

Genome-Wide Identification and Co-Expression Analysis of WRKY Genes Unveil Their Role in Regulating Anthocyanin Accumulation During Euscaphis japonica Fruit Maturation
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of WRKY Transcription Factors in E. japonica
2.2. Phylogenetic Classification
2.3. Gene Structure and Conserved Motif Characterization
2.4. Genomic Distribution and Duplication Events
2.5. Promoter Cis-Element Analysis
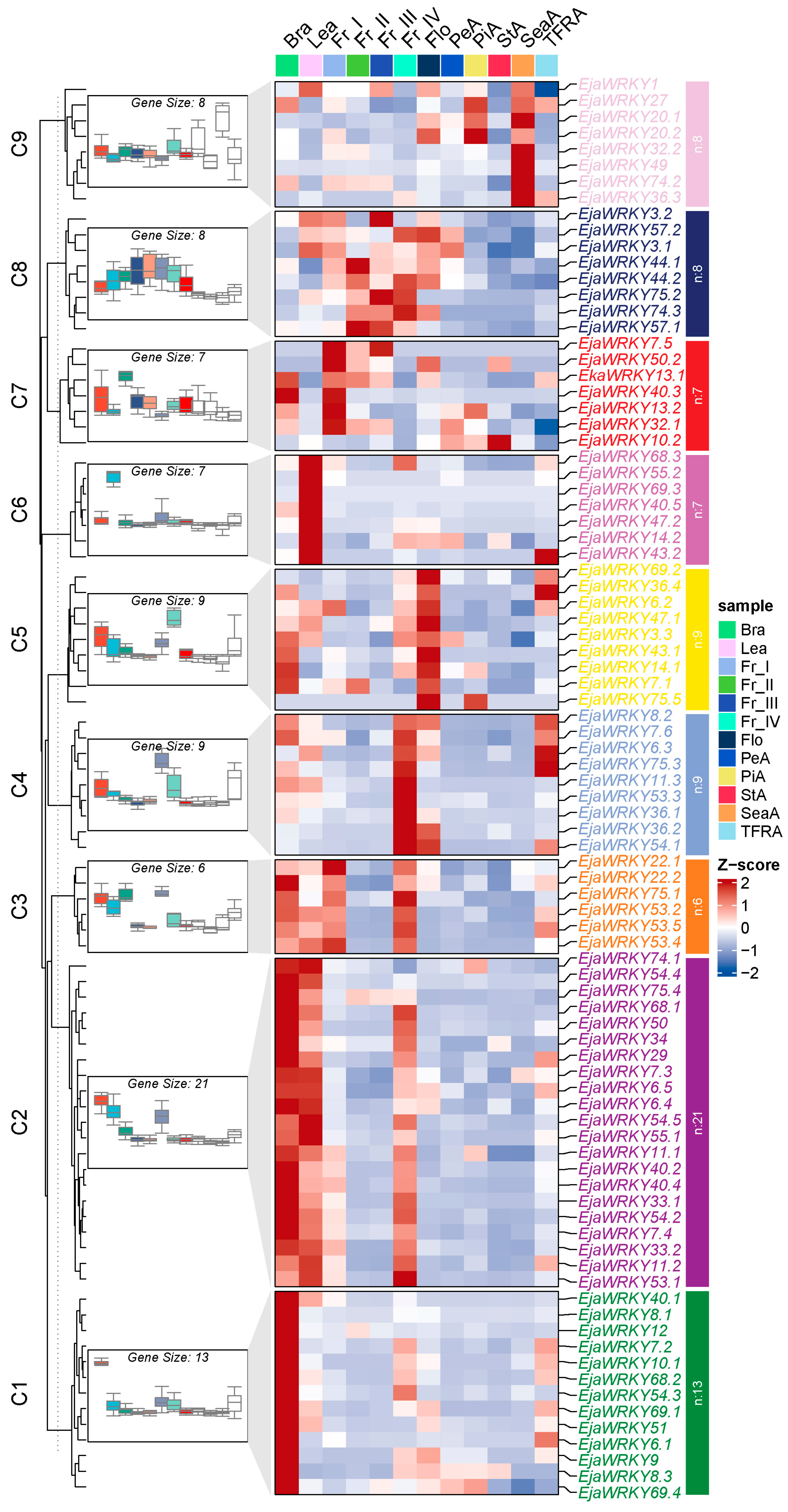
2.6. Expression Profiling
2.7. Correlation Analysis and RT-qPCR Verification
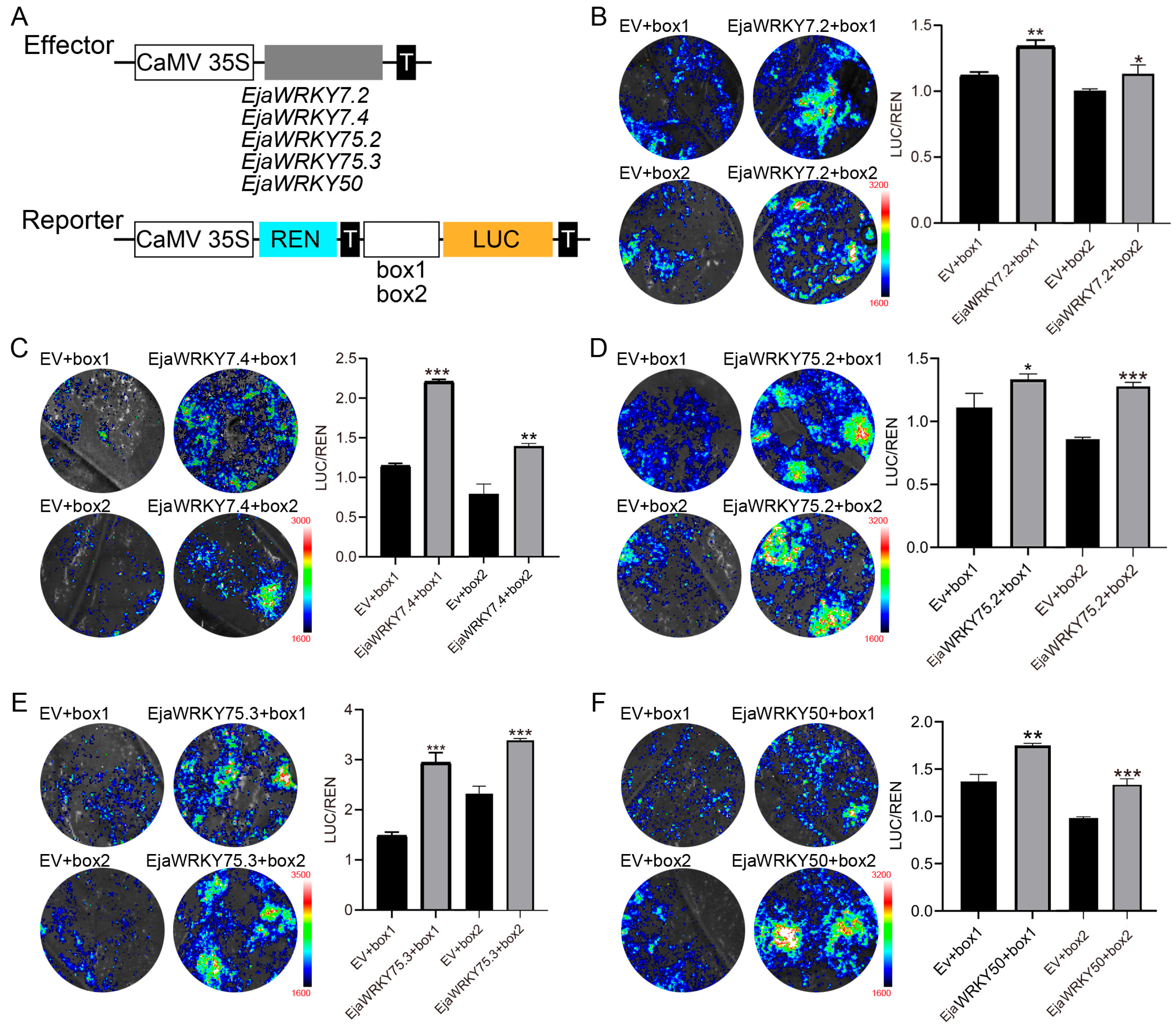
2.8. DNA Binding and Transactivation Assay
3. Results
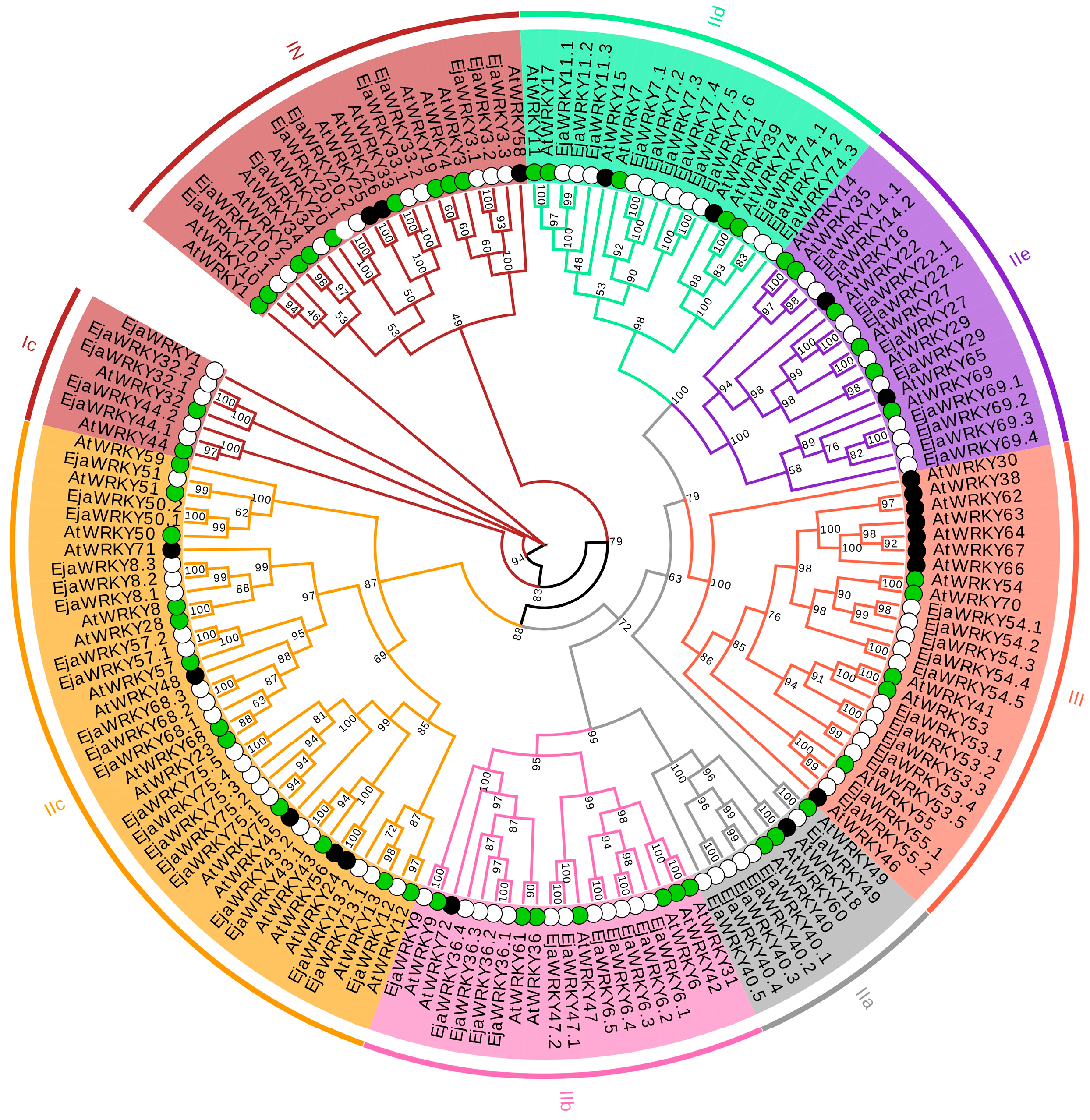
3.1. Identification and Phylogenetic Analysis of the WRKY Family in E. japonica
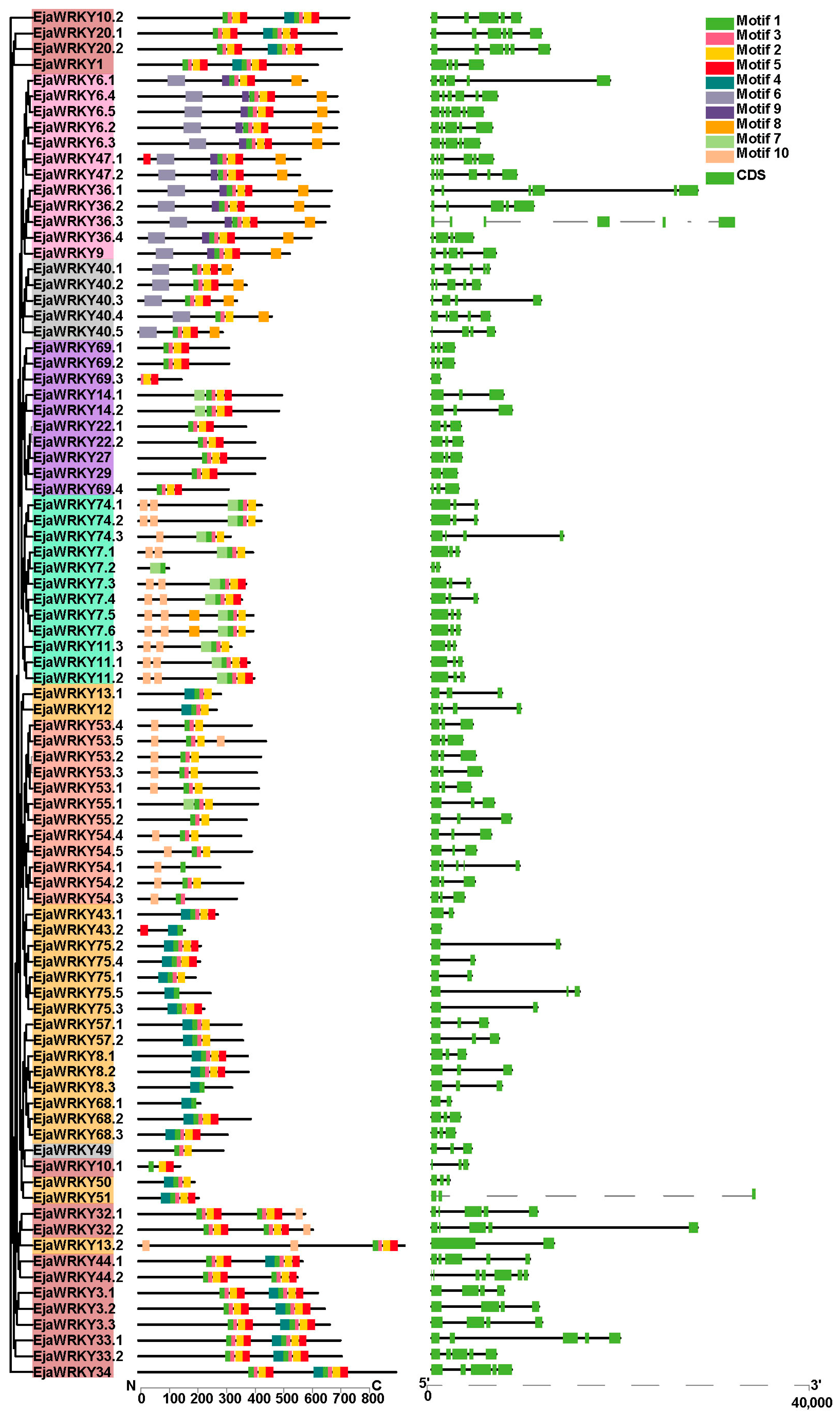
3.2. EjaWRKY Protein Motif Composition and Corresponding Gene Structure Analysis
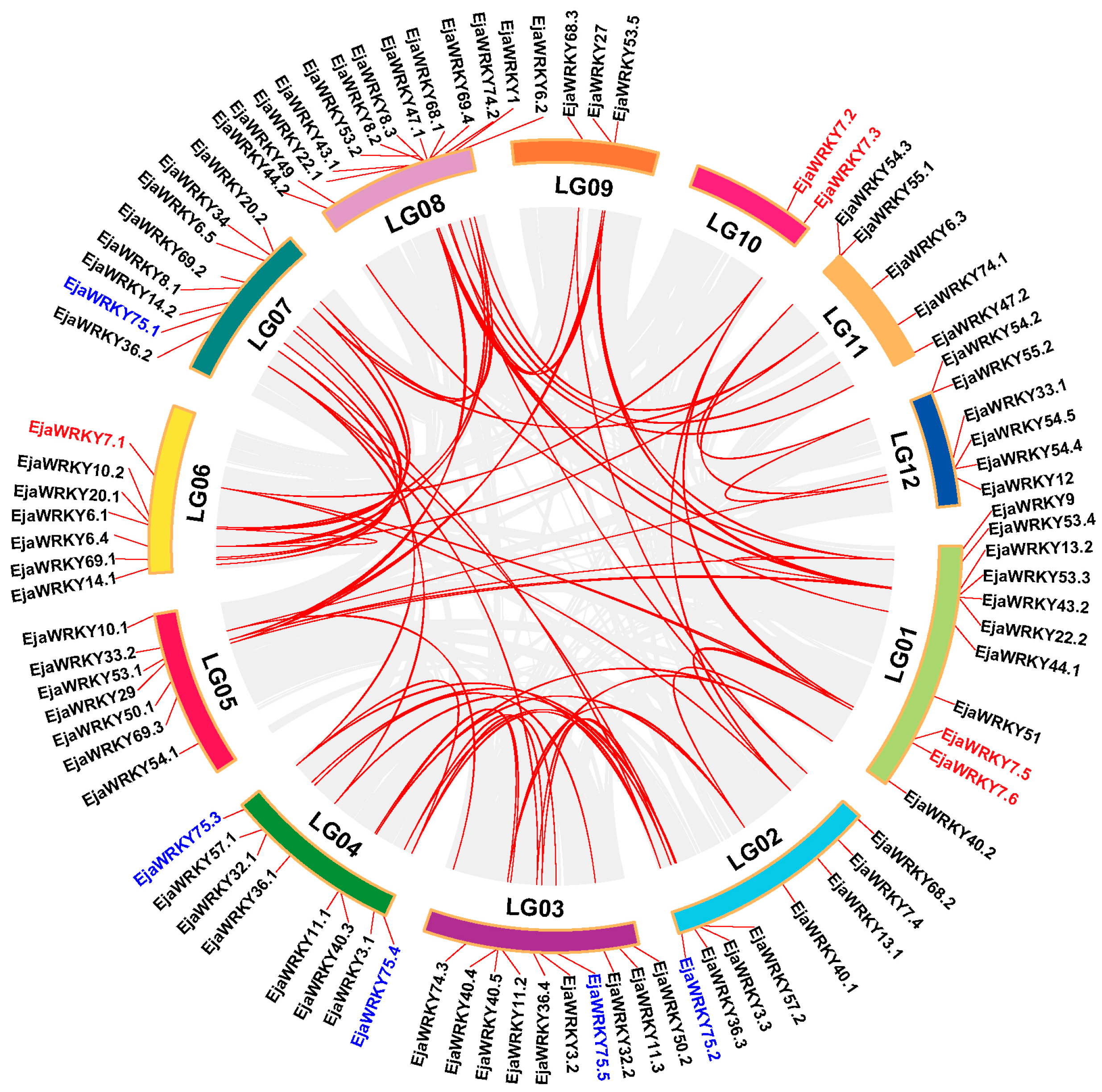
3.3. Chromosomal Organization of EjaWRKY Genes
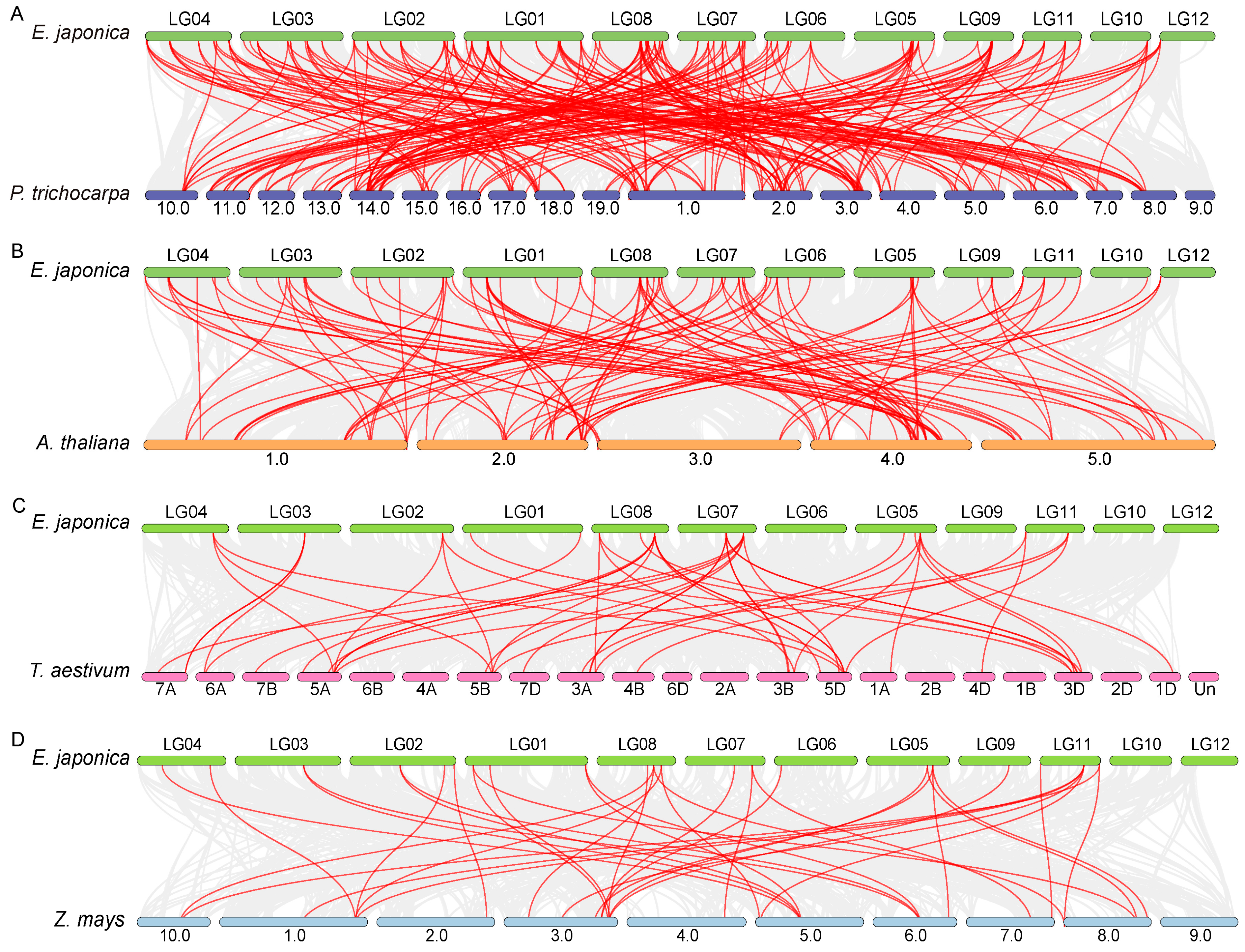
3.4. Comparative Synteny Analysis of EjaWRKY Genes
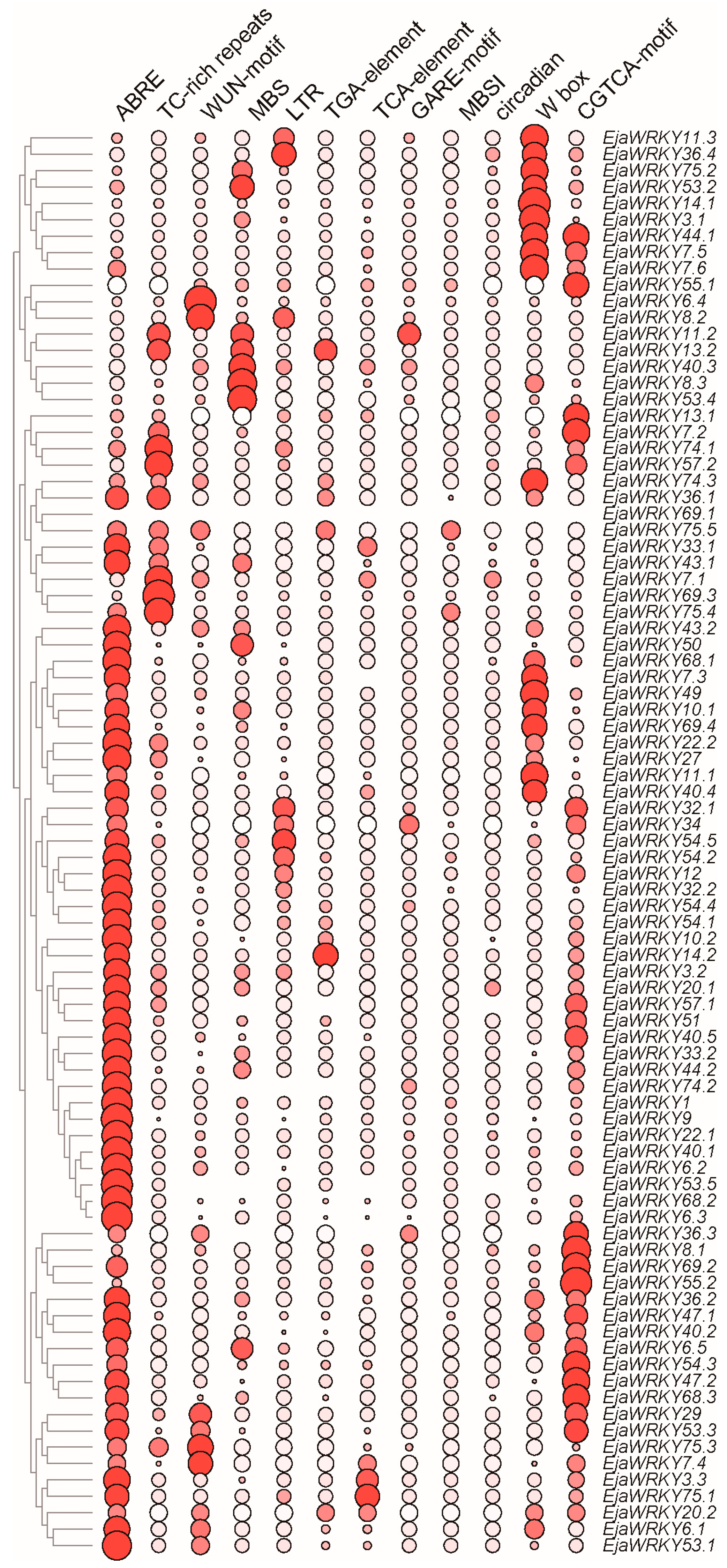
3.5. Cis-Regulatory Element Analysis of EjaWRKY Promoters
Spatial Expression Patterns of EjaWRKY Genes
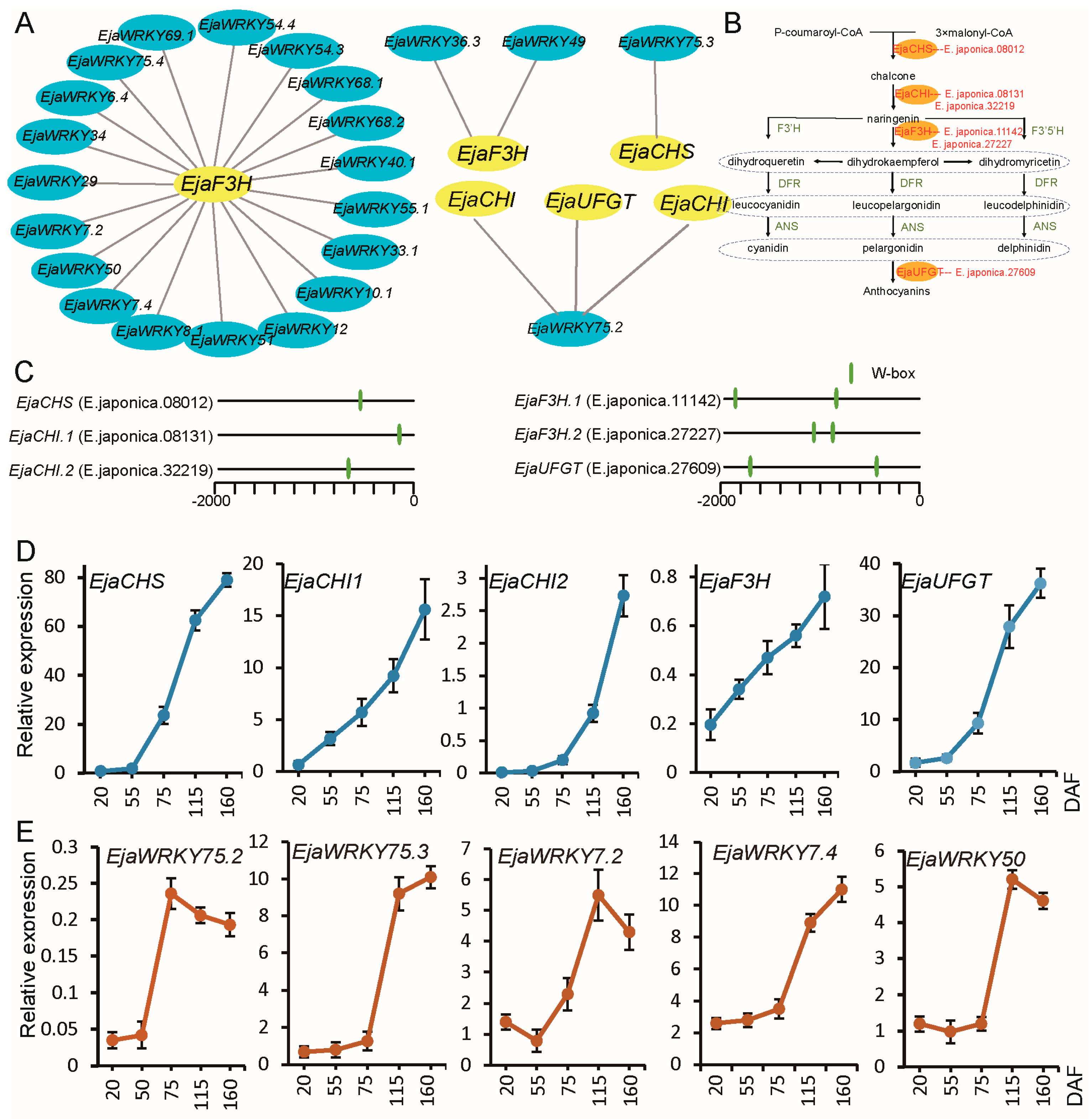
