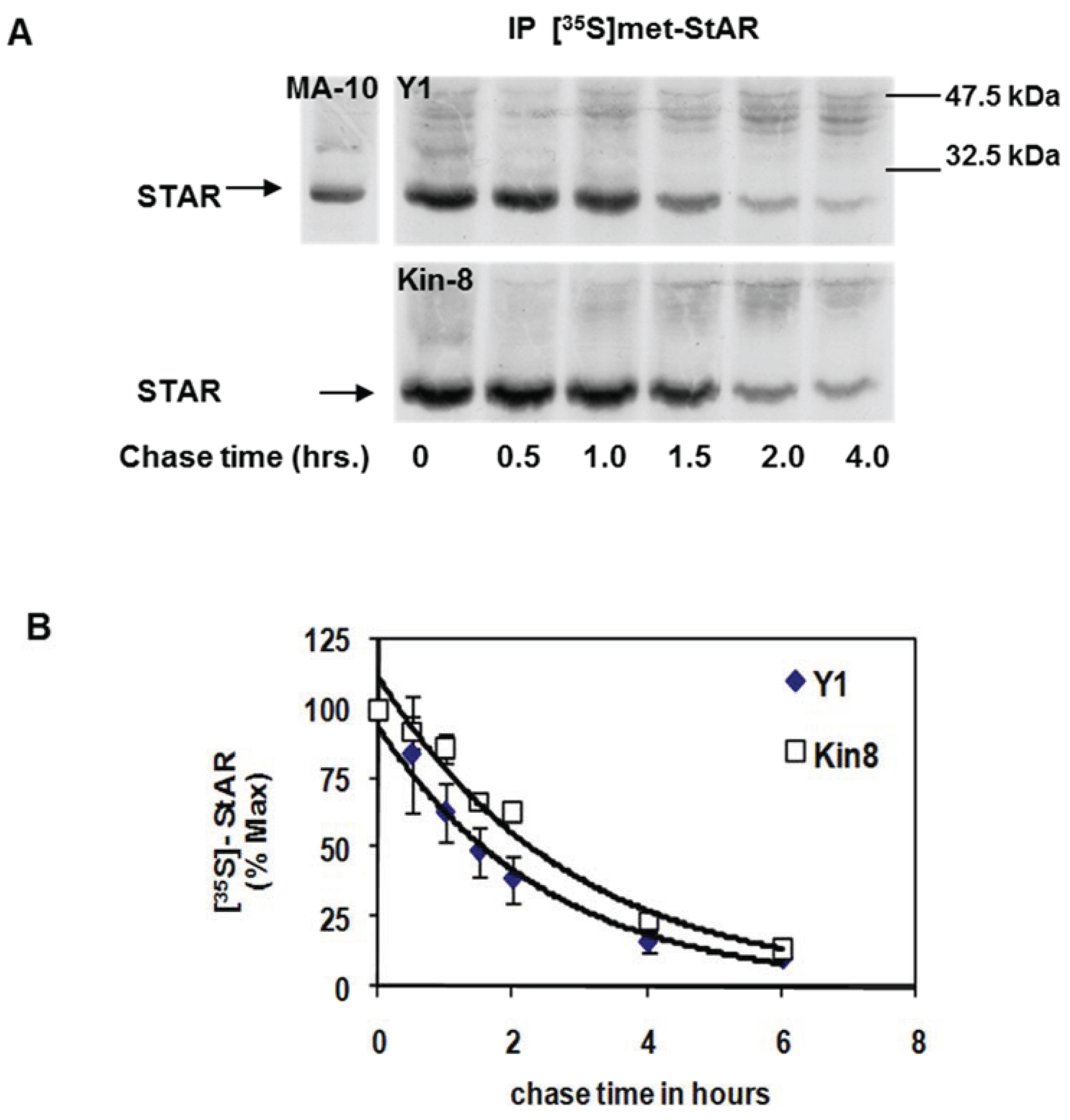
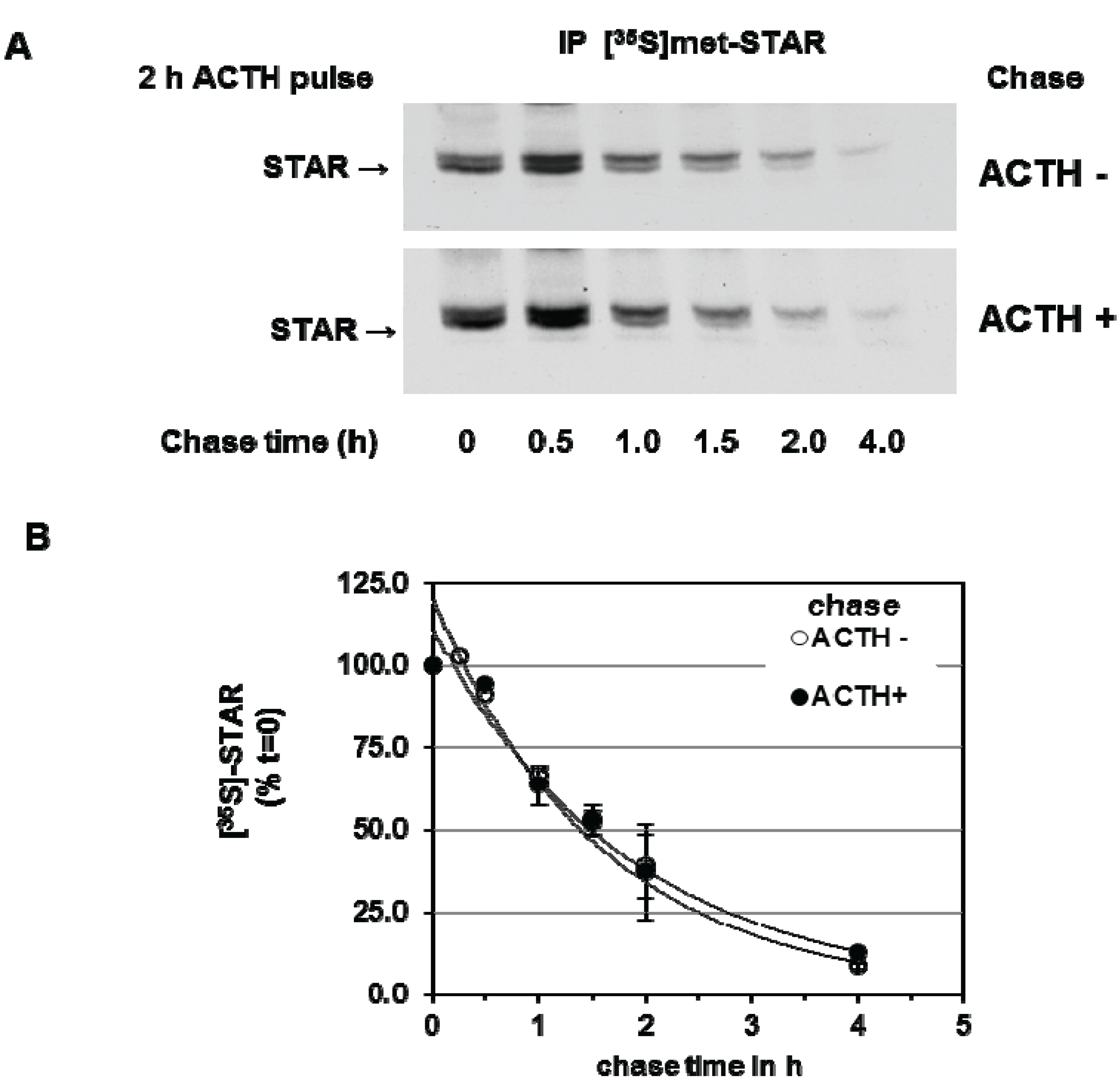
3.2. Precursor STAR Expression in Y1 Cells
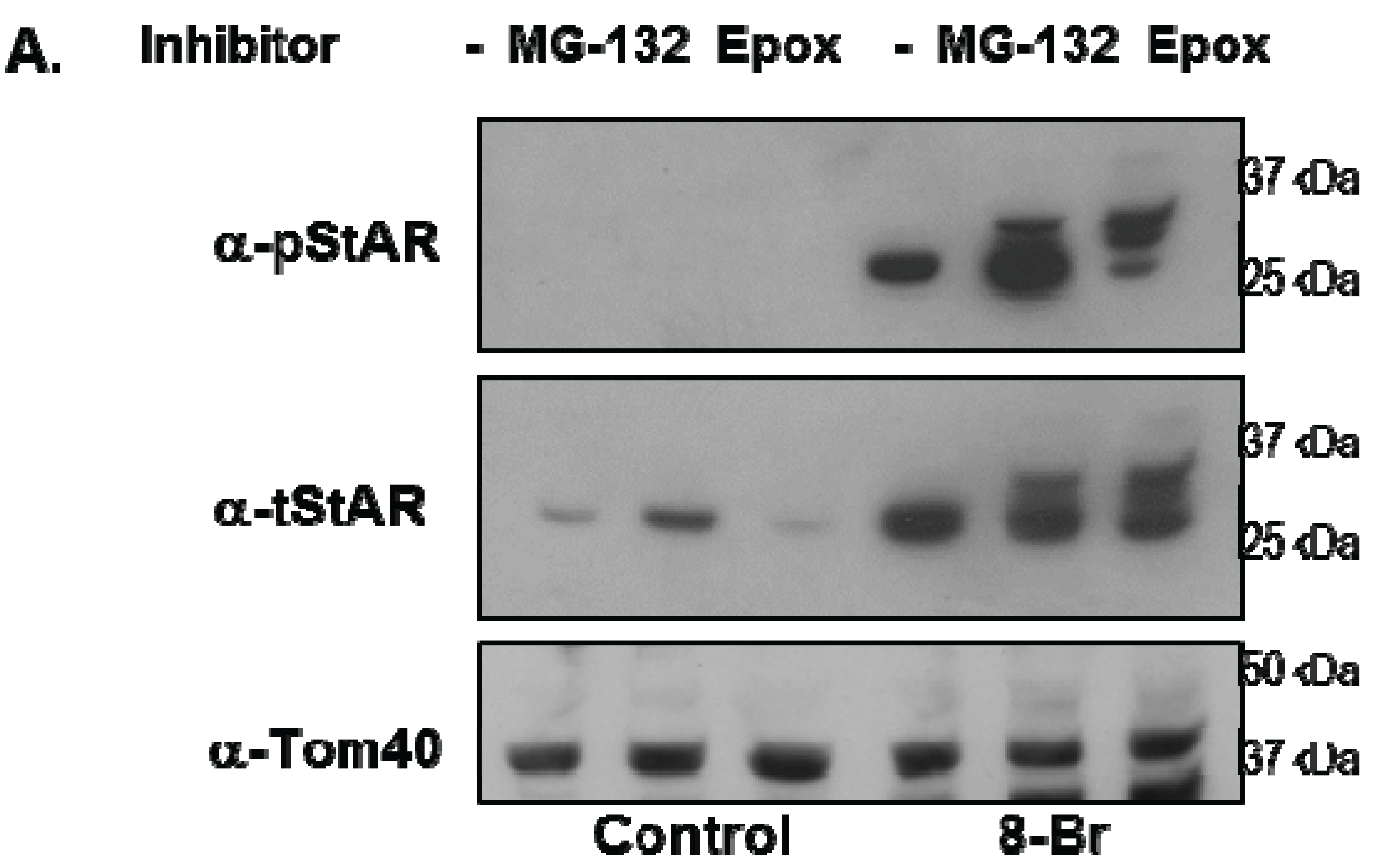
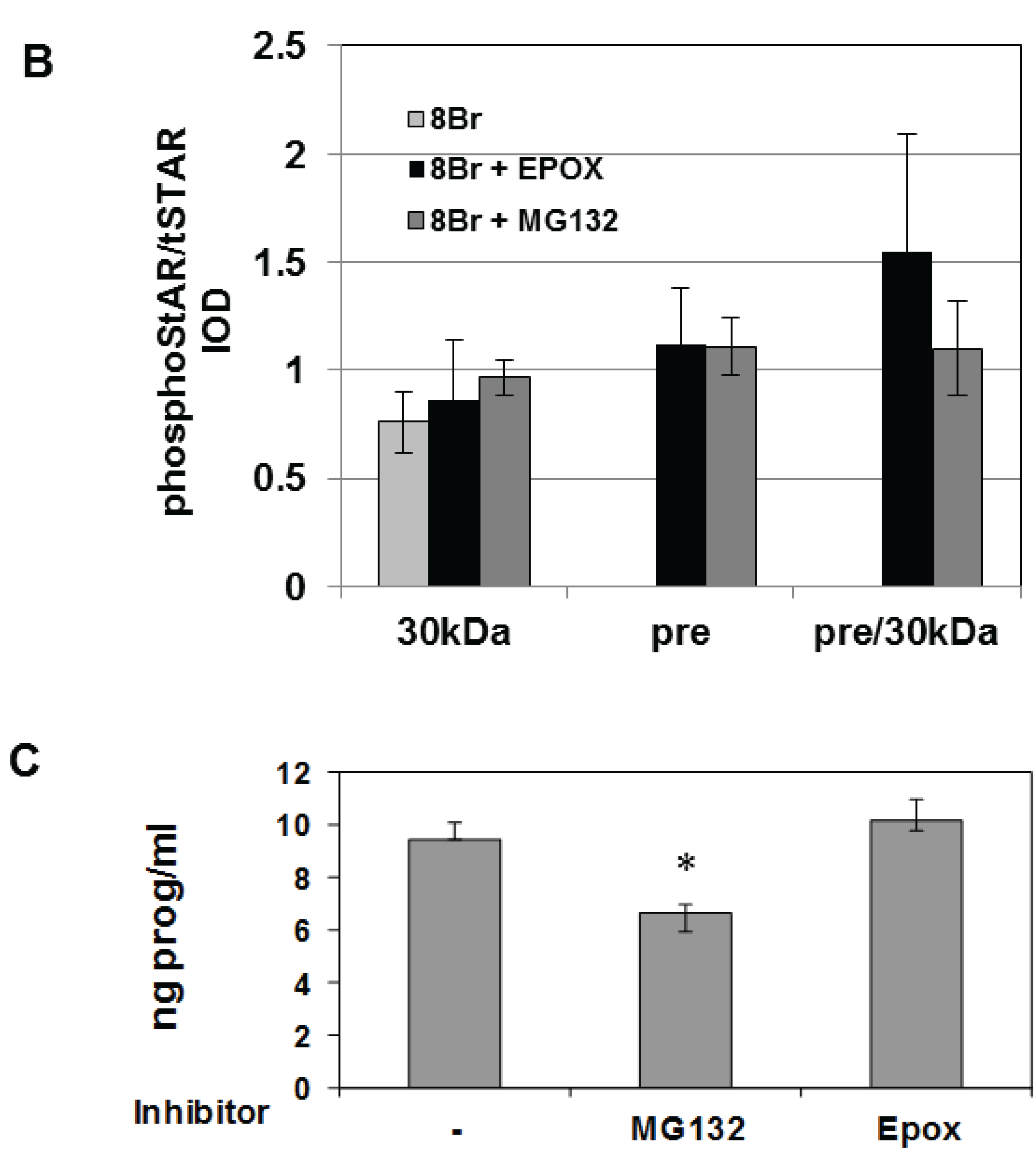
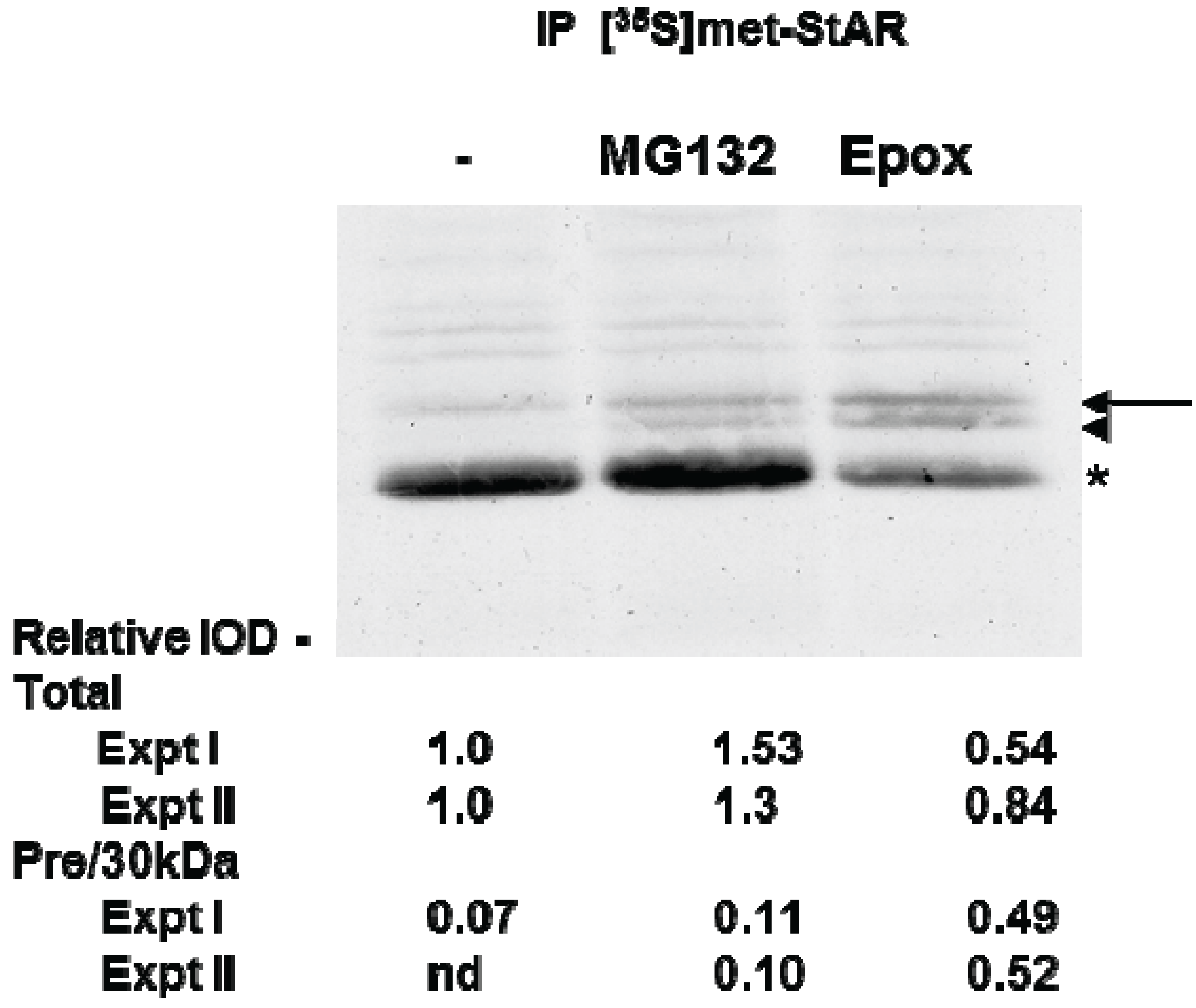
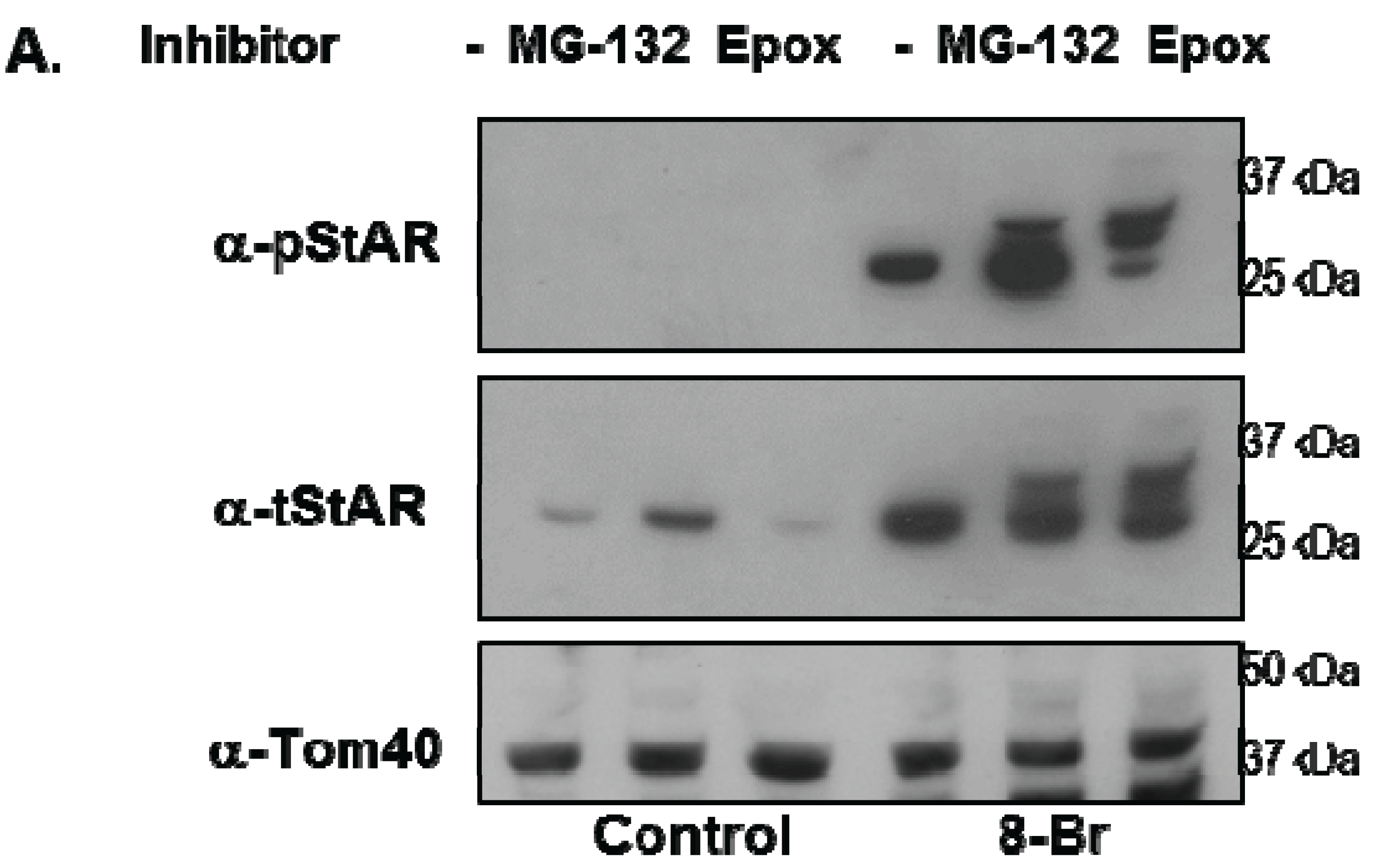
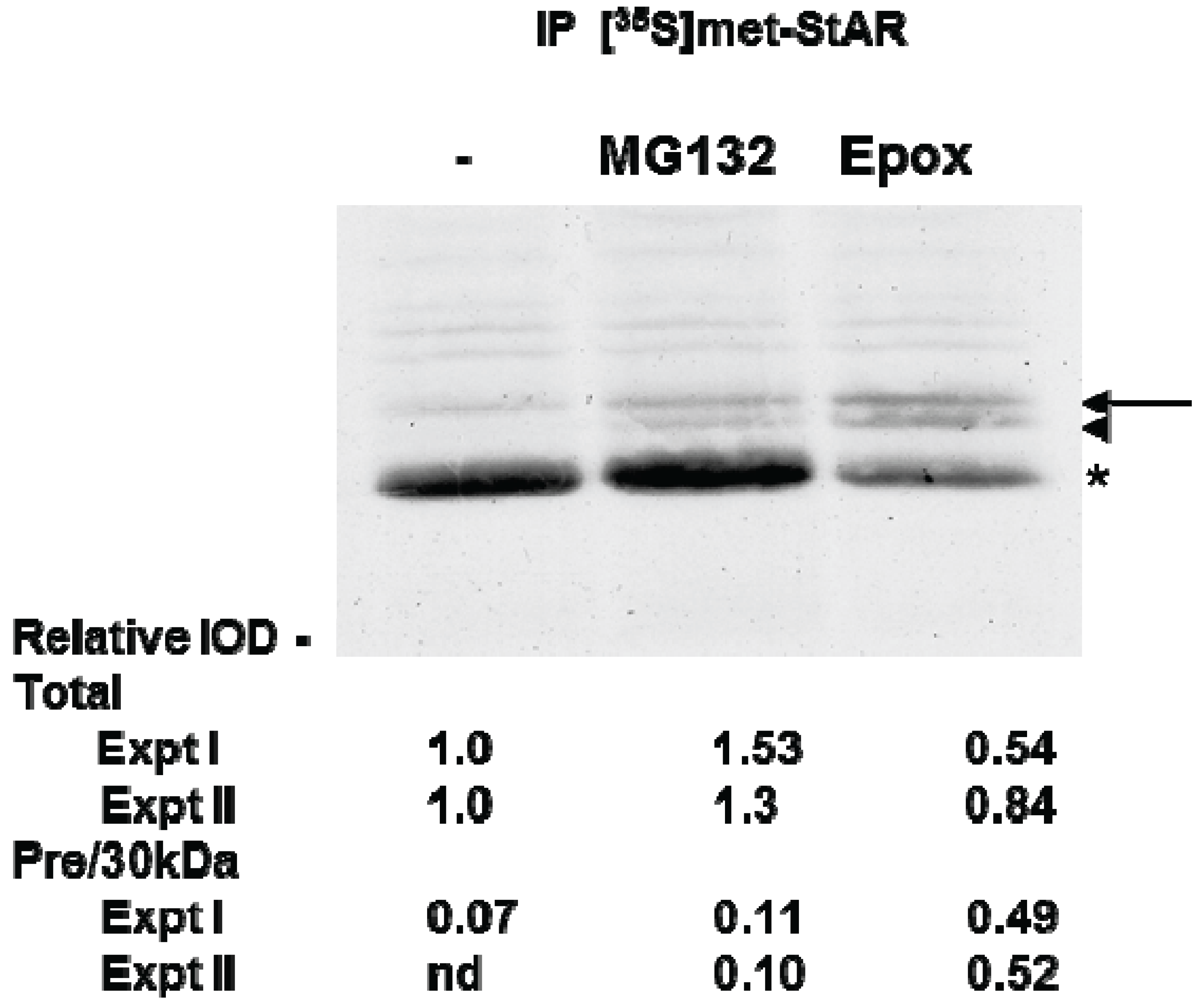
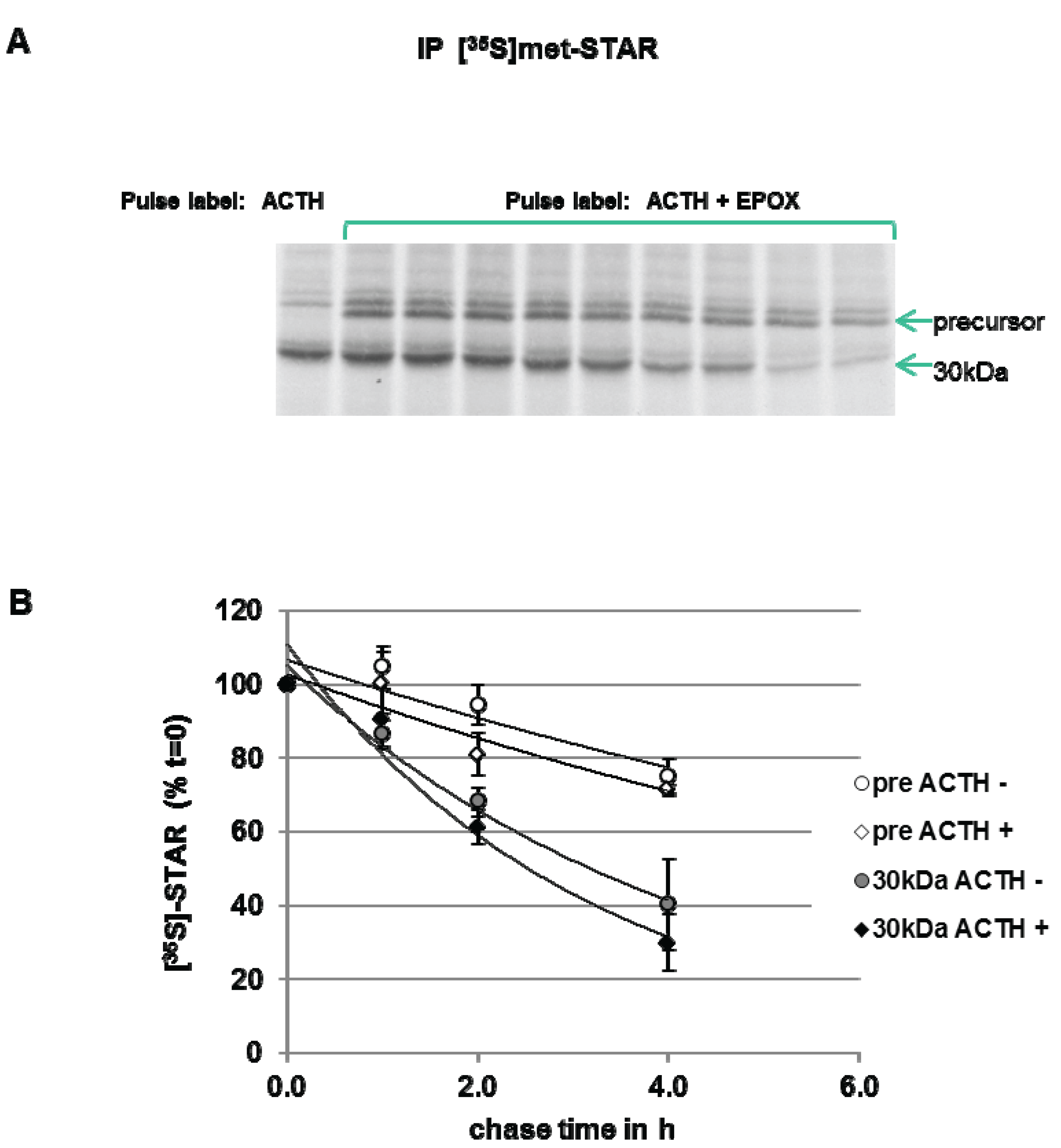
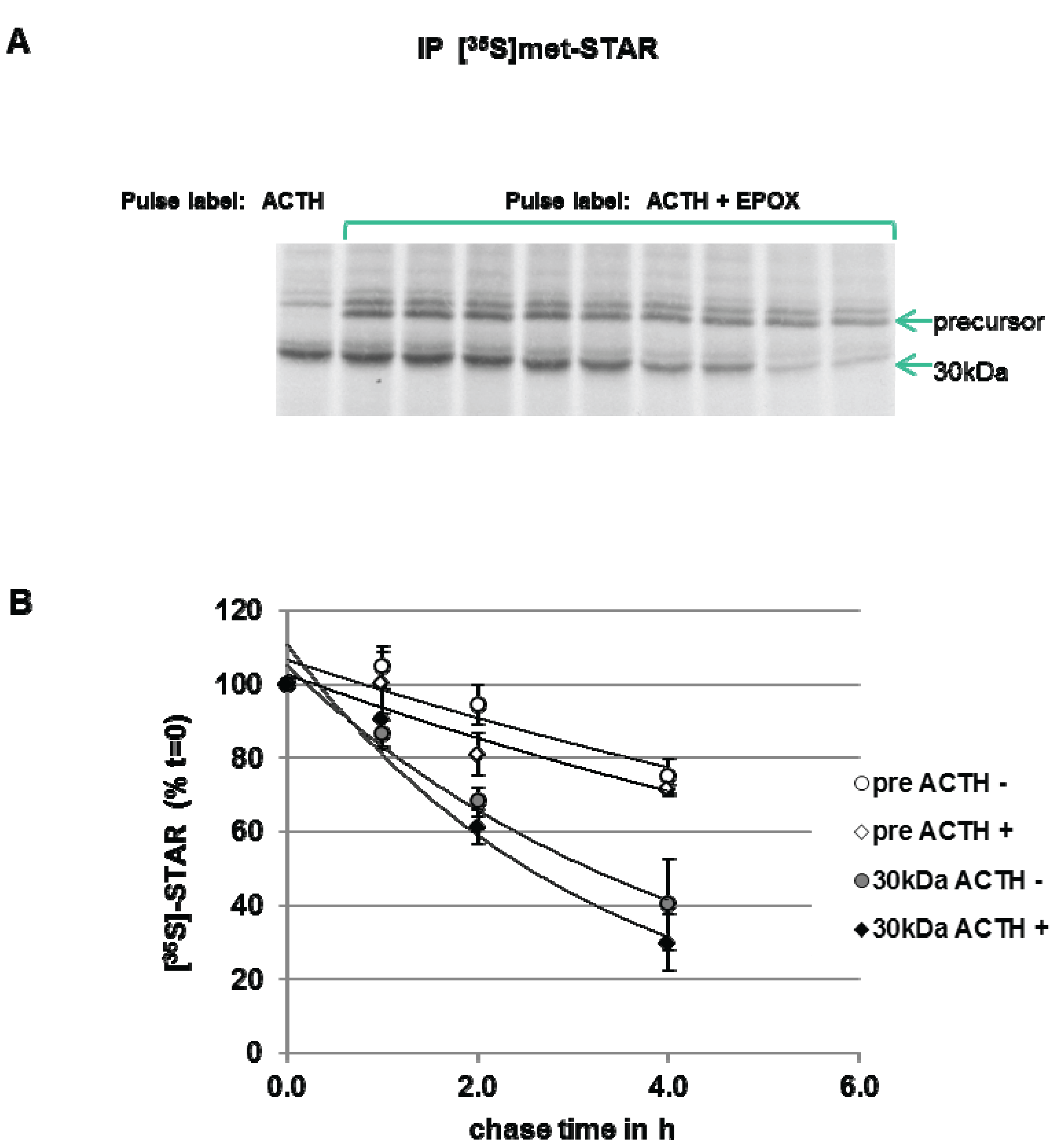
3.3. Precursor STAR Stability in Epox-Treated Y1 Cells
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflict of Interest
References
- Sewer, M.B.; Waterman, M.R. ACTH modulation of transcription factors responsible for steroid hydroxylase gene expression in the adrenal cortex. Microsc. Res. Tech. 2003, 61, 300–307. [Google Scholar]
- Clark, B.J.; Wells, J.; King, S.R.; Stocco, D.M. The purification, cloning, and expression of a novel luteinizing hormone-induced mitochondrial protein in MA-10 mouse leydig tumor cells. Characterization of the steroidogenic acute regulatory protein (StAR). J. Biol. Chem. 1994, 269, 28314–28322. [Google Scholar]
- Lin, D.; Sugawara, T.; Strauss, J.F., 3rd; Clark, B.J.; Stocco, D.M.; Saenger, P.; Rogol, A.; Miller, W.L. Role of steroidogenic acute regulatory protein in Adrenal and Gonadal Steroidogenesis. Science 1995, 267, 1828–1831. [Google Scholar]
