
Host Shaping Associated Microbiota in Hydrothermal Vent Snails from the Indian Ocean Ridge

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
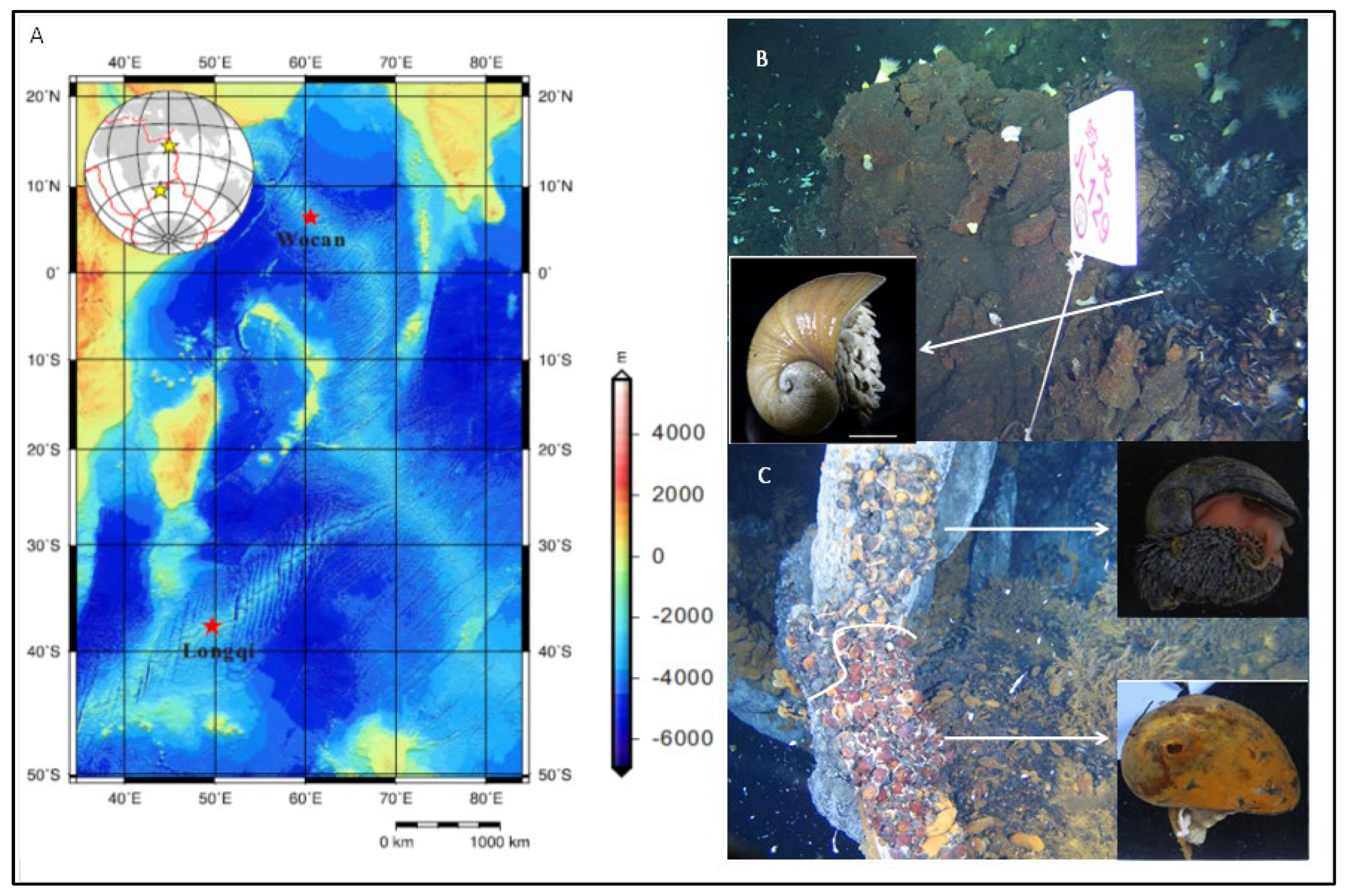
2.1. Sample Collection
2.2. 16S rRNA Gene Library Preparation, Sequencing and Analysis for 17 Samples from C. squamiferum (BC, WC) and G. aegis (G)
2.3. Metagenomic Sequencing, Assembly and Binning for Six Samples from C. squamiferum (BC, WC) and G. aegis (G)
2.4. Gene Annotation and Metabolic Analysis of MAGs and Metagenomes
2.5. Phylogenomic Analysis of MAGs
2.6. Data Availability
3. Results
3.1. Distributions of Snails on Indian Ocean Ridges and Ultrastructural Characterization of Scaly-Foot Snails by Scanning Electron Microscopy (SEM)
3.2. Characterization of Microbial Communities Associated with Hydrothermal Snails
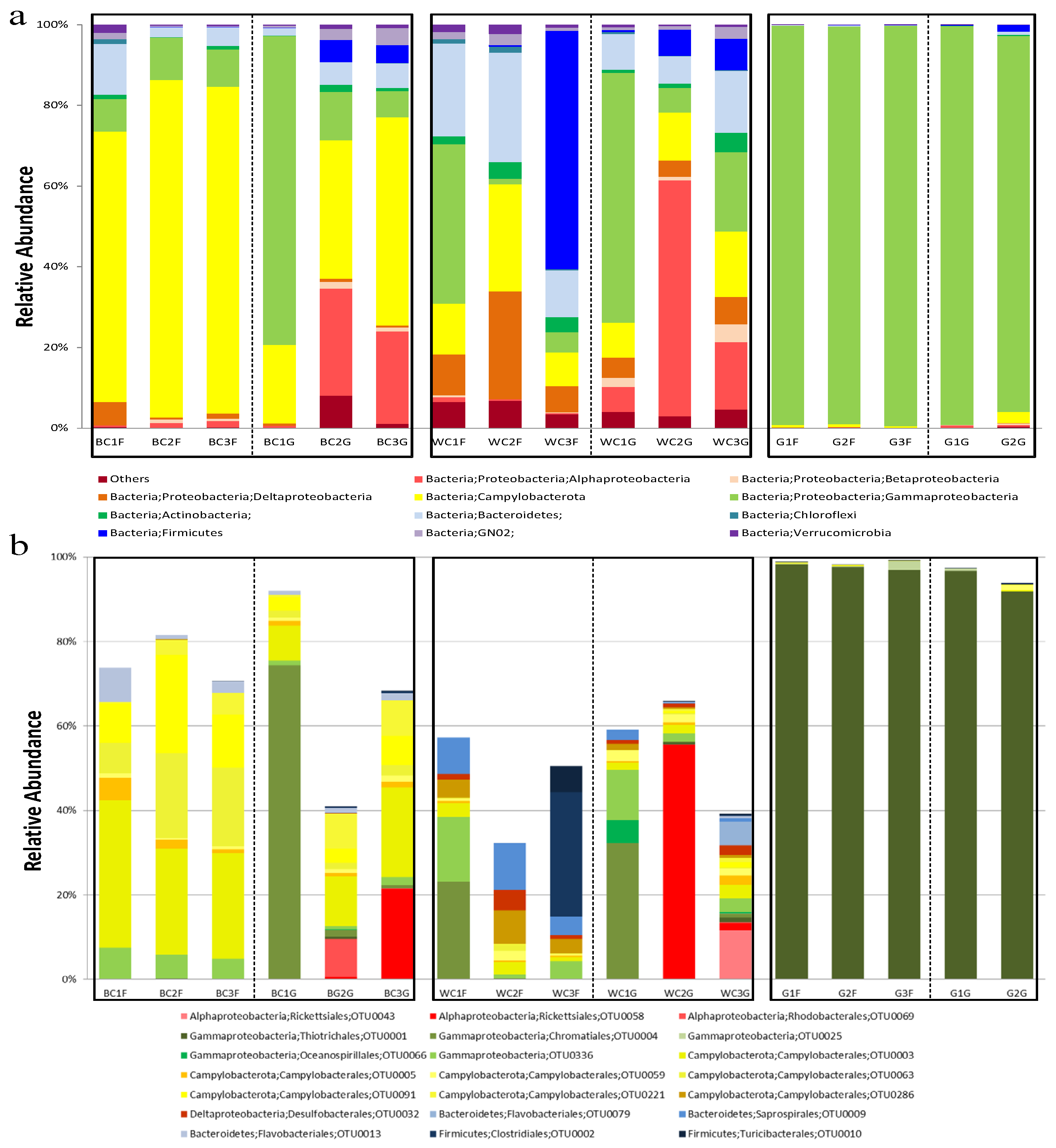
3.2.1. Microbial Community Structure Revealed by High Throughput Sequencing
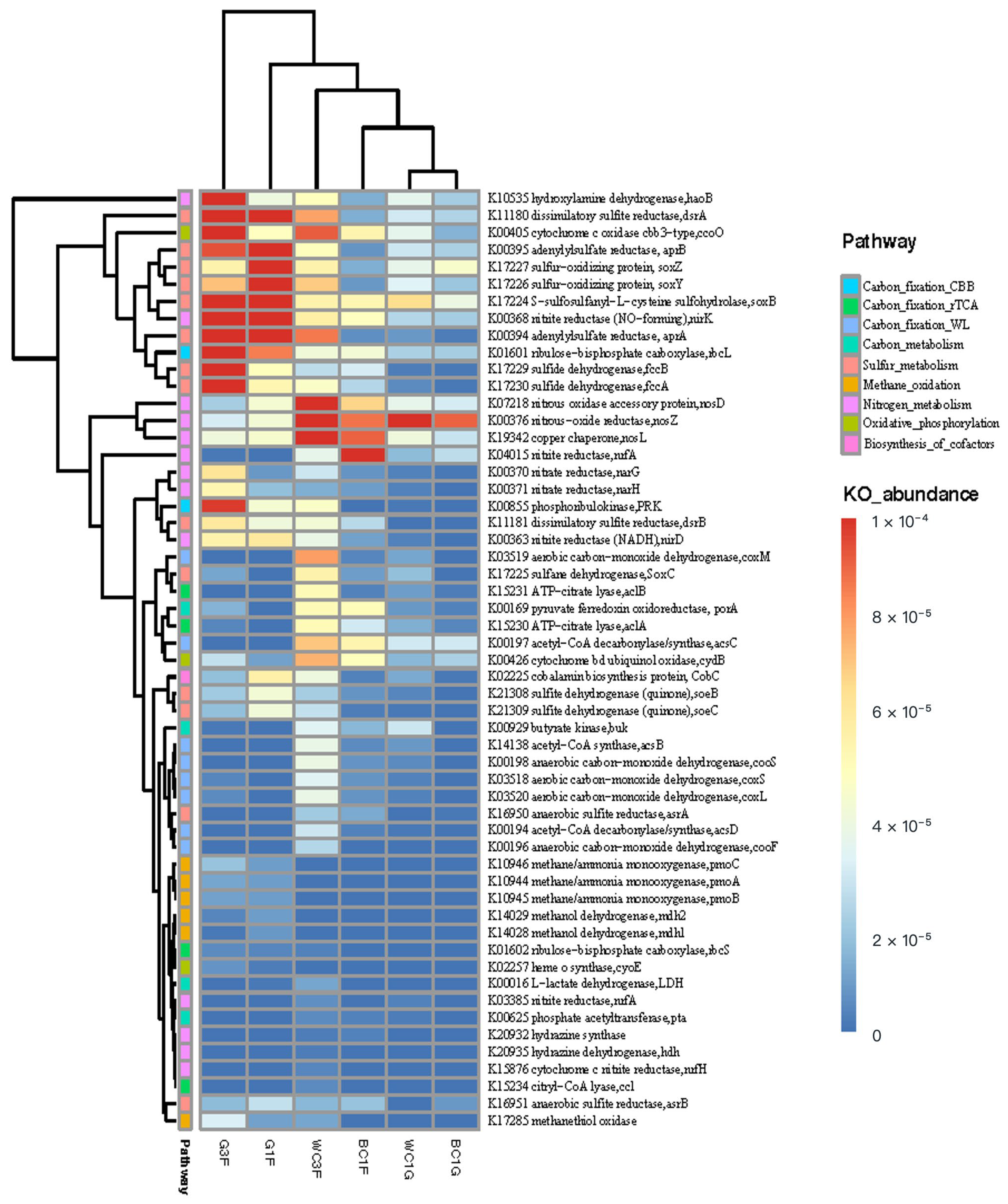
3.2.2. Metabolic Potential of the Microbial Community Based on Metagenomic Analysis
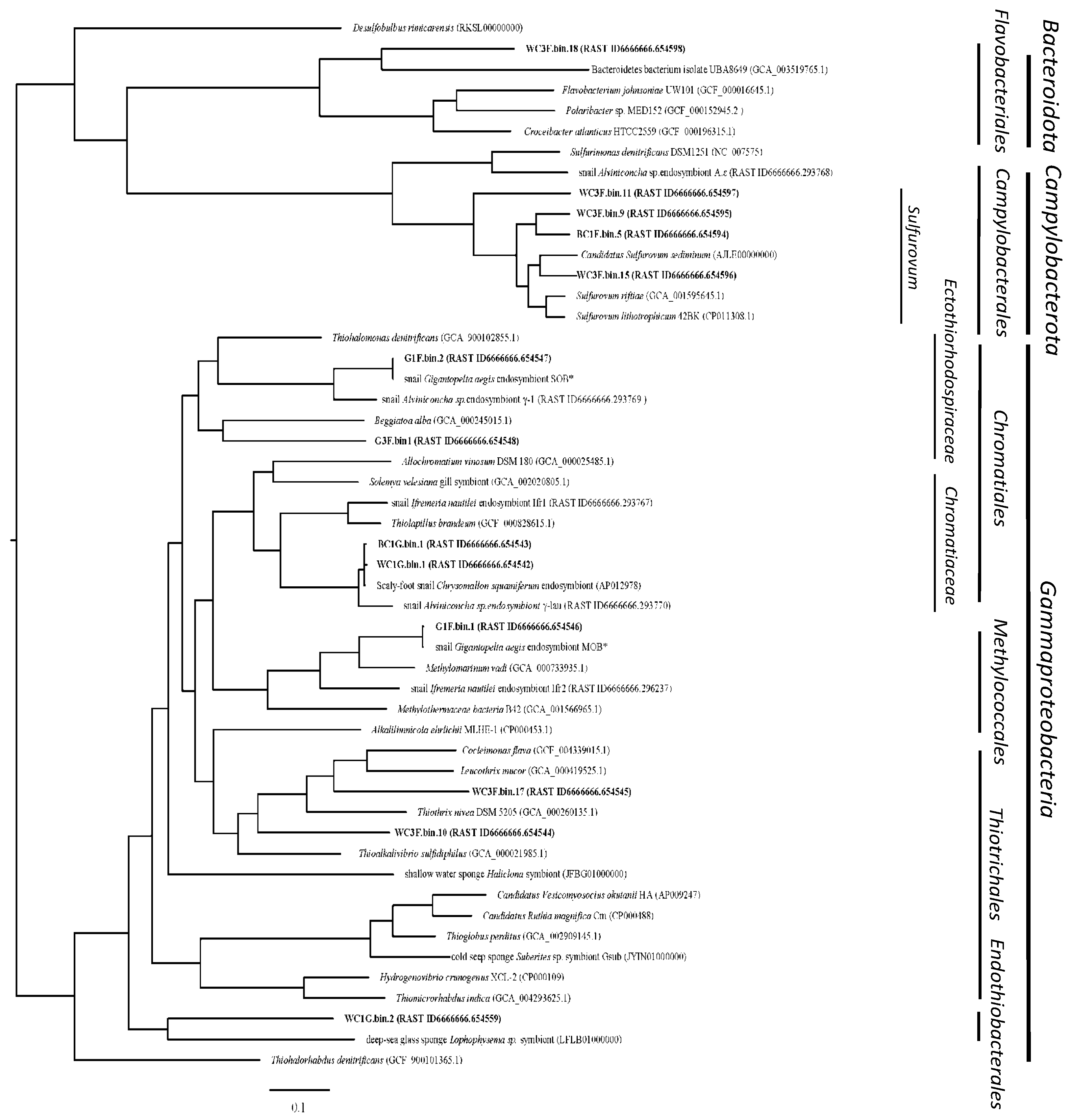
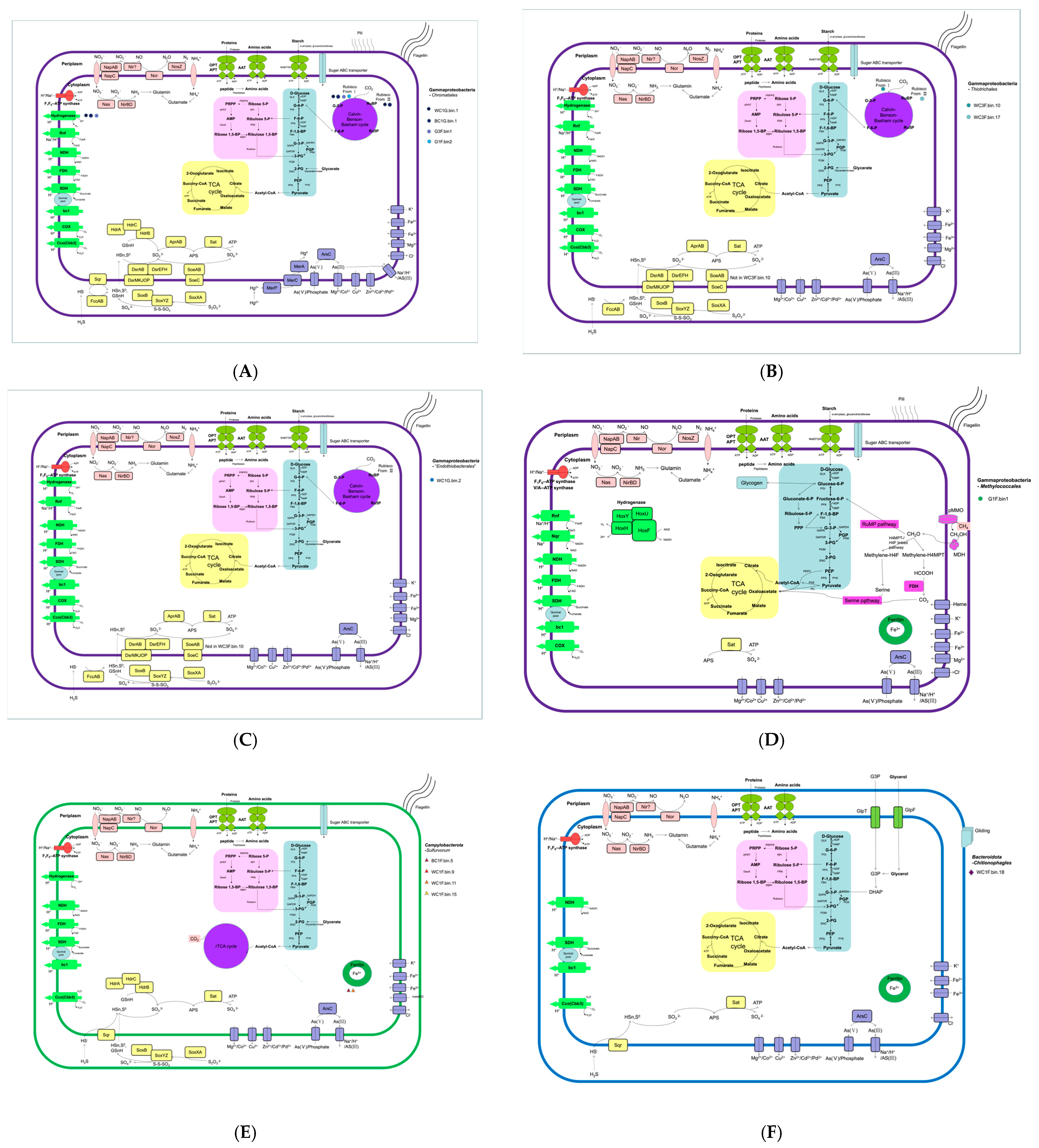
3.2.3. Phylogenomics and Predicted Metabolic Capabilities of Dominant Metagenome-Assembled Genomes (MAGs)
Chromatiales of Gammaproteobacteria
Thiotrichales of Gammaproteobacteria
Methylococcales of Gammaproteobacteria
Candidatus Endothiobacterales of Gammaproteobacteria
Campylobacterales of Campylobacterota
Flavobacteriales of Bacteroidetes
4. Discussion
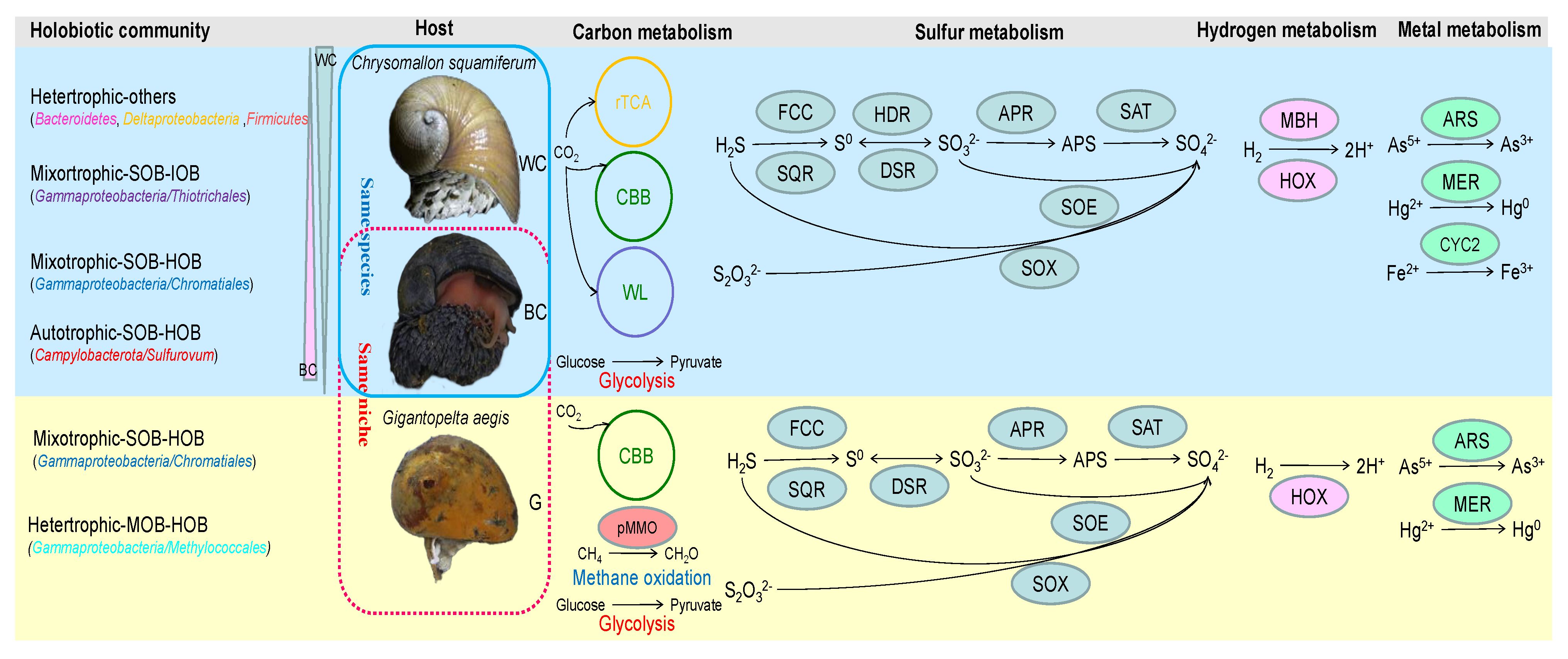
4.1. Host-Specific Symbiont Type in Hydrothermal Snails
4.2. “Core” Microbial Community with Abundance Variation in the Hydrothermal Snail C. squamiferum
4.3. The Association of Hydrothermal Snails and Their Symbionts to Adapt to the Environment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| ANI | Average Nucleotide Identity |
| PCA | Principal Component Analysis |
| MAG | Metagenome-Assembled Genome |
| ASV | Amplicon Sequence Variant |
References
- Dubilier, N.; Bergin, C.; Lott, C. Symbiotic diversity in marine animals: The art of harnessing chemosynthesis. Nat. Rev. Microbiol. 2008, 6, 725–740. [Google Scholar] [CrossRef]
- Dick, G.J. The microbiomes of deep-sea hydrothermal vents: Distributed globally, shaped locally. Nat. Rev. Microbiol. 2019, 17, 271–283. [Google Scholar] [CrossRef]
- Ramírez, G.A.; Mara, P.; Sehein, T.; Wegener, G.; Chambers, C.R.; Joye, S.B.; Peterson, R.N.; Philippe, A.; Burgaud, G.; Edgcomb, V.P.; et al. Environmental factors shaping bacterial, archaeal and fungal community structure in hydrothermal sediments of Guaymas Basin, Gulf of California. PLoS ONE 2021, 16, e0256321. [Google Scholar] [CrossRef]
- Olins, H.C.; Rogers, D.R.; Preston, C.; Ussler, W.; Pargett, D.; Jensen, S.; Roman, B.; Birch, J.M.; Scholin, C.A.; Haroon, M.F.; et al. Co-registered geochemistry and metatranscriptomics reveal unexpected distributions of microbial activity within a hydrothermal vent field. Front. Microbiol. 2017, 8, 1042. [Google Scholar] [CrossRef]
- Chomicki, G.; Kiers, E.T.; Renner, S.S. The evolution of mutualistic dependence. Annu. Rev. Ecol. Evol. Syst. 2020, 51, 409–432. [Google Scholar] [CrossRef]
