
Population Genetics of the Asian Buffalo Leech (Hirudinaria manillensis) in Southern China Based on Mitochondrial Protein-Coding Genes
 ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
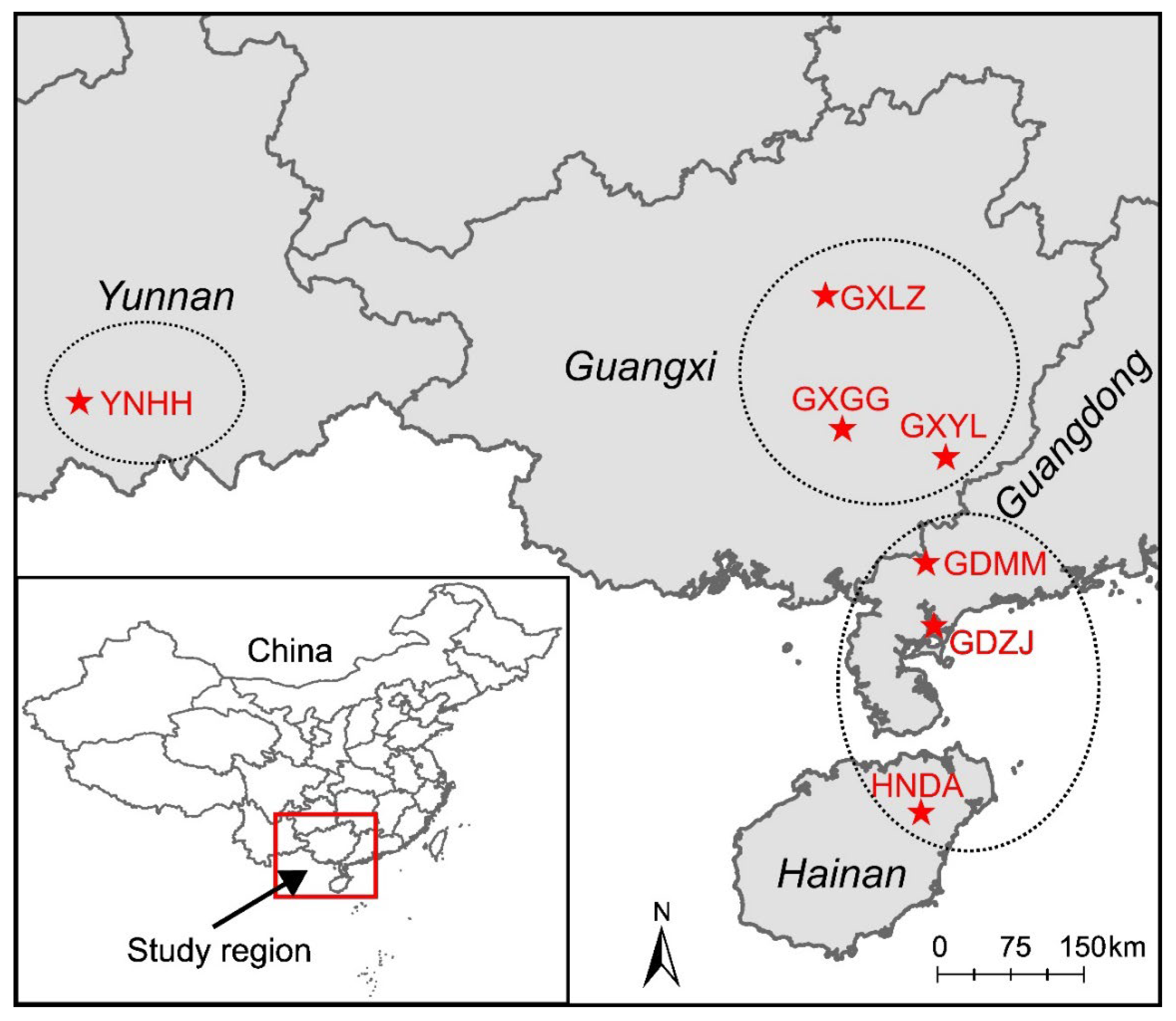
2.1. Sampling and Sequencing
2.2. MtPCGs Sequence Extraction
2.3. Genetic Diversity Analysis
2.4. Network and Phylogenetic Analysis of Haplotypes
2.5. Population Genetic Structure Analysis
2.6. Population Dynamics
3. Results
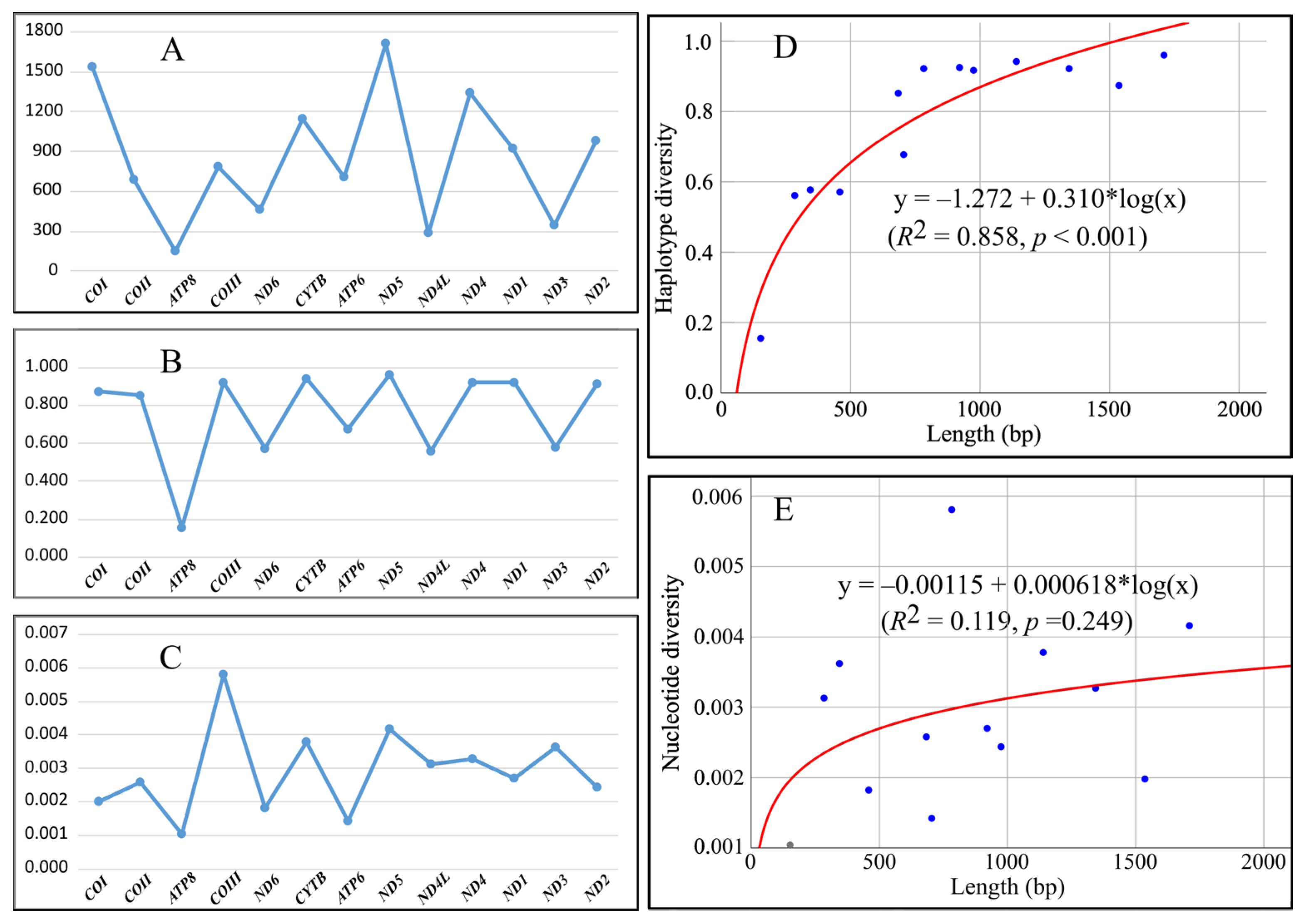
3.1. Sequence Variation in Different Genes
3.2. Population Genetic Diversity
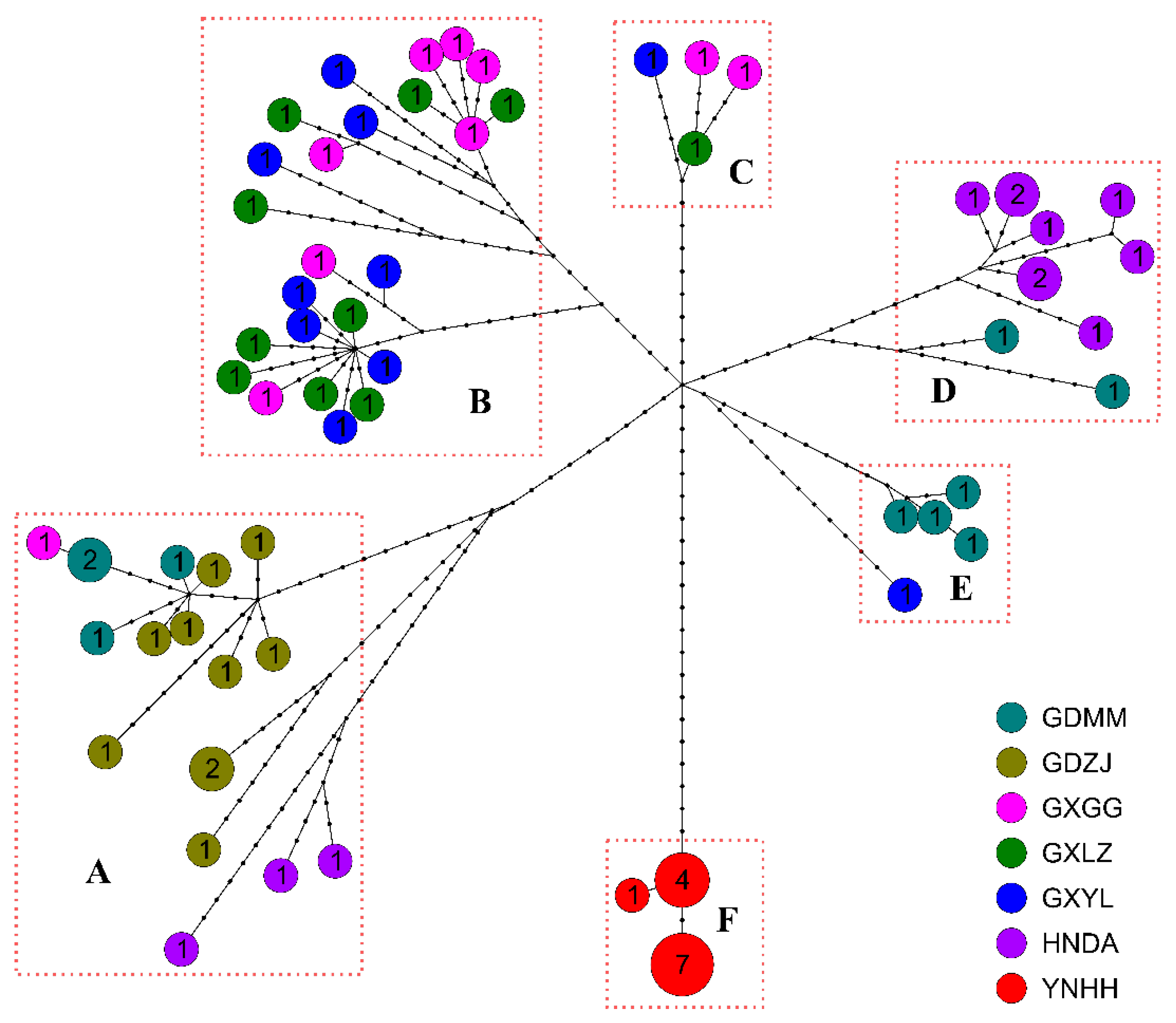
3.3. Haplotype Network Analysis
3.4. Phylogenetic Analysis
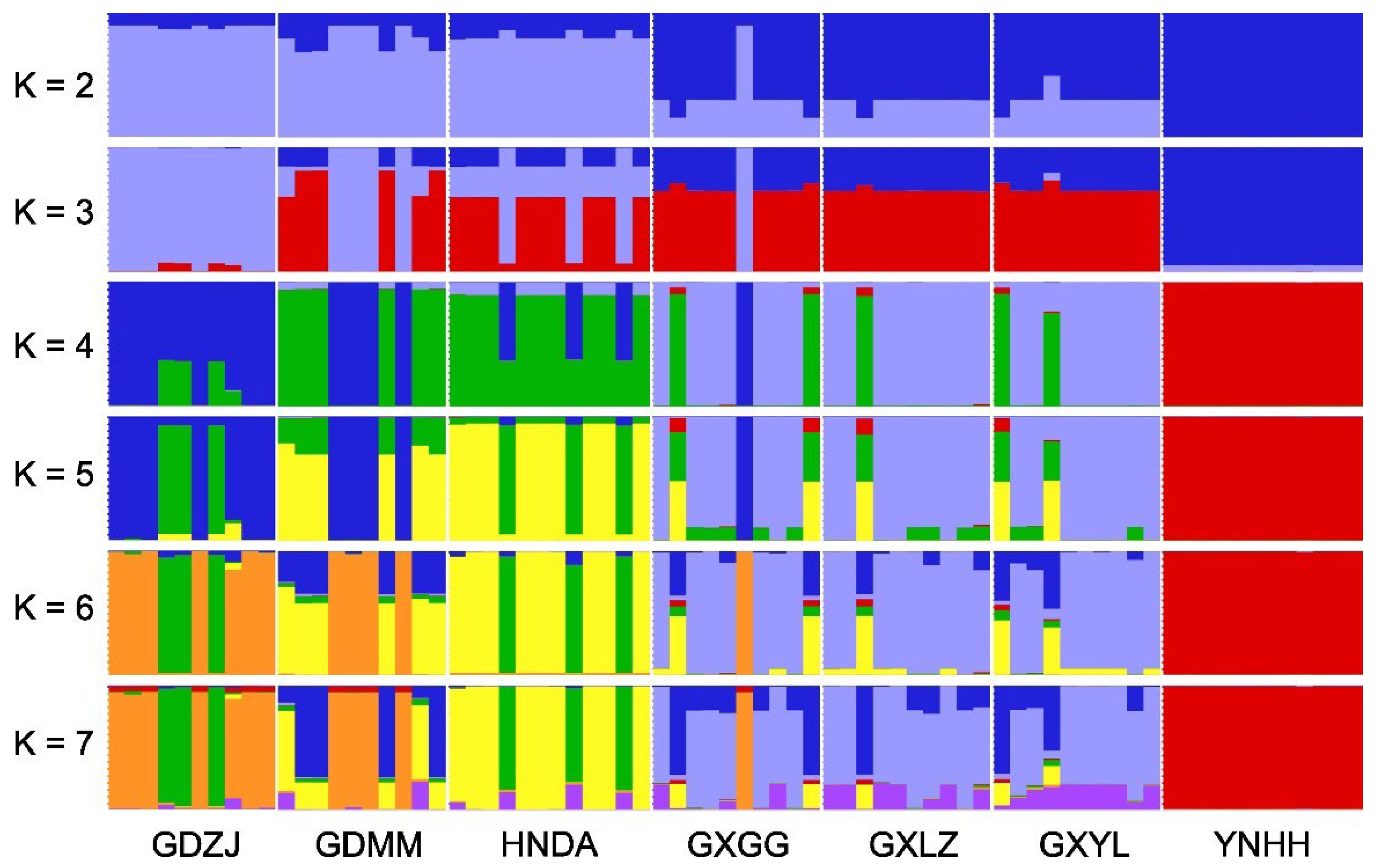
3.5. Genetic Structure and Genetic Differentiation
3.6. Historical Population Dynamics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MitPCGs | mitochondrial protein-coding genes |
| VS | number of variable sites |
| HN | number of haplotypes |
| Hd | haplotype diversity |
| Pi | nucleotide diversity |
| SD | standard deviation |
References
- Sket, B.; Trontelj, P. Global diversity of leeches (Hirudinea) in freshwater. Hydrobiologia 2008, 595, 129–137. [Google Scholar] [CrossRef]
- Yang, T. Fauna Sinica (Annelida Hirudinea); Science Press: Beijing, China, 1996. [Google Scholar]
- Ma, C.J.; Li, X.; Chen, H. Research progress in the use of leeches for medical purposes. Tradit. Med. Res. 2021, 6, 56–69. [Google Scholar] [CrossRef]