- Stocco, D.M. Star protein and the regulation of steroid hormone biosynthesis. Annu. Rev. Physiol. 2001, 63, 193–213. [Google Scholar]
- Arakane, F.; Sugawara, T.; Nishino, H.; Liu, Z.; Holt, J.A.; Pain, D.; Stocco, D.M.; Miller, W.L.; Strauss, J.F., 3rd. Steroidogenic acute regulatory protein (StAR) retains activity in the absence of its mitochondrial import sequence: Implications for the mechanism of star action. Proc. Natl. Acad. Sci. USA 1996, 93, 13731–13736. [Google Scholar]
- Bose, H.S.; Lingappa, V.R.; Miller, W.L. Rapid regulation of steroidogenesis by mitochondrial protein import. Nature 2002, 417, 87–91. [Google Scholar]
- Miller, W.L.; Bose, H.S. Early steps in steroidogenesis: Intracellular cholesterol trafficking: Thematic review series: Genetics of human lipid diseases. J. Lipid Res. 2011, 52, 2111–2135. [Google Scholar]
- Jefcoate, C. High-Flux Mitochondrial cholesterol trafficking, a specialized function of the adrenal cortex. J. Clin. Investig. 2002, 110, 881–890. [Google Scholar]
- Mathieu, A.P.; Fleury, A.; Ducharme, L.; Lavigne, P.; LeHoux, J.G. Insights into steroidogenic acute regulatory protein (StAR)-dependent cholesterol transfer in mitochondria: Evidence from molecular modeling and structure-based thermodynamics supporting the existence of partially unfolded states of StAR. J. Mol. Endocrinol. 2002, 29, 327–345. [Google Scholar]
- Manna, P.R.; Dyson, M.T.; Stocco, D.M. Regulation of the steroidogenic acute regulatory protein gene expression: Present and future perspectives. Mol. Hum. Reprod. 2009, 15, 321–333. [Google Scholar]
- Arakane, F.; King, S.R.; Du, Y.; Kallen, C.B.; Walsh, L.P.; Watari, H.; Stocco, D.M.; Strauss, J.F., 3rd. Phosphorylation of steroidogenic acute regulatory protein (StAR) modulates its steroidogenic activity. J. Biol. Chem. 1997, 272, 32656–32662. [Google Scholar]
- Fleury, A.; Mathieu, A.P.; Ducharme, L.; Hales, D.B.; LeHoux, J.-G. Phosphorylation and function of the hamster adrenal steroidogenic acute regulatory protein (StAR). J. Steroid Biochem. Mol. Biol. 2004, 91, 259–271. [Google Scholar]
- Jo, Y.; Stocco, D.M. Regulation of steroidogenesis and steroidogenic acute regulatory protein in R2C cells by Dax-1 (dosage-sensitive sex reversal, adrenal hypoplasia congenita, critical region on the X chromosome, gene-1). Endocrinology 2004, 145, 5629–5637. [Google Scholar]