- Prazeres, M.A.T.; Roberts, T.E.; Pandolfi, J.M.; Leggat, W. Symbiosis and microbiome flexibility in calcifying benthic foraminifera of the Great Barrier Reef. Microbiome 2017, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Beinart, R.A.; Sanders, J.G.; Faure, B.; Sylva, S.P.; Lee, R.W.; Becker, E.L.; Gartman, A.; Luther, G.W.; Seewald, J.S.; Fisher, C.R.; et al. Evidence for the role of endosymbionts in regional-scale habitat partitioning by hydrothermal vent symbioses. Proc. Natl. Acad. Sci. USA 2012, 109, E3241–E3250. [Google Scholar] [CrossRef] [PubMed]
- Meier, D.P.P.; Bach, W.; Hourdez, S.; Girguis, P.R.; Vidoudez, C.; Amann, R.; Meyerdierks, A. Niche partitioning of diverse sulfur-oxidizing bacteria at hydrothermal vents. ISME J. 2017, 11, 1545–1558. [Google Scholar] [CrossRef]
- Osvatic, J.T.; Wilkins, L.G.E.; Leibrecht, L.; Leray, M.; Zauner, S.; Polzin, J.; Camacho, Y.; Gros, O.; van Gils, J.A.; Eisen, J.A.; et al. Global biogeography of chemosynthetic symbionts reveals both localized and globally distributed symbiont groups. Proc. Natl. Acad. Sci. USA 2021, 118, e2104378118. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Linse, K.; Copley, J.; Rogers, A.D. The ‘scaly-foot gastropod’: A new genus and species of hydrothermal vent-endemic gastropod (Neomphalina: Peltospiridae) from the Indian Ocean. J. Molluscan Stud. 2015, 81, 322–334. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, Y.; Meng, L.; Fan, G.; Bai, J.; Chen, J.; Song, Y.; Seim, I.; Wang, C.; Shao, Z.; et al. Genome sequencing of deep-sea hydrothermal vent snails reveals adaptions to extreme environments. Gigascience 2020, 9, giaa139. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Chen, C.; Watsuji, T.O.; Nishizawa, M.; Suzuki, Y.; Sano, Y.; Bissessur, D.; Deguchi, S.; Takai, K. The making of natural iron sulfide nanoparticles in a hot vent snail. Proc. Natl. Acad. Sci. USA 2019, 116, 20376–20381. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Sun, J.; Chen, C.; Sun, Y.; Zhou, Y.; Yang, Y.; Zhang, W.; Li, R.; Zhou, K.; Wong, W.C.; et al. Hologenome analysis reveals dual symbiosis in the deep-sea hydrothermal vent snail Gigantopelta aegis. Nat. Commun. 2021, 12, 1165. [Google Scholar] [CrossRef]
- Chen, C.; Uematsu, K.; Linse, K.; Sigwart, J.D. By more ways than one: Rapid convergence at hydrothermal vents shown by 3D anatomical reconstruction of Gigantopelta (Mollusca: Neomphalina). BMC Evol. Biol. 2017, 17, 62. [Google Scholar] [CrossRef]
- Goffredi, S.K.; Waren, A.; Orphan, V.J.; Van Dover, C.L.; Vrijenhoek, R.C. Novel forms of structural integration between microbes and a hydrothermal vent gastropod from the indian ocean. Appl. Environ. Microbiol. 2004, 70, 3082–3090. [Google Scholar] [CrossRef]
- Lan, Y.; Sun, J.; Chen, C.; Wang, H.; Xiao, Y.; Perez, M.; Yang, Y.; Kwan, Y.H.; Sun, Y.; Zhou, Y.; et al. Endosymbiont population genomics sheds light on transmission mode, partner specificity, and stability of the scaly-foot snail holobiont. ISME J. 2022, 16, 2132–2143. [Google Scholar] [CrossRef]
- Ji, F.; Zhou, H.; Yang, Q.; Gao, H.; Wang, H.; Lilley, M.D. Geochemistry of hydrothermal vent fluids and its implications for subsurface processes at the active Longqi hydrothermal field, Southwest Indian Ridge. Deep Sea Res. Part I Oceanogr. Res. Pap. 2017, 122, 41–47. [Google Scholar] [CrossRef]