- Sig, A.K.; Guney, M.; Uskudar Guclu, A.; Ozmen, E. Medicinal leech therapy—An overall perspective. Integr. Med. Res. 2017, 6, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Yuan, Z.; Zhou, Y.; Zhang, Y.; Ye, X.; Hu, B. Comparative studies on anti-thrombus and anti-coagulation effects of Hirudo of different species. Chin. J. Hosp. Pharm. 2012, 32, 1093–1096. [Google Scholar] [CrossRef]
- Wang, L. 300 Effective Prescriptions for Cardiovascular Diseases: A Collection of Effective Prescriptions in Various Clinical Departments; Science and Technology Literature Press: Beijing, China, 2000. [Google Scholar]
- Xu, S.; Wang, X. A Collection of Effective Formulas for Various Clinical Departments—400 Effective Formulas for Peripheral Vascular Diseases; Science and Technology Literature Press: Beijing, China, 2006. [Google Scholar]
- Gillespie, J.H. Population Genetics: A Concise Guide, 2nd ed.; Johns Hopkins University Press: Baltimore, MD, USA, 2004. [Google Scholar]
- Jensen, A.J.; Cove, M.V.; Goldstein, B.R.; Kays, R.; Mcshea, W.; Pacifici, K.; Rooney, B.; Kierepka, E.J.G.E. Geographic barriers but not life history traits shape the phylogeography of North American mammals. Glob. Ecol. Biogeogr. 2024, 33, e13875. [Google Scholar] [CrossRef]
- Li, Z.; Fan, H.; Liao, Z.; Wang, Y.; Wei, F. Global spatiotemporal patterns of demographic fluctuations in terrestrial vertebrates during the Late Pleistocene. Sci. Adv. 2025, 11, eadq3938. [Google Scholar] [CrossRef]
- Bielecki, A.; Polok, K. Genetic variation and species identification among selected leeches (Hirudinea) revealed by RAPD markers. Biologia 2012, 67, 721–730. [Google Scholar] [CrossRef]
