
Gut Microbiota Dysbiosis Remodels the Lysine Acetylome of the Mouse Cecum in Early Life
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
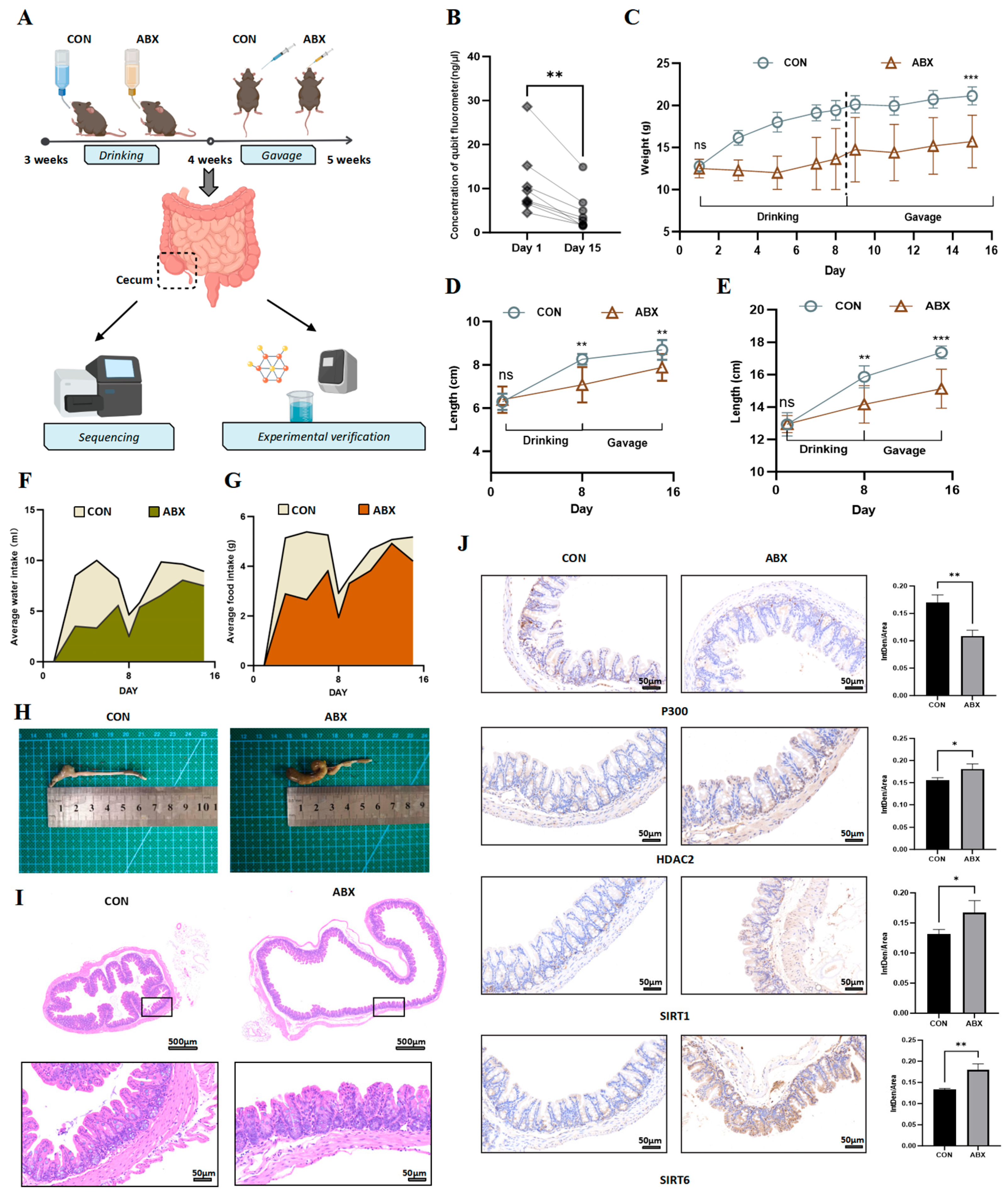
2.1. Animals
2.2. ABX Protocol
2.3. Hematoxylin–Eosin (HE) Staining
2.4. Immunohistochemical (IHC) Staining
2.5. Protein Extraction and Trypsin Digestion
2.6. Acetylated Peptide Enrichment and High-Performance Liquid Chromatography (HPLC) Separation and Mass Spectrometry (MS)
2.7. Statistical Analysis
3. Results
3.1. Gut Microbiota Dysbiosis in Mice Leads to Fluctuations in the Levels of Acetyltransferases and Deacetylases
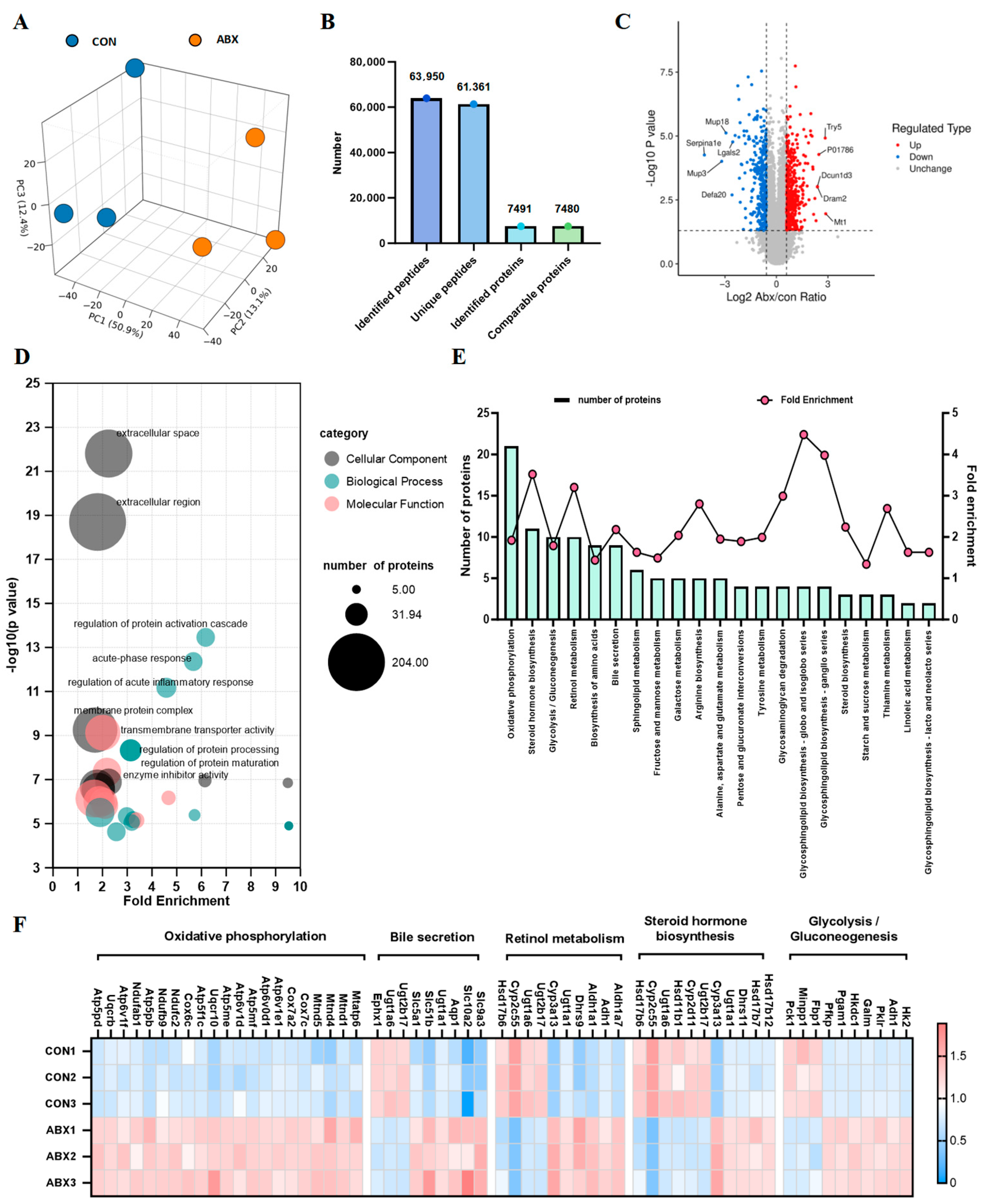
3.2. Gut Microbiota Dysbiosis in the Intestinal Microbiota of Mice Leads to Changes in the Cecal Proteome
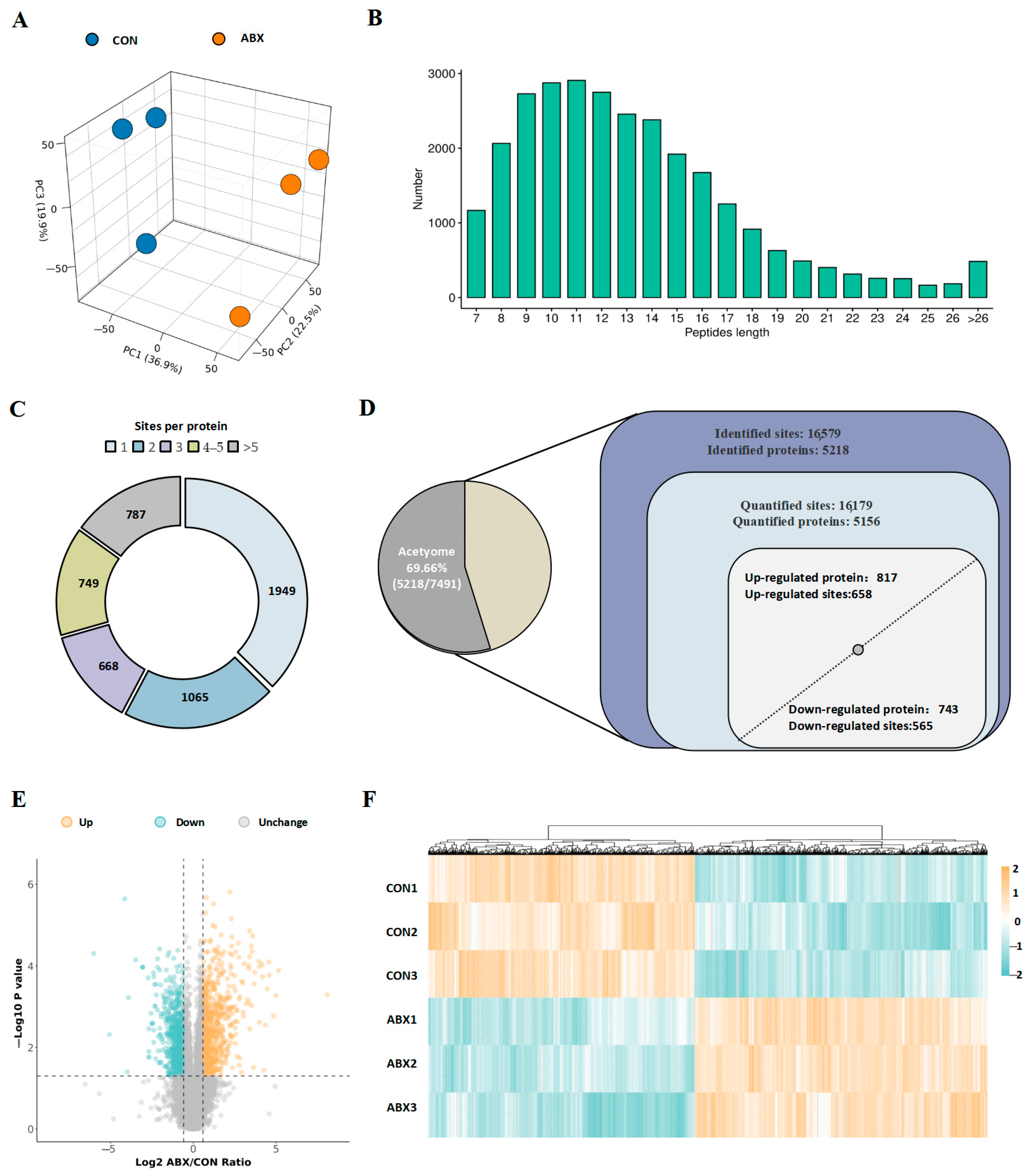
3.3. Identification of Lysine-Acetylated Proteins and Sites in the Cecal Tissue of Mice Induced by Gut Microbiota Dysbiosis
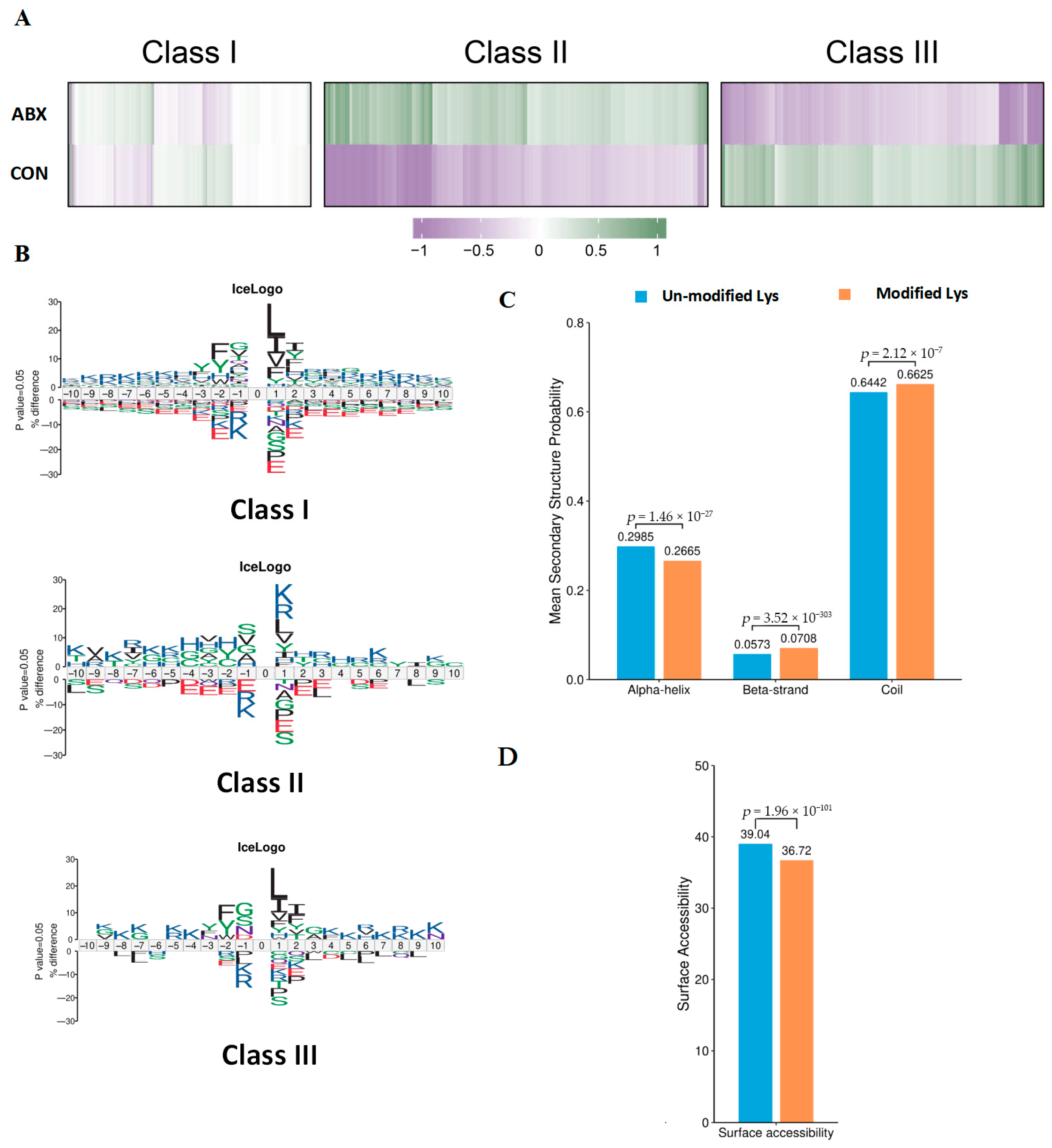
3.4. Characteristics of Acetylation Sites in Mouse Cecal Tissue Induced by Gut Microbiota Dysbiosis
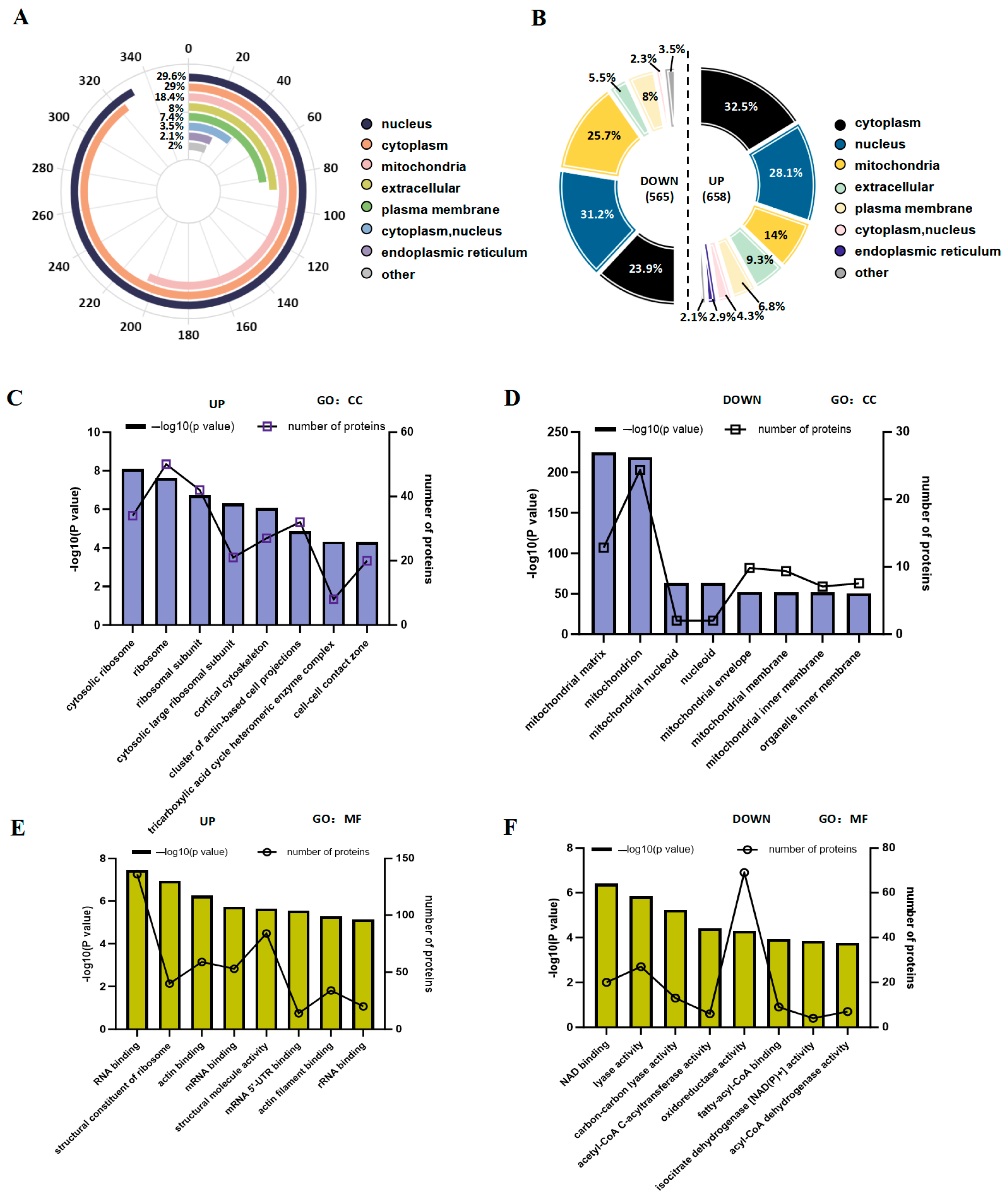
3.5. Analysis of the Subcellular Distribution and Gene Ontology of Acetylated Proteins That Are Significantly up- and Downregulated in Gut Microbiota Dysbiosis
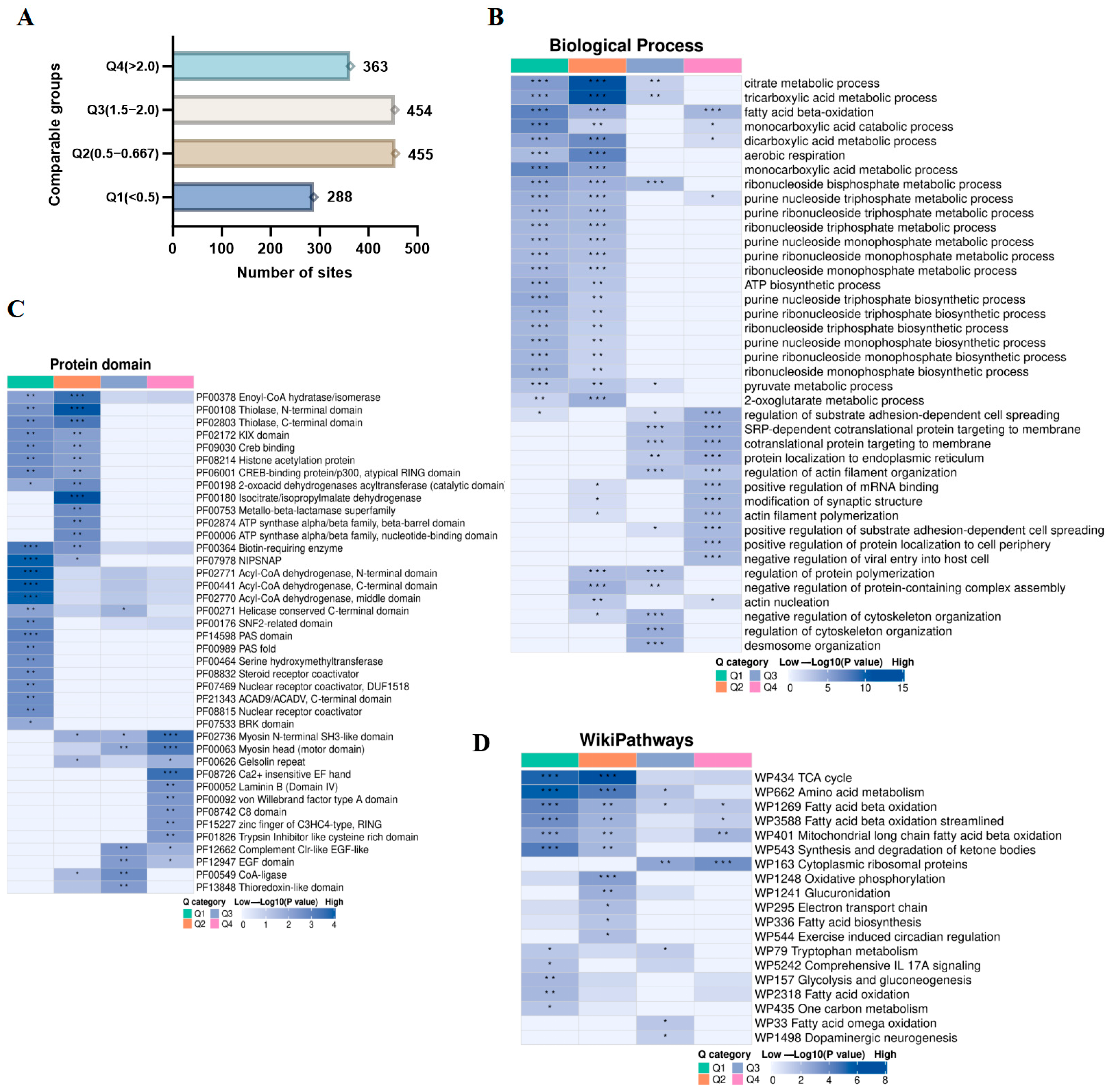