- Li, C.; Tan, X.; Bai, J.; Xu, Q.; Liu, S.; Guo, W.; Yu, C.; Fan, G.; Lu, Y.; Zhang, H.; et al. A survey of the sperm whale (Physeter catodon) commensal microbiome. Peer J. 2019, 7, e7257. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; Gonzalez, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Microbiol. 2012, 27, 10.7.1–10.7.20. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Sondergaard, D.; Pedersen, C.N.S.; Greening, C. HydDB: A web tool for hydrogenase classification and analysis. Sci. Rep. 2016, 6, 34212. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.I.; Nealson, K.H.; Okamoto, A.; McAllister, S.M.; Merino, N. FeGenie: A comprehensive tool for the identification of iron genes and iron gene neighborhoods in genome and metagenome assemblies. Front. Microbiol. 2020, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.F.; Rodland, E.A.; Staerfeldt, H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.O.K.Y.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, D.; Zhang, R.; Liu, Z.; Tao, C.; Lu, B.; Sun, D.; Xu, P.; Lin, R.; Wang, J.; et al. Characterization of vent fauna at three hydrothermal vent fields on the Southwest Indian Ridge: Implications for biogeography and interannual dynamics on ultraslow-spreading ridges. Deep Sea Res. Part I Oceanogr. Res. Pap. 2018, 137, 1–12. [Google Scholar] [CrossRef]
- Zeng, X.; Alain, K.; Shao, Z. Microorganisms from deep-sea hydrothermal vents. Mar. Life Sci. Technol. 2021, 3, 204–230. [Google Scholar] [CrossRef]
- Tian, R.; Wang, Y.; Bougouffa, S.; Gao, Z.; Cai, L.; Bajic, V.B.; Qian, P. Genomic analysis reveals versatile heterotrophic capacity of a potentially symbiotic sulfur-oxidizing bacterium in sponge. Environ. Microbiol. 2014, 16, 3548–3561. [Google Scholar] [CrossRef]
- Lavy, A.; Keren, R.; Yu, K.; Thomas, B.C.; Alvarez-Cohen, L.; Banfield, J.F.; Ilan, M. A novel Chromatiales bacterium is a potential sulfide oxidizer in multiple orders of marine sponges. Environ. Microbiol. 2018, 20, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Shimamura, S.; Takaki, Y.; Suzuki, Y.; Murakami, S.; Watanabe, T.; Fujiyoshi, S.; Mino, S.; Sawabe, T.; Maeda, T.; et al. Allying with armored snails: The complete genome of gammaproteobacterial endosymbiont. ISME J. 2014, 8, 40–51. [Google Scholar] [CrossRef]
- Zvi-Kedem, T.; Shemesh, E.; Tchernov, D.; Rubin-Blum, M. The worm affair: Fidelity and environmental adaptation in symbiont species that co-occur in vestimentiferan tubeworms. Environ. Microbiol. Rep. 2021, 13, 744–752. [Google Scholar] [CrossRef]
- Badger, M.R.; Bek, E.J. Multiple Rubisco forms in proteobacteria: Their functional significance in relation to CO2 acquisition by the CBB cycle. J. Exp. Bot. 2008, 59, 1525–1541. [Google Scholar] [CrossRef]
- Dahl, C.; Franz, B.; Hensen, D.; Kesselheim, A.L.; Zigann, R. Sulfite oxidation in the purple sulfur bacterium Allochromatium vinosum: Identification of SoeABC as a major player and relevance of SoxYZ in the process. Microbiology 2013, 159, 2626–2638. [Google Scholar] [CrossRef] [PubMed]
- Quatrini, R.; Appiaayme, C.; Denis, Y.; Jedlicki, E.; Holmes, D.S.; Bonnefoy, V. Extending the models for iron and sulfur oxidation in the extreme acidophile Acidithiobacillus ferrooxidans. BMC Genom. 2009, 10, 394. [Google Scholar] [CrossRef]