- Liu, F.; Guo, Q.S.; Shi, H.Z.; Cheng, B.X.; Lu, Y.X.; Gou, L.; Wang, J.; Shen, W.B.; Yan, S.M.; Wu, M.J. Genetic variation in Whitmania pigra, Hirudo nipponica and Poecilobdella manillensis, three endemic and endangered species in China using SSR and TRAP markers. Gene 2016, 579, 172–182. [Google Scholar] [CrossRef]
- Struck, T.H.; Golombek, A.; Hoesel, C.; Dimitrov, D.; Elgetany, A.H. Mitochondrial genome evolution in annelida-A systematic study on conservative and variable gene orders and the factors influencing its evolution. Syst. Biol. 2023, 72, 925–945. [Google Scholar] [CrossRef]
- Sodhi, M.; Sobti, R.C.; Mukesh, M. Chapter 3—Mitochondrial DNA: A tool for elucidating molecular phylogenetics and population. In Advances in Animal Experimentation and Modeling; Sobti, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 27–38. [Google Scholar]
- Jin, P.; Tian, Y.; Zang, E.; Zeng, L.; Zhang, Z.; Liu, J.; Shi, L. Complete mitochondrial DNA sequence of Alboglossiphonia lata Oka, 1910 (Rhynchobdellida: Glossiphoniidae) and its phylogenetic analysis. Mitochondrial DNA Part B 2024, 9, 652–656. [Google Scholar] [CrossRef]
- Yue, L.; Xiong, L.; Wang, S.; Wang, Q.; Wang, M.; Chen, H. Genetic diversity analysis of three populations of Whitmania pigra Whitman based on mitochondrial Cytb gene. J. Shanghai Ocean Univ. 2020, 29, 9–16. [Google Scholar] [CrossRef]