3.6. Enrichment Clustering of Protein Domains and Biological Function Analysis of the Kac Proteome in Gut Microbiota Dysbiosis
3.7. The Impact of Acetylated Lysine on Gut Microbiota Dysbiosis During Early Life Can Disrupt Metabolic Processes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iliev, I.D.; Ananthakrishnan, A.N. Microbiota in inflammatory bowel disease: Mechanisms of disease and therapeutic opportunities. Nat. Rev. Microbiol. 2025, 23, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Pepke, M.L.; Hansen, S.B. Unraveling host regulation of gut microbiota through the epigenome-microbiome axis. Trends Microbiol. 2024, 32, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.R.; Barile, D.; Underwood, M.A.; Mills, D.A. The impact of the milk glycobiome on the neonate gut microbiota. Annu. Rev. Anim. Biosci. 2015, 3, 419–445. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; He, K.; Gu, Z.; Zhao, X. Emerging chemophysiological diversity of gut microbiota metabolites. Trends Pharmacol. Sci. 2024, 45, 824–838. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 2020, 11, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, Y.; Li, Y.; Bai, G.; Pang, J.; Wu, M.; Li, J.; Zhao, X.; Xia, Y. Implications of gut microbiota-mediated epigenetic modifications in intestinal diseases. Gut Microbes 2025, 17, 2508426. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zheng, X.; Fan, J.; Cheng, W.; Yan, T.M.; Lai, Y.; Zhang, N.; Lu, Y.; Qi, J.; Huo, Z.; et al. Antibiotic-Induced Gut Microbiota Dysbiosis Modulates Host Transcriptome and m(6)A Epitranscriptome via Bile Acid Metabolism. Adv. Sci. 2024, 11, e2307981. [Google Scholar] [CrossRef] [PubMed]
- Mann, E.R.; Lam, Y.K.; Uhlig, H.H. Short-chain fatty acids: Linking diet, the microbiome and immunity. Nat. Rev. Immunol. 2024, 24, 577–595. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Woo, V.; Alenghat, T. Epigenetic regulation by gut microbiota. Gut Microbes 2022, 14, 2022407. [Google Scholar] [CrossRef] [PubMed]
- Stricker, S.H.; Köferle, A.; Beck, S. From profiles to function in epigenomics. Nat. Rev. Genet. 2017, 18, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Gates, L.A.; Reis, B.S.; Lund, P.J.; Paul, M.R.; Leboeuf, M.; Djomo, A.M.; Nadeem, Z.; Lopes, M.; Vitorino, F.N.; Unlu, G.; et al. Histone butyrylation in the mouse intestine is mediated by the microbiota and associated with regulation of gene expression. Nat. Metab. 2024, 6, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Ansari, I.; Raddatz, G.; Gutekunst, J.; Ridnik, M.; Cohen, D.; Abu-Remaileh, M.; Tuganbaev, T.; Shapiro, H.; Pikarsky, E.; Elinav, E.; et al. The microbiota programs DNA methylation to control intestinal homeostasis and inflammation. Nat. Microbiol. 