- Wang, R.; Lin, J.Q.; Liu, X.M.; Pang, X.; Zhang, C.J.; Yang, C.L.; Gao, X.Y.; Lin, C.M.; Li, Y.Q.; Li, Y.; et al. Sulfur oxidation in the acidophilic autotrophic Acidithiobacillus spp. Front. Microbiol. 2019, 9, 3290. [Google Scholar]
- Nunoura, T.; Takaki, Y.; Kazama, H.; Kakuta, J.; Shimamura, S.; Makita, H.; Hirai, M.; Miyazaki, M.; Takai, K. Physiological and genomic features of a novel sulfur-oxidizing gammaproteobacterium belonging to a previously uncultivated symbiotic lineage isolated from a hydrothermal vent. PLoS ONE 2014, 9, e104959. [Google Scholar] [CrossRef] [PubMed]
- Beinart, R.A.; Luo, C.; Konstantinidis, K.T.; Stewart, F.J.; Girguis, P.R. The Bacterial symbionts of closely related hydrothermal vent snails with distinct geochemical habitats show broad similarity in chemoautotrophic gene content. Front. Microbiol. 2019, 10, 1818. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.M.; Muntyan, M.S.; Yu, L.V.; Ustiyan, V.S.; Dubinina, G.A. Lithoheterotrophic growth and electron transfer chain components of the filamentous gliding bacterium Leucothrix mucor DSM 2157 during oxidation of sulfur compounds. FEMS Microbiol. Lett. 1999, 175, 155–161. [Google Scholar] [CrossRef]
- Tanaka, N.; Romanenko, L.A.; Iino, T.; Frolova, G.M.; Mikhailov, V.V. Cocleimonas flava gen. nov., sp. nov., a gammaproteobacterium isolated from sand snail (Umbonium costatum). Int. J. Syst. Evol. Microbiol. 2011, 61, 412. [Google Scholar] [CrossRef]
- Larkin, J.M.; Shinabarger, D.L. Characterization of Thiothrix nivea. Int. J. Syst. Bacteriol. 1983, 33, 841–846. [Google Scholar] [CrossRef]
- Rossetti, S.; Blackall, L.L.; Levantesi, C.; Uccelletti, D.; Tandoi, V. Phylogenetic and physiological characterization of a heterotrophic, chemolithoautotrophic Thiothrix strain isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 2003, 53, 1271–1276. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Muntyan, M.S.; Panteleeva, A.N.; Muyzer, G. Thioalkalivibrio sulfidiphilus sp. nov., a haloalkaliphilic, sulfur-oxidizing gammaproteobacterium from alkaline habitats. Int. J. Syst. Evol. Microbiol. 2012, 62, 1884–1889. [Google Scholar] [CrossRef]
- Brigmon, R.L.; De Ridder, C. Symbiotic relationship of Thiothrix spp. with an Echinoderm. Appl. Environ. Microbiol. 1998, 64, 3491–3495. [Google Scholar] [CrossRef]
- McAllister, S.M.; Polson, S.W.; Butterfield, D.A.; Glazer, B.T.; Sylvan, J.B.; Chan, C.S. Validating the Cyc2 neutrophilic iron oxidation pathway using meta-omics of Zetaproteobacteria iron mats at marine hydrothermal vents. mSystems 2020, 5, e00553-19. [Google Scholar] [CrossRef]
- Barco, R.A.H.C.; Ramírez, G.A.; Toner, B.M.; Edwards, K.J.; Sylvan, J.B. In-situ incubation of iron-sulfur mineral reveals a diverse chemolithoautotrophic community and a new biogeochemical role for Thiomicrospira. Environ. Microbiol. 2017, 19, 1322–1337. [Google Scholar] [CrossRef] [PubMed]
- Neely, C.; Bou Khalil, C.; Cervantes, A.; Diaz, R.; Escobar, A.; Ho, K.; Hoefler, S.; Smith, H.H.; Abuyen, K.; Savalia, P.; et al. Genome sequence of Hydrogenovibrio sp. strain SC-1, a chemolithoautotrophic sulfur and iron oxidizer. Genome Announc. 2018, 6, e01581-17. [Google Scholar] [CrossRef]