- Popa, O.P.; Ștefan, A.; Baltag, E.Ș.; Stratan, A.A.; Popa, L.O.; Surugiu, V. High genetic diversity of Hirudo verbana Carena, 1820 (Annelida: Hirudinea: Hirudinidae) in Romania confirms that the Balkans are refugia within refugium. Diversity 2024, 16, 726. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, B.; Gong, Y.; Yu, X.; Lv, J. Anticoagulant active substances extraction and anti-thrombin activity analysis of several species of leeches. Acta Sci. Nat. Univ. Sunyatseni 2012, 51, 92–96. [Google Scholar]
- Liu, Z.; Zhao, F.; Huang, Z.; Hu, Q.; Meng, R.; Lin, Y.; Qi, J.; Lin, G. Revisiting the Asian buffalo leech (Hirudinaria manillensis) genome: Focus on antithrombotic genes and their corresponding proteins. Genes 2023, 14, 2068. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinf. 2009, 10, 421. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Chevreux, B.; Wetter, T.; Suhai, S. Genome sequence assembly using trace signals and additional aequence information. In Proceedings of the German Conference on Bioinformatics, Hannover, Germany, 4–6 October 1999. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Vences, M.; Patmanidis, S.; Schmidt, J.C.; Matschiner, M.; Miralles, A.; Renner, S.S. Hapsolutely: A user-friendly tool integrating haplotype phasing, network construction, and haploweb calculation. Bioinf. Adv. 2024, 4, vbae083. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Hill, V.; Baele, G. Bayesian estimation of past population dynamics in BEAST 1.10 using the skygrid coalescent model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Brower, A.V. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6491–6495. [Google Scholar] [CrossRef]