3.6. Association Analysis Between EjaWRKY Genes and Anthocyanin Biosynthesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andersen, Ø.M.; Jordheim, M.; Byamukama, R.; Mbabazi, A.; Ogweng, G.; Skaar, I.; Kiremire, B. Anthocyanins with unusual furanose sugar (apiose) from leaves of Synadenium grantii (Euphorbiaceae). Phytochemistry 2010, 71, 1558–1563. [Google Scholar] [CrossRef] [PubMed]
- Alappat, B.; Alappat, J. Anthocyanin Pigments: Beyond Aesthetics. Molecules 2020, 25, 5500. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.; Tattini, M.; Gould, K.S. Multiple functional roles of anthocyanins in plant-environment interactions. Environ. Exp. Bot. 2015, 119, 4–17. [Google Scholar] [CrossRef]
- Glover, B.J.; Martin, C. Anthocyanins. Curr. Biol. CB 2012, 22, R147–R150. [Google Scholar] [CrossRef]
- Agarwal, P.; Holland, T.M.; Wang, Y.; Bennett, D.A.; Morris, M.C. Association of Strawberries and Anthocyanidin Intake with Alzheimer’s Dementia Risk. Nutrients 2019, 11, 3060. [Google Scholar] [CrossRef]
- Hughes, N.M.; Morley, C.B.; Smith, W.K. Coordination of anthocyanin decline and photosynthetic maturation in juvenile leaves of three deciduous tree species. New Phytol. 2007, 175, 675–685. [Google Scholar] [CrossRef]
- Mekapogu, M.; Vasamsetti, B.M.K.; Kwon, O.K.; Ahn, M.S.; Lim, S.H.; Jung, J.A. Anthocyanins in Floral Colors: Biosynthesis and Regulation in Chrysanthemum Flowers. Int. J. Mol. Sci. 2020, 21, 6537. [Google Scholar] [CrossRef]