- Hirayama, H.; Fuse, H.; Abe, M.; Miyazaki, M.; Nakamura, T.; Nunoura, T.; Furushima, Y.; Yamamoto, H.; Takai, K. Methylomarinum vadi gen. nov., sp. nov., a methanotroph isolated from two distinct marine environments. Int. J. Syst. Evol. Microbiol. 2013, 63, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Orata, F.D.; Meier-Kolthoff, J.P.; Sauvageau, D.; Stein, L.Y. Phylogenomic analysis of the Gammaproteobacterial Methanotrophs (Order Methylococcales) calls for the reclassification of members at the genus and species levels. Front. Microbiol. 2018, 9, 3162. [Google Scholar] [CrossRef] [PubMed]
- Rubin-Blum, M.; Antony, C.P.; Sayavedra, L.; Martinez-Perez, C.; Birgel, D.; Peckmann, J.L.; Wu, Y.; Cardenas, P.; Macdonald, I.R.; Marcon, Y. Fueled by methane: Deep-sea sponges from asphalt seeps gain their nutrition from methane-oxidizing symbionts. ISME J. 2019, 13, 1209–1225. [Google Scholar] [CrossRef]
- Flynn, J.D.; Hirayama, H.; Sakai, Y.; Dunfield, P.F.; Klotz, M.G.; Knief, C.; Op den Camp, H.J.M.; Jetten, M.S.M.; Khmelenina, V.N.; Trotsenko, Y.A.; et al. Draft Genome sequences of gammaproteobacterial methanotrophs isolated from marine ecosystems. Microbiol. Resour. Announc. 2016, 4, e01629-15. [Google Scholar] [CrossRef]
- Tian, R.M.; Sun, J.; Cai, L.; Zhang, W.P.; Zhou, G.W.; Qiu, J.W.; Qian, P.Y. The deep-sea glass sponge Lophophysema eversa harbours potential symbionts responsible for the nutrient conversions of carbon, nitrogen and sulfur. Environ. Microbiol. 2016, 18, 2481–2494. [Google Scholar] [CrossRef]
- Nakagawa, S.; Takai, K. Deep-sea vent chemoautotrophs: Diversity, biochemistry and ecological significance. FEMS Microbiol. Ecol. 2008, 65, 1–14. [Google Scholar] [CrossRef]
- Inagaki, F.; Takai, K.; Nealson, K.H.; Horikoshi, K. Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the ε-Proteobacteria isolated from Okinawa Trough hydrothermal sediments. Int. J. Syst. Evol. Microbiol. 2004, 54, 1477–1482. [Google Scholar] [CrossRef]
- Park, S.; Ghai, R.; Martin-Cuadrado, A.; Rodriguez-Valera, F.; Jung, M.; Kim, J.; Rhee, S. Draft genome sequence of the sulfur-oxidizing bacterium “Candidatus Sulfurovum sediminum” AR, which belongs to the Epsilonproteobacteria. J. Bacteriol. 2012, 194, 4128–4129. [Google Scholar] [CrossRef]
- Jeon, W.; Priscilla, L.; Park, G.; Lee, H.; Lee, N.; Lee, D.; Kwon, H.; Ahn, I.S.; Lee, C.; Lee, H. Complete genome sequence of the sulfur-oxidizing chemolithoautotrophic Sulfurovum lithotrophicum 42BKTT. Stand. Genom. Sci. 2017, 12, 54. [Google Scholar] [CrossRef]
- Fernandez-Gomez, B.; Richter, M.; Schuler, M.; Pinhassi, J.; Acinas, S.G.; Gonzalez, J.M.; Pedros-Alio, C. Ecology of marine Bacteroidetes: A comparative genomics approach. ISME J. 2013, 7, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Stokke, R.; Dahle, H.; Roalkvam, I.; Wissuwa, J.; Daae, F.L.; Tooming-Klunderud, A.; Thorseth, I.H.; Pedersen, R.B.; Steen, I.H. Functional interactions among filamentous Epsilonproteobacteria and Bacteroidetes in a deep-sea hydrothermal vent biofilm. Environ. Microbiol. 2015, 17, 4063–4077. [Google Scholar] [CrossRef]
- Cavanaugh, C.M. Microbial symbiosis: Patterns of diversity in the marine environment. Integr. Comp. Biol. 2015, 34, 79–89. [Google Scholar] [CrossRef]