- Pons, J.; Ribera, I.; Bertranpetit, J.; Balke, M. Nucleotide substitution rates for the full set of mitochondrial protein-coding genes in Coleoptera. Mol. Phylogenet. Evol. 2010, 56, 796–807. [Google Scholar] [CrossRef]
- NEEM Community Members. Eemian interglacial reconstructed from a Greenland folded ice core. Nature 2013, 493, 489–494. [Google Scholar] [CrossRef]
- McGee, D. Glacial-interglacial precipitation changes. Annu. Rev. Mar. Sci. 2020, 12, 525–557. [Google Scholar] [CrossRef]
- Clark, P.U.; Dyke, A.S.; Shakun, J.D.; Carlson, A.E.; Clark, J.; Wohlfarth, B.; Mitrovica, J.X.; Hostetler, S.W.; McCabe, A.M. The Last Glacial Maximum. Science 2009, 325, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Osman, M.B.; Tierney, J.E.; Zhu, J.; Tardif, R.; Hakim, G.J.; King, J.; Poulsen, C.J. Globally resolved surface temperatures since the Last Glacial Maximum. Nature 2021, 599, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Zhou, J.; Jiang, K.; Chen, J.; Ye, Z.; Xue, H.; Bu, W. The anthropogenic effect of land use on population genetics of Malcus inconspicuus. Evol. Appl. 2023, 16, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA; Chichester, UK, 1987. [Google Scholar]
- Grant, W.A.S.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered. 1998, 89, 415–426. [Google Scholar] [CrossRef]
- Losapio, G.; Lee, J.R.; Fraser, C.I.; Gillespie, M.A.K.; Kerr, N.R.; Zawierucha, K.; Hamilton, T.L.; Hotaling, S.; Kaufmann, R.; Kim, O.-S.; et al. Impacts of deglaciation on biodiversity and ecosystem function. Nat. Rev. Biodivers. 2025, 1, 371–385. [Google Scholar] [CrossRef]