- Ushimaru, A.; Watanabe, T.; Nakata, K. Colored floral organs influence pollinator behavior and pollen transfer in Commelina communis (Commelinaceae). Am. J. Bot. 2007, 94, 249–258. [Google Scholar] [CrossRef]
- Liu, B.; Yang, Q.; Xin, G.L.; Wang, X.; Zhang, L.; He, D.; Zhang, S.; Pan, Y.; Zou, S.Q.; Zhang, J.; et al. A comprehensive proteomic map revealing the regulation of the development of long-duration, red butterfly-shaped fruit in Euscaphis japonica. Int. J. Biol. Macromol. 2025, 292, 139061. [Google Scholar] [CrossRef]
- Steyn, W.J.; Wand, S.J.; Jacobs, G.; Rosecrance, R.C.; Roberts, S.C. Evidence for a photoprotective function of low-temperature-induced anthocyanin accumulation in apple and pear peel. Physiol. Plant. 2009, 136, 461–472. [Google Scholar] [CrossRef]
- Boo, H.O.; Heo, B.G.; Gorinstein, S.; Chon, S.U. Positive effects of temperature and growth conditions on enzymatic and antioxidant status in lettuce plants. Plant Sci. Int. J. Exp. Plant Biol. 2011, 181, 479–484. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Q.; Escalona Contreras, V.H.; Huang, T.; Jiang, H.; Song, B.; Duan, Z.; Li, Y.; Yang, X.; Song, H.; et al. Short-term high-light intensity and low temperature improve the quality and flavor of lettuce grown in plant factory. J. Sci. Food Agric. 2024, 104, 9046–9055. [Google Scholar] [CrossRef]
- Xu, Z.; Rothstein, S.J. ROS-Induced anthocyanin production provides feedback protection by scavenging ROS and maintaining photosynthetic capacity in Arabidopsis. Plant Signal. Behav. 2018, 13, e1451708. [Google Scholar] [CrossRef]