- Chen, C.; Copley, J.T.; Linse, K.; Rogers, A.D. Low connectivity between ‘scaly-foot gastropod’ (Mollusca: Peltospiridae) populations at hydrothermal vents on the Southwest Indian Ridge and the Central Indian Ridge. Org. Divers. Evol. 2015, 15, 663–670. [Google Scholar] [CrossRef]
- Tokuda, G.; Yamada, A.; Nakano, K.; Arita, N.O.; Yamasaki, H. Colonization of Sulfurovum sp. on the gill surfaces of Alvinocaris longirostris, a deep-sea hydrothermal vent shrimp. Mar. Ecol. 2010, 29, 106–114. [Google Scholar] [CrossRef]
- Sylvan, J.B.; Toner, B.M.; Edwards, K.J. Life and death of deep-sea vents: Bacterial diversity and ecosystem succession on inactive hydrothermal sulfides. mBio 2012, 3, e00279-11. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Sievert, S.M.; Wang, Y.; Seewald, J.S.; Xiao, X. Microbial succession during the transition from active to inactive stages of deep-sea hydrothermal vent sulfide chimneys. Microbiome 2020, 8, 102. [Google Scholar] [CrossRef]
- Cao, W.; Wang, L.; Saren, G.; Yu, X.; Li, Y. Variable microbial communities in the non-hydrothermal sediments of the Mid-Okinawa Trough. Geomicrobiol. J. 2020, 37, 881–889. [Google Scholar] [CrossRef]
- Zhang, L.; Kang, M.; Xu, J.; Xu, J.; Shuai, Y.; Zhou, X.; Yang, Z.; Ma, K. Bacterial and archaeal communities in the deep-sea sediments of inactive hydrothermal vents in the Southwest India Ridge. Sci. Rep. 2016, 6, 25982. [Google Scholar] [CrossRef]
- Miyazaki, J.; Ikuta, T.; Watsuji, T.O.; Abe, M.; Yamamoto, M.; Nakagawa, S.; Takaki, Y.; Nakamura, K.; Takai, K. Dual energy metabolism of the Campylobacterota endosymbiont in the chemosynthetic snail Alviniconcha marisindica. ISME J. 2020, 14, 1273–1289. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | The Black Scaly Chrysomallon squamiferum (BC) | The White Scaly Chrysomallon squamiferum (WC) | Gigantopelta aegis (G) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Samples | Scale Foot | Gland | Scale Foot | Gland | Foot | Gland | |||||||||||
| ID | BC1F | BC2F | BC3F | BC1G | BC2G | BC3G | WC1F | WC2F | WC3F | WC1G | WC2G | WC3G | G1F | G2F | G3F | G1G | G2G |
| Effective sequence tags | 83,413 | 41,705 | 48,417 | 102,478 | 86,262 | 77,152 | 72,329 | 15,542 | 71,883 | 72,329 | 58,503 | 65,641 | 95,143 | 89,073 | 84,091 | 101,179 | 100,557 |
| ASV | 187 | 284 | 326 | 206 | 417 | 264 | 291 | 197 | 323 | 469 | 344 | 336 | 98 | 145 | 99 | 126 | 204 |
| Shannon (H’) | 2.67 | 2.45 | 2.77 | 1.32 | 3.40 | 3.27 | 3.39 | 3.68 | 3.38 | 3.45 | 2.81 | 4.60 | 0.14 | 0.19 | 0.19 | 0.25 | 0.84 |
| Simpson | 0.15 | 0.16 | 0.13 | 0.56 | 0.05 | 0.11 | 0.09 | 0.04 | 0.10 | 0.13 | 0.31 | 0.03 | 0.97 | 0.95 | 0.94 | 0.94 | 0.84 |
| Chao1 | 202.79 | 387.46 | 475.24 | 226.04 | 458.17 | 277.00 | 340.40 | 230.91 | 395.55 | 508.00 | 378.50 | 493.50 | 152.09 | 224.69 | 147.46 | 140.25 | 246.86 |
| Ace | 206.55 | 386.90 | 546.24 | 233.05 | 541.17 | 273.87 | 318.05 | 232.01 | 382.70 | 500.82 | 358.77 | 504.86 | 143.58 | 201.53 | 171.82 | 134.41 | 224.84 |
| Taxonomy | Gammaproteobacteria | Campylobacterota | Bacteroidota | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromatiales | Thiotrichales | Methy- Lococcales | “Endothio- Bacterales” | Campylobacterales /Sulfurovum | Chitin- Ophagales | ||||||||