- Poderoso, C.; Maloberti, P.; Duarte, A.; Neuman, I.; Paz, C.; Maciel, F.C.; Podesta, E.J. Hormonal activation of a kinase cascade localized at the mitochondria is required for StAR protein activity. Mol. Cell. Endocrinol. 2009, 300, 37–42. [Google Scholar]
- Sasaki, G.; Zubair, M.; Ishii, T.; Mitsui, T.; Hasegawa, T.; Auchus, R.J. The contribution of Serine 194 phosphorylation to steroidogenic acute regulatory protein function. Mol. Endocrinol. 2014, 28, 1088–1096. [Google Scholar]
- Dyson, M.T.; Jones, J.K.; Kowalewski, M.P.; Manna, P.R.; Alonso, M.; Gottesman, M.E.; Stocco, D.M. Mitochondrial a-kinase anchoring protein 121 binds type II protein kinase aand enhances steroidogenic acute regulatory protein-mediated steroidogenesis in MA-10 mouse leydig tumor cells. Biol. Reprod. 2008, 78, 267–277. [Google Scholar]
- Dyson, M.T.; Kowalewski, M.P.; Manna, P.R.; Stocco, D.M. The differential regulation of steroidogenic acute regulatory protein-mediated steroidogenesis by type I and type II PKA in MA-10 Cells. Mol. Cell. Endocrinol. 2009, 300, 94–103. [Google Scholar]
- Grozdanov, P.N.; Stocco, D.M. Short RNA molecules with high binding affinity to the KH motif of A-kinase anchoring protein 1 (AKAP1): Implications for the regulation of steroidogenesis. Mol. Endocrinol. 2012, 26, 2104–2117. [Google Scholar]