- Zhu, H. Biogeographical evidences help revealing the origin of Hainan island. PLoS ONE 2016, 11, e0151941. [Google Scholar] [CrossRef]
- Lambeck, K.; Chappell, J. Sea level change through the last glacial cycle. Science 2001, 292, 679–686. [Google Scholar] [CrossRef]
- Wang, C.; Mo, W.; Hu, J.; Zhang, L.; Hu, D. Reconstruction of the Cenozoic paleocoastline and evolution of the Qiongzhou strait in the Beibu Gulf—Leizhou Peninsula—Hainan Island. Geol. Rev. 2024, 70, 2400–2410. [Google Scholar] [CrossRef]
- Zhao, F.; Tang, L.; He, B.; Liu, Z.; Wu, Q.; Huang, Z.; Lin, G. Intraspecific variation in the hirudin gene family of the Asian buffalo leech (Hirudinaria manillensis). J. Jinggangshan Univ. (Nat. Sci.) 2024, 45, 38–45. [Google Scholar] [CrossRef]
- Wang, D.; Tong, X.; Wang, B.; Guo, Q.; Yang, L.; Shen, F. The importance of the species and faunna studies on Hirudinea in Yunnan Province. J. Kunming Univ. 2009, 31, 49–51. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locality | City, Province | Longitude | Latitude | Sample Size |
|---|---|---|---|---|
| GDMM | Maoming, Guangdong | 110.393 | 21.829 | 10 |
| GDZJ | Zhanjiang, Guangdong | 110.463 | 21.250 | 10 |
| GXGG | Guigang, Guangxi | 109.620 | 23.065 | 10 |
| GXLZ | Liuzhou, Guangxi | 109.465 | 24.300 | 10 |
| GXYL | Yulin, Guangxi | 110.574 | 22.805 | 10 |
| HNDA | Dingan, Hainan | 110.348 | 19.525 | 12 |
| YNHH | Honghe, Yunnan | 102.580 | 23.315 | 12 |
| Gene | Length | VS | HN | Hd ± SD | Pi ± SD |
|---|---|---|---|---|---|
| COI | 1536 | 34 | 34 | 0.874 ± 0.029 | 0.00198 ± 0.00015 |