| MAGs | WC1G. bin.1 | BC1G. bin.1 | G3F. bin1 | G1F. bin2 | WC3F. bin.10 | WC3F. bin.17 | G1F. bin.1 | WC1G. bin.2 | BC1F. bin.5 | WC3F. bin.9 | WC3F. bin.15 | WC3F. bin.11 | WC3F. bin.18 |
| Completeness | 97.59 | 99.74 | 92.76 | 98.55 | 80.28 | 80.16 | 98.13 | 88.48 | 82.77 | 87.89 | 85.38 | 89.95 | 95.32 |
| Genome size (bp) | 2,472,218 | 2,782,074 | 4,909,376 | 3,525,970 | 3,222,582 | 2,694,131 | 2,407,989 | 2,093,704 | 1,424,152 | 1,383,753 | 1,614,001 | 2,030,818 | 3,087,810 |
| GC (%) | 65.54 | 64.89 | 61.05 | 40.24 | 53.60 | 42.37 | 45.43 | 57.7 | 44.77 | 46.10 | 38.91 | 31.71 | 29.11 |
| No. protein coding gene | 2468 | 2668 | 4289 | 3256 | 3031 | 2369 | 2409 | 2187 | 1478 | 1438 | 1640 | 1906 | 2503 |
| Coding density (%) | 89.50 | 89.66 | 76.25 | 85.74 | 77.26 | 76.37 | 88.36 | 86.88 | 79.77 | 88.01 | 83.29 | 77.03 | 76.98 |
| Carbon Fixation | |||||||||||||
| CBB (form I) | + | + | - | - | + | - | - | - | - | - | - | - | - |
| CBB (form II) | + | + | + | + | - | + | - | + | - | - | - | - | - |
| rTCA | - | - | - | - | - | - | - | - | + | + | + | + | - |
| Sulfur oxidation | |||||||||||||
| SoxBAZYX | + | + | + | + | + | + | - | + | - | - | - | + | - |
| SoxCDYZ | - | - | - | - | - | - | - | - | + | + | + | + | - |
| Sqr | + | + | + | + | + | + | - | + | + | + | + | + | + |
| Fcc | - | + | + | + | + | + | - | + | - | - | - | - | - |
| HdrABC | + | + | - | - | - | - | - | - | - | - | - | - | - |
| DsrAB | + | + | + | + | + | + | - | + | - | - | - | - | - |
| AprAB | + | + | + | - | + | + | - | + | - | - | - | - | - |
| sat | + | + | + | + | + | + | + | + | + | + | + | + | + |
| SoeABC | + | + | - | + | + | - | - | - | - | - | - | - | - |
| Hydrogen oxidation | |||||||||||||
| MBHL | + | + | + | - | - | + | - | - | - | - | - | - | - |
| Hox | + | - | + | - | + | - | + | - | - | - | - | - | - |
| Methano oxidation | |||||||||||||
| pMMO | - | - | - | - | - | - | + | - | - | - | - | - | - |
| CO oxidation | |||||||||||||
| Coo | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Metal utilization and resistance | |||||||||||||
| Iron oxidase Cyc2 | - | - | - | - | + | + | - | - | - | + | - | - | - |
| Iron reduction genes | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Iron storage genes | - | - | - | - | - | - | + | + | + | - | + | - | + |
| Arsenite oxidation genes | - | - | - | - | - | - | - | - | - | - | - | - | - |
| ArsC | + | + | + | + | + | + | + | + | + | + | + | + | + |
| MerA | + | + | + | + | + | - | - | - | - | - | - | - | - |
| Copper resistance | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Oxygen respiration | |||||||||||||
| Cox | + | + | + | + | - | - | + | + | - | - | - | - | + |
| Cco | + | + | + | - | + | + | - | - | + | + | + | + | + |
| Glc | + | + | + | - | + | + | - | - | - | - | - | - | - |
| Nitrate and nitrite ammonification | |||||||||||||
| NapAB | + | + | + | + | + | + | + | + | - | + | + | + | + |
| NasA | + | + | + | + | + | - | + | + | + | - | + | - | + |
| NirBD | + | + | + | + | + | - | + | + | + | - | + | - | + |
| Electron transport chain | |||||||||||||
| ATP synthase | F-type | F-type | F-type; V-type | F-type | F-type | F-type | F-type; V-type | F-type | F-type | F-type | F-type | F-type | F-type |