- Chalker-Scott, L. Environmental Significance of Anthocyanins in Plant Stress Responses. Photochem. Photobiol. 1999, 70, 1–9. [Google Scholar] [CrossRef]
- LaFountain, A.M.; Yuan, Y.W. Repressors of anthocyanin biosynthesis. New Phytol. 2021, 231, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ma, S.; Ye, N.; Jiang, M.; Cao, J.; Zhang, J. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Bakshi, M.; Oelmüller, R. WRKY transcription factors: Jack of many trades in plants. Plant Signal. Behav. 2014, 9, e27700. [Google Scholar] [CrossRef]
- Lloyd, A.; Brockman, A.; Aguirre, L.; Campbell, A.; Bean, A.; Cantero, A.; Gonzalez, A. Advances in the MYB-bHLH-WD Repeat (MBW) Pigment Regulatory Model: Addition of a WRKY Factor and Co-option of an Anthocyanin MYB for Betalain Regulation. Plant Cell Physiol. 2017, 58, 1431–1441. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Y.; Yu, L.; Jiang, H.; Guo, Z.; Xu, H.; Jiang, S.; Fang, H.; Zhang, J.; Su, M.; et al. MdWRKY11 Participates in Anthocyanin Accumulation in Red-Fleshed Apples by Affecting MYB Transcription Factors and the Photoresponse Factor MdHY5. J. Agric. Food Chem. 2019, 67, 8783–8793. [Google Scholar] [CrossRef]
- Alabd, A.; Ahmad, M.; Zhang, X.; Gao, Y.; Peng, L.; Zhang, L.; Ni, J.; Bai, S.; Teng, Y. Light-responsive transcription factor PpWRKY44 induces anthocyanin accumulation by regulating PpMYB10 expression in pear. Hortic. Res. 2022, 9, uhac199. [Google Scholar] [CrossRef]