| COII | 684 | 17 | 19 | 0.852 ± 0.029 | 0.00258 ± 0.00023 |
| ATP8 | 153 | 3 | 4 | 0.155 ± 0.056 | 0.00104 ± 0.00039 |
| COIII | 783 | 33 | 25 | 0.922 ± 0.014 | 0.00581 ± 0.00024 |
| ND6 | 459 | 14 | 12 | 0.571 ± 0.067 | 0.00182 ± 0.00031 |
| CYTB | 1140 | 35 | 31 | 0.942 ± 0.013 | 0.00378 ± 0.00021 |
| ATP6 | 705 | 13 | 15 | 0.677 ± 0.054 | 0.00142 ± 0.00018 |
| ND5 | 1710 | 66 | 36 | 0.960 ± 0.010 | 0.00416 ± 0.00023 |
| ND4L | 285 | 9 | 9 | 0.561 ± 0.064 | 0.00313 ± 0.00064 |
| ND4 | 1344 | 41 | 29 | 0.922 ± 0.017 | 0.00327 ± 0.00015 |
| ND1 | 921 | 20 | 23 | 0.925 ± 0.013 | 0.00270 ± 0.00012 |
| ND3 | 345 | 8 | 7 | 0.577 ± 0.058 | 0.00362 ± 0.00051 |
| ND2 | 975 | 25 | 24 | 0.917 ± 0.015 | 0.00244 ± 0.00015 |
| Population | VS | HN | Haplotypes | Hd ± SD | Pi ± SD |
|---|---|---|---|---|---|
| GDMM | 68 | 9 | H01–H09 | 0.978 ± 0.054 | 0.00245 ± 0.00026 |
| GDZJ | 59 | 9 | H10–H18 | 0.978 ± 0.054 | 0.00180 ± 0.00035 |
| GXGG | 98 | 10 | H19–H28 | 1.000 ± 0.045 | 0.00236 ± 0.00040 |
| GXLZ | 71 | 10 | H29–H38 | 1.000 ± 0.045 | 0.00183 ± 0.00026 |
| GXYL | 88 | 10 | H39–H48 | 1.000 ± 0.045 | 0.00214 ± 0.00030 |
| HNDA | 75 | 10 | H49–H58 | 0.970 ± 0.044 | 0.00200 ± 0.00047 |
| YNHH | 3 | 3 | H59–H61 | 0.591 ± 0.108 | 0.00011 ± 0.00002 |
| Total | 318 | 61 | — | 0.989 ± 0.006 | 0.00309 ± 0.00008 |
| Population | GDMM | GDZJ | GXGG | GXLZ | GXYL | HNDA | YNHH |
|---|---|---|---|---|---|---|---|
| GDMM | — | 0.008 | 0.009 | <0.001 | <0.001 | <0.001 | <0.001 |