- Granot, Z.; Geiss-Friedlander, R.; Melamed-Book, N.; Eimerl, S.; Timberg, R.; Weiss, A.M.; Hales, K.H.; Hales, D.B.; Stocco, D.M.; Orly, J. Proteolysis of normal and mutated steroidogenic acute regulatory proteins in the mitochondria: The fate of unwanted proteins. Mol. Endocrinol. 2003, 17, 2461–2476. [Google Scholar]
- Granot, Z.; Kobiler, O.; Melamed-Book, N.; Eimerl, S.; Bahat, A.; Lu, B.; Braun, S.; Maurizi, M.R.; Suzuki, C.K.; Oppenheim, A.B.; et al. Turnover of mitochondrial steroidogenic acute regulatory (StAR) protein by lon protease: The unexpected effect of proteasome inhibitors. Mol. Endocrinol. 2007, 21, 2164–2177. [Google Scholar]
- Artemenko, I.P.; Zhao, D.; Hales, D.B.; Hales, K.H.; Jefcoate, C.R. Mitochondrial processing of newly synthesized steroidogenic acute regulatory protein (StAR), but not total StAR, mediates cholesterol transfer to cytochrome P450 side chain cleavage enzyme in adrenal cells. J. Biol. Chem. 2001, 276, 46583–46596. [Google Scholar]
- Epstein, L.F.; Orme-Johnson, N.R. Regulation of steroid hormone biosynthesis. Identification of precursors of a phosphoprotein targeted to the mitochondrion in stimulated rat adrenal cortex cells. J. Biol. Chem. 1991, 266, 19739–19745. [Google Scholar]