- Wang, N.; Song, G.; Zhang, F.; Shu, X.; Cheng, G.; Zhuang, W.; Wang, T.; Li, Y.; Wang, Z. Characterization of the WRKY Gene Family Related to Anthocyanin Biosynthesis and the Regulation Mechanism under Drought Stress and Methyl Jasmonate Treatment in Lycoris radiata. Int. J. Mol. Sci. 2023, 24, 2423. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Qu, Y.; Sha, G.; Zhang, S.; Ma, Y.; Chen, M.; Zhai, R.; Yang, C.; Xu, L.; Wang, Z. PbWRKY75 promotes anthocyanin synthesis by activating PbDFR, PbUFGT, and PbMYB10b in pear. Physiol. Plant. 2021, 173, 1841–1849. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Wang, J.; Gao, C.; Jin, C.; Li, D.; Peng, D.; Du, G.; Li, Y.; Chen, M. Functional characterization of a heterologously expressed Brassica napus WRKY41-1 transcription factor in regulating anthocyanin biosynthesis in Arabidopsis thaliana. Plant Sci. Int. J. Exp. Plant Biol. 2018, 268, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.; Cavallini, E.; Walker, A.R.; Pezzotti, M.; Bliek, M.; Quattrocchio, F.; Koes, R.; Ruperti, B.; Bertini, E.; Zenoni, S.; et al. The MYB5-driven MBW complex recruits a WRKY factor to enhance the expression of targets involved in vacuolar hyper-acidification and trafficking in grapevine. Plant J. Cell Mol. Biol. 2019, 99, 1220–1241. [Google Scholar] [CrossRef]