| GDZJ | 0.189 | — | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| GXGG | 0.215 | 0.411 | — | 0.309 | 0.345 | <0.001 | <0.001 |
| GXLZ | 0.342 | 0.528 | 0.010 | — | 0.980 | <0.001 | <0.001 |
| GXYL | 0.298 | 0.490 | 0.009 | 0.000 | — | <0.001 | <0.001 |
| HNDA | 0.279 | 0.445 | 0.351 | 0.435 | 0.395 | — | <0.001 |
| YNHH | 0.675 | 0.792 | 0.667 | 0.738 | 0.700 | 0.725 | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, G.; Yin, J.; Zhang, W.; Huang, Z.; Liu, Z.; Chen, H.; Tang, L.; Zhao, F. Population Genetics of the Asian Buffalo Leech (Hirudinaria manillensis) in Southern China Based on Mitochondrial Protein-Coding Genes. Biology 2025, 14, 926. https://doi.org/10.3390/biology14080926
Lin G, Yin J, Zhang W, Huang Z, Liu Z, Chen H, Tang L, Zhao F. Population Genetics of the Asian Buffalo Leech (Hirudinaria manillensis) in Southern China Based on Mitochondrial Protein-Coding Genes. Biology. 2025; 14(8):926. https://doi.org/10.3390/biology14080926
Chicago/Turabian StyleLin, Gonghua, Jingjing Yin, Wenting Zhang, Zuhao Huang, Zichao Liu, Huanhuan Chen, Lizhou Tang, and Fang Zhao. 2025. "Population Genetics of the Asian Buffalo Leech (Hirudinaria manillensis) in Southern China Based on Mitochondrial Protein-Coding Genes" Biology 14, no. 8: 926. https://doi.org/10.3390/biology14080926
APA StyleLin, G., Yin, J., Zhang, W., Huang, Z., Liu, Z., Chen, H., Tang, L., & Zhao, F. (2025). Population Genetics of the Asian Buffalo Leech (Hirudinaria manillensis) in Southern China Based on Mitochondrial Protein-Coding Genes. Biology, 14(8), 926. https://doi.org/10.3390/biology14080926