- Allen, J.A.; Shankara, T.; Janus, P.; Buck, S.; Diemer, T.; Hales, K.H.; Hales, D.B. Energized, polarized, and actively respiring mitochondria are required for acute leydig cell steroidogenesis. Endocrinology 2006, 147, 3924–3935. [Google Scholar]
- King, S.R.; Liu, Z.; Soh, J.; Eimerl, S.; Orly, J.; Stocco, D.M. Effects of disruption of the mitochondrial electrochemical gradient on steroidogenesis and the steroidogenic acute regulatory (StAR) protein. J. Steroid Biochem. Mol. Biol. 1999, 69, 143–154. [Google Scholar]
- Tajima, K.; Babich, S.; Yoshida, Y.; Dantes, A.; Strauss, J.F., 3rd; Amsterdam, A. The proteasome inhibitor MG132 promotes accumulation of the steroidogenic acute regulatory protein (StAR) and steroidogenesis. FEBS Lett. 2001, 490, 59–64. [Google Scholar]
- Rae, P.A.; Gutmann, N.S.; Tsao, J.; Schimmer, B.P. Mutations in cyclic AMP-dependent protein kinase and corticotropin (ACTH)-sensitive adenylate cyclase affect adrenal steroidogenesis. Proc. Natl. Acad. Sci. USA 1979, 76, 1896–1900. [Google Scholar]