- Yue, M.; Jiang, L.; Zhang, N.; Zhang, L.; Liu, Y.; Wang, Y.; Li, M.; Lin, Y.; Zhang, Y.; Zhang, Y.; et al. Importance of FaWRKY71 in Strawberry (Fragaria × ananassa) Fruit Ripening. Int. J. Mol. Sci. 2022, 23, 12483. [Google Scholar] [CrossRef]
- Ishiguro, S.; Nakamura, K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and beta-amylase from sweet potato. Mol. Gen. Genet. MGG 1994, 244, 563–571. [Google Scholar] [CrossRef]
- Wu, K.L.; Guo, Z.J.; Wang, H.H.; Li, J. The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2005, 12, 9–26. [Google Scholar] [CrossRef]
- Xie, T.; Chen, C.; Li, C.; Liu, J.; Liu, C.; He, Y. Genome-wide investigation of WRKY gene family in pineapple: Evolution and expression profiles during development and stress. BMC Genom. 2018, 19, 490. [Google Scholar] [CrossRef]
- Li, J.; Islam, F.; Huang, Q.; Wang, J.; Zhou, W.; Xu, L.; Yang, C. Genome-wide characterization of WRKY gene family in Helianthus annuus L. and their expression profiles under biotic and abiotic stresses. PLoS ONE 2020, 15, e0241965. [Google Scholar] [CrossRef]
- Yuan, X.; Sun, W.; Zou, X.; Liu, B.; Huang, W.; Chen, Z.; Li, Y.; Qiu, M.Y.; Liu, Z.J.; Mao, Y.; et al. Sequencing of Euscaphis konishii Endocarp Transcriptome Points to Molecular Mechanisms of Endocarp Coloration. Int. J. Mol. Sci. 2018, 19, 3209. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.H.; Li, Z.; Xiang, S.; Ni, L.; Zhang, D.; Chen, D.Q.; Qiu, M.Y.; Zhang, Q.G.; Xiao, L.; Din, L.; et al. The Euscaphis japonica genome and the evolution of malvids. Plant J. Cell Mol. Biol. 2021, 108, 1382–1399. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat. Commun. 2022, 13, 6968. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Subramanian, B.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.H. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019, 47, W270–W275. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Ni, L.; Carballar-Lejarazú, R.; Zou, X.; Sun, W.; Wu, L.; Yuan, X.; Mao, Y.; Huang, W.; Zou, S. Comparative transcriptome among Euscaphis konishii Hayata tissues and analysis of genes involved in flavonoid biosynthesis and accumulation. BMC Genom. 2019, 20, 24. [Google Scholar] [CrossRef]
- Zhang, J. ClusterGVis: One-Step to Cluster and Visualize Gene Expression Matrix. 2022. Available online: https://cran.r-project.org/web/packages/ClusterGVis/index.html (accessed on 4 January 2024). [CrossRef]
- Liu, B.; Zhu, J.; Lin, L.; Yang, Q.; Hu, B.; Wang, Q.; Zou, X.X.; Zou, S.Q. Genome-Wide Identification and Co-Expression Analysis of ARF and IAA Family Genes in Euscaphis konishii: Potential Regulators of Triterpenoids and Anthocyanin Biosynthesis. Front. Genet. 2022, 12, 737293. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing, Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 11 May 2022).