| Nuo | + | + | + | + | + | + | - | + | + | + | + | + | + |
| Fdh | + | + | + | - | - | - | + | - | - | - | - | - | - |
| Sdh | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Nqr | - | - | - | - | - | - | + | - | - | - | - | - | - |
| Rnf | + | + | + | + | - | + | + | + | - | - | - | - | - |
| Motility | |||||||||||||
| Flagellum | + | + | - | + | + | - | + | - | - | - | - | - | - |
| Pili | + | + | + | + | + | + | + | + | - | - | - | - | - |
| Gliding | - | - | - | - | - | - | - | - | - | - | - | - | + |
| Vitamin and cofactor | |||||||||||||
| Thiamine (Vitamin B1) | + | + | + | + | - | + | + | + | + | + | + | + | - |
| Riboflavin (Vitamin B2) | + | + | + | - | - | + | - | + | + | + | + | + | + |
| pyridoxine (Vitamin B6) | + | + | - | + | - | + | + | - | + | + | - | + | - |
| Biotin (Vitamin B7) | + | + | + | + | + | - | + | + | + | - | - | + | - |
| Folic acid (Vitamin B9) | + | + | + | + | + | - | + | + | + | + | + | + | + |
| Cobalamin (vitamin B12) | - | - | - | + | + | - | + | + | - | - | - | - | - |
| Host | Habitat | Symbiont Location | Symbiont Type | Main Symbiont Community | Refs |
|---|---|---|---|---|---|
| Alviniconcha spp. /Provannidae | SWIR, CR, Indian Ocean; Western Pacific Ocean | Intracellular; Gill; | SOB; HOB; | Sulfurimonas/Sulfurovum, Helicobacteraceae, Campylobacterales, Campylobacterota | [7,71] |
| SOB; HOB; | Ectothiorhodospiraceae, Chromatiales, Gammaproteobacteria Chromatiaceae, Chromatiales, Gammaproteobacteria | ||||
| Gigantopelta spp. /Peltospiridae | SWIR, Indian Ocean; Southern Ocean | Intracellular; Gland; | SOB; MOB | Ectothiorhodospiraceae, Chromatiales, Gammaproteobacteria Methylococcaceae, Methylococcales, Gammaproteobacteria | [13] This study |
| Chrysomallon squamiferum /Peltospiridae | SWIR, CIR, CR, Indian Ocean | Scale; | SOB; HOB; HB | Sulfurovum, Helicobacteraceae, Campylobacterales, Campylobacterota Thiotrichaceae, Thiotrichales, Gammaproteobacteria Flavobacteriales, Bacteroidota | [16,36] This study |
| Intracellular; Gland; | SOB; HOB; | Chromatiaceae, Chromatiales, Gammaproteobacteria Thiotrichaceae, Thiotrichales, Gammaproteobacteria “Endothiobacterales”, Gammaproteobacteria |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Chen, J.; Liu, G.; Zhou, Y.; Wang, L.; Zhang, Y.; Liu, S.; Shao, Z. Host Shaping Associated Microbiota in Hydrothermal Vent Snails from the Indian Ocean Ridge. Biology 2025, 14, 954. https://doi.org/10.3390/biology14080954
Zeng X, Chen J, Liu G, Zhou Y, Wang L, Zhang Y, Liu S, Shao Z. Host Shaping Associated Microbiota in Hydrothermal Vent Snails from the Indian Ocean Ridge. Biology. 2025; 14(8):954. https://doi.org/10.3390/biology14080954
Chicago/Turabian StyleZeng, Xiang, Jianwei Chen, Guilin Liu, Yadong Zhou, Liping Wang, Yaolei Zhang, Shanshan Liu, and Zongze Shao. 2025. "Host Shaping Associated Microbiota in Hydrothermal Vent Snails from the Indian Ocean Ridge" Biology 14, no. 8: 954. https://doi.org/10.3390/biology14080954
APA StyleZeng, X., Chen, J., Liu, G., Zhou, Y., Wang, L., Zhang, Y., Liu, S., & Shao, Z. (2025). Host Shaping Associated Microbiota in Hydrothermal Vent Snails from the Indian Ocean Ridge. Biology, 14(8), 954. https://doi.org/10.3390/biology14080954