- Olson, M.F.; Krolczyk, A.J.; Gorman, K.B.; Steinberg, R.A.; Schimmer, B.P. Molecular basis for the 3',5'-cyclic adenosine monophosphate resistance of Kin mutant Y1 adrenocortical tumor cells. Mol. Endocrinol. 1993, 7, 477–487. [Google Scholar]
- Wong, M.; Rice, D.A.; Parker, K.L.; Schimmer, B.P. The roles of cAMP and cAMP-dependent protein kinase in the expression of cholesterol side chain cleavage and steroid 11 beta-hydroxylase genes in mouse adrenocortical tumor cells. J. Biol. Chem. 1989, 264, 12867–12871. [Google Scholar]
- Clark, B.J.; Ranganathan, V.; Combs, R. Steroidogenic acute regulatory protein expression is dependent upon post-translational effects of cAMP-dependent protein kinase A. Mol. Cell. Endocrinol. 2001, 173, 183–192. [Google Scholar]
- Clark, B.J.; Combs, R.; Hales, K.H.; Hales, D.B.; Stocco, D.M. Inhibition of transcription affects synthesis of steroidogenic acute regulatory protein and steroidogenesis in MA-10 mouse leydig tumor cells. Endocrinology 1997, 138, 4893–4901. [Google Scholar]
- Jo, Y.; King, S.R.; Khan, S.A.; Stocco, D.M. Involvement of protein kinase C and cyclic adenosine 3',5'-monophosphate-dependent kinase in steroidogenic acute regulatory protein expression and steroid biosynthesis in leydig cells. Biol. Reprod. 2005, 73, 244–255. [Google Scholar]