- Prion, S.; Haerling, K.A. Making Sense of Methods and Measurement: Pearson Product-Moment Correlation Coefficient. Clin. Simul. Nurs. 2014, 10, 587–588. [Google Scholar] [CrossRef]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, J.; Yang, Z.; Matsui, A.; Seki, M.; Li, S.; Yan, X.; Kohnen, M.V.; Gu, L.; Prasad, K.; et al. PtWOX11 acts as master regulator conducting the expression of key transcription factors to induce de novo shoot organogenesis in poplar. Plant Mol. Biol. 2018, 98, 389–406. [Google Scholar] [CrossRef]
- Zuntini, A.R.; Carruthers, T.; Maurin, O.; Bailey, P.C.; Leempoel, K.; Brewer, G.E.; Epitawalage, N.; Françoso, E.; Gallego-Paramo, B.; McGinnie, C.; et al. Phylogenomics and the rise of the angiosperms. Nature 2024, 629, 843–850. [Google Scholar] [CrossRef]
- Yin, G.; Xu, H.; Xiao, S.; Qin, Y.; Li, Y.; Yan, Y.; Hu, Y. The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol. 2013, 13, 148. [Google Scholar] [CrossRef]
- Ma, J.; Lu, J.; Xu, J.; Duan, B.; He, X.; Liu, J. Genome-wide Identification of WRKY Genes in the Desert Poplar Populus euphratica and Adaptive Evolution of the Genes in Response to Salt Stress. Evol. Bioinform. Online 2015, 11, 47–55. [Google Scholar] [CrossRef]
- Chen, C.; Chen, X.; Han, J.; Lu, W.; Ren, Z. Genome-wide analysis of the WRKY gene family in the cucumber genome and transcriptome-wide identification of WRKY transcription factors that respond to biotic and abiotic stresses. BMC Plant Biol. 2020, 20, 443. [Google Scholar] [CrossRef] [PubMed]
- Nan, H.; Gao, L.Z. Genome-Wide Analysis of WRKY Genes and Their Response to Hormone and Mechanic Stresses in Carrot. Front. Genet. 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Guo, J.; Zhang, T.; Bai, S.; He, K.; Wang, Z. Genome-Wide Analysis of WRKY Gene Family and the Dynamic Responses of Key WRKY Genes Involved in Ostrinia furnacalis Attack in Zea mays. Int. J. Mol. Sci. 2021, 22, 13045. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.; Sato, Y.; Sinclair, R.; Tsukamoto, K.; Nishida, M. Rapid genome reshaping by multiple-gene loss after whole-genome duplication in teleost fish suggested by mathematical modeling. Proc. Natl. Acad. Sci. USA 2015, 112, 14918–14923. [Google Scholar] [CrossRef]
- Almeida-Silva, F.; Van de Peer, Y. Whole-genome Duplications and the Long-term Evolution of Gene Regulatory Networks in Angiosperms. Mol. Biol. Evol. 2023, 40, msad141. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Wang, Q.; He, D.; Wang, X.; Xin, G.; Zou, X.; Zhang, D.; Zou, S.; Liao, J. Genome-Wide Identification and Co-Expression Analysis of WRKY Genes Unveil Their Role in Regulating Anthocyanin Accumulation During Euscaphis japonica Fruit Maturation. Biology 2025, 14, 958. https://doi.org/10.3390/biology14080958
Liu B, Wang Q, He D, Wang X, Xin G, Zou X, Zhang D, Zou S, Liao J. Genome-Wide Identification and Co-Expression Analysis of WRKY Genes Unveil Their Role in Regulating Anthocyanin Accumulation During Euscaphis japonica Fruit Maturation. Biology. 2025; 14(8):958. https://doi.org/10.3390/biology14080958
Chicago/Turabian StyleLiu, Bobin, Qingying Wang, Dongmei He, Xiaqin Wang, Guiliang Xin, Xiaoxing Zou, Daizhen Zhang, Shuangquan Zou, and Jiakai Liao. 2025. "Genome-Wide Identification and Co-Expression Analysis of WRKY Genes Unveil Their Role in Regulating Anthocyanin Accumulation During Euscaphis japonica Fruit Maturation" Biology 14, no. 8: 958. https://doi.org/10.3390/biology14080958
APA StyleLiu, B., Wang, Q., He, D., Wang, X., Xin, G., Zou, X., Zhang, D., Zou, S., & Liao, J. (2025). Genome-Wide Identification and Co-Expression Analysis of WRKY Genes Unveil Their Role in Regulating Anthocyanin Accumulation During Euscaphis japonica Fruit Maturation. Biology, 14(8), 958. https://doi.org/10.3390/biology14080958