2020, 5, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Fellows, R.; Denizot, J.; Stellato, C.; Cuomo, A.; Jain, P.; Stoyanova, E.; Balázsi, S.; Hajnády, Z.; Liebert, A.; Kazakevych, J.; et al. Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat. Commun. 2018, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sun, J.; Cao, H.; Tian, R.; Cai, L.; Ding, W.; Qian, P.Y. Post-translational modifications are enriched within protein functional groups important to bacterial adaptation within a deep-sea hydrothermal vent environment. Microbiome 2016, 4, 49. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Liu, P.; Wang, J.; Wang, Y.; Hou, L.; Gu, W.; Wang, W. Systematic analysis of the lysine acetylome of the pathogenic bacterium Spiroplasma eriocheiris reveals acetylated proteins related to metabolism and helical structure. J. Proteom. 2016, 148, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.G.; Baumgartner, J.T.; Xie, X.; Jew, K.M.; Basisty, N.; Schilling, B.; Kuhn, M.L.; Wolfe, A.J. Mechanisms, Detection, and Relevance of Protein Acetylation in Prokaryotes. mBio 2019, 10, e02708-18. [Google Scholar] [CrossRef] [PubMed]
- Lund, P.J.; Gates, L.A.; Leboeuf, M.; Smith, S.A.; Chau, L.; Lopes, M.; Friedman, E.S.; Saiman, Y.; Kim, M.S.; Shoffler, C.A.; et al. Stable isotope tracing in vivo reveals a metabolic bridge linking the microbiota to host histone acetylation. Cell Rep. 2022, 41, 111809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, K.; Ning, Z.; Mayne, J.; Walker, K.; Chi, H.; Farnsworth, C.L.; Lee, K.; Figeys, D. Exploring the Microbiome-Wide Lysine Acetylation, Succinylation, and Propionylation in Human Gut Microbiota. Anal. Chem. 2021, 93, 6594–6598. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Pei, J.; Lan, J.; Sang, H.; Chen, H.; Yuan, H.; Wu, D.; Zhang, Y.; Wang, Y.; Wang, D.; et al. A SNP of bacterial blc disturbs gut lysophospholipid homeostasis and induces inflammation through epithelial barrier disruption. eBioMedicine 2020, 52, 102652. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yan, W.; Chen, C.; Zeng, Y.; Kong, Y.; He, X.; Pei, P.; Wang, S.; Zhang, T. Acetylome analyses provide novel insights into the effects of chronic intermittent hypoxia on hippocampus-dependent cognitive impairment. Front. Mol. Neurosci. 2024, 17, 1324458. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.L.; Rakus, J.F.; Quenet, D. Acetylation, ADP-ribosylation and methylation of malate dehydrogenase. Essays Biochem. 2024, 68, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Zhang, Y.; Chen, Q.; Wang, Y.; Zhang, D.; Guo, J.; Zhang, Q.; Zhang, W.; Gong, Z. The acetylation of MDH1 and IDH1 is associated with energy metabolism in acute liver failure. iScience 2024, 27, 109678. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, D.; Zhu, Y.; Li, D.; Shen, L.; Wang, Q.; Gao, Y.; Li, X.; Yu, M. Protein lysine acetylation played an important role in NH(3)-induced AEC2 damage and pulmonary fibrosis in piglets. Sci. Total Environ. 2024, 908, 168303. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Xu, Y.; Chen, B.; Ju, K.; Jin, Y.; Chen, H.; Zhang, S.; Luan, X. Structural and biochemical mechanism of short-chain enoyl-CoA hydratase (ECHS1) substrate recognition. Commun. Biol. 2025, 8, 619. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Long, Q.; Wu, H.; Zhou, Y.; Duan, L.; Yuan, H.; Ding, Y.; Huang, Y.; Wu, Y.; Huang, J.; et al. Nuclear localization of mitochondrial TCA cycle enzymes modulates pluripotency via histone acetylation. Nat. Commun. 2022, 13, 7414. [Google Scholar] [CrossRef] [PubMed]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- D’Aquila, P.; Carelli, L.L.; De Rango, F.; Passarino, G.; Bellizzi, D. Gut Microbiota as Important Mediator Between Diet and DNA Methylation and Histone Modifications in the Host. Nutrients 2020, 12, 597. [Google Scholar] [CrossRef] [PubMed]