- Bahat, A.; Perlberg, S.; Melamed-Book, N.; Lauria, I.; Langer, T.; Orly, J. StAR enhances transcription of genes encoding the mitochondrial proteases involved in its own degradation. Mol. Endocrinol. 2014, 28, 208–224. [Google Scholar]
- Alberta, J.A.; Epstein, L.F.; Pon, L.A.; Orme-Johnson, N.R. Mitochondrial localization of a phosphoprotein that rapidly accumulates in adrenal cortex cells exposed to adrenocorticotropic hormone or to cAMP. J. Biol. Chem. 1989, 264, 2368–2372. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clark, B.J.; Hudson, E.A. StAR Protein Stability in Y1 and Kin-8 Mouse Adrenocortical Cells. Biology 2015, 4, 200-215. https://doi.org/10.3390/biology4010200
Clark BJ, Hudson EA. StAR Protein Stability in Y1 and Kin-8 Mouse Adrenocortical Cells. Biology. 2015; 4(1):200-215. https://doi.org/10.3390/biology4010200
Chicago/Turabian StyleClark, Barbara J., and Elizabeth A. Hudson. 2015. "StAR Protein Stability in Y1 and Kin-8 Mouse Adrenocortical Cells" Biology 4, no. 1: 200-215. https://doi.org/10.3390/biology4010200
APA StyleClark, B. J., & Hudson, E. A. (2015). StAR Protein Stability in Y1 and Kin-8 Mouse Adrenocortical Cells. Biology, 4(1), 200-215. https://doi.org/10.3390/biology4010200