- Hays, K.E.; Pfaffinger, J.M.; Ryznar, R. The interplay between gut microbiota, short-chain fatty acids, and implications for host health and disease. Gut Microbes 2024, 16, 2393270. [Google Scholar] [CrossRef] [PubMed]
- Nshanian, M.; Gruber, J.J.; Geller, B.S.; Chleilat, F.; Lancaster, S.M.; White, S.M.; Alexandrova, L.; Camarillo, J.M.; Kelleher, N.L.; Zhao, Y.; et al. Short-chain fatty acid metabolites propionate and butyrate are unique epigenetic regulatory elements linking diet, metabolism and gene expression. Nat. Metab. 2025, 7, 196–211. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, Q.; Zhu, H.; Chen, Y.; Lin, G.; Zhang, H.; Chen, W.; Wang, G.; Tian, P. Bifidobacteria with indole-3-lactic acid-producing capacity exhibit psychobiotic potential via reducing neuroinflammation. Cell Rep. Med. 2024, 5, 101798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, Q.; Li, T.; Lu, L.; Wang, F.; Zhang, H.; Liu, Z.; Ma, H.; Zhu, Q.; Wang, J.; et al. Lactobacillus plantarum-derived indole-3-lactic acid ameliorates colorectal tumorigenesis via epigenetic regulation of CD8(+) T cell immunity. Cell Metab. 2023, 35, 943–960.e9. [Google Scholar] [CrossRef] [PubMed]
- Eshleman, E.M.; Rice, T.; Potter, C.; Waddell, A.; Hashimoto-Hill, S.; Woo, V.; Field, S.; Engleman, L.; Lim, H.W.; Schumacher, M.A. Microbiota-derived butyrate restricts tuft cell differentiation via histone deacetylase 3 to modulate intestinal type 2 immunity. Immunity 2024, 57, 319–332.e6. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Wang, Y.; Yu, C.; Qiu, R.; Jiang, Y.; Jia, J.; Tao, Z.; Zhang, L.; Zou, B.; Tang, D. Gut microbiota mediates the inhibition of lymphopoiesis in dietary-restricted mice by suppressing glycolysis. Gut Microbes 2022, 14, 2117509. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, J.; Hou, Q.; Xu, X.; Ren, J.; Ma, L.; Yan, X. Core-predominant gut fungus Kazachstania slooffiae promotes intestinal epithelial glycolysis via lysine desuccinylation in pigs. Microbiome 2023, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Duan, L. The role of microbiota-mitochondria crosstalk in pathogenesis and therapy of intestinal diseases. Pharmacol. Res. 2022, 186, 106530. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, R.; Wang, M.; Peng, R.; Fu, S.; Fu, A.; Le, J.; Yao, Q.; Yuan, T.; Chi, H.; et al. 3β-Hydroxysteroid dehydrogenase expressed by gut microbes degrades testosterone and is linked to depression in males. Cell Host Microbe 2022, 30, 329–339.e5. [Google Scholar] [CrossRef] [PubMed]
- Gudas, L.J. Retinoid metabolism: New insights. J. Mol. Endocrinol. 2022, 69, T37–T49. [Google Scholar] [CrossRef] [PubMed]
- Mayneris-Perxachs, J.; Castells-Nobau, A.; Arnoriaga-Rodríguez, M.; Martin, M.; de la Vega-Correa, L.; Zapata, C.; Burokas, A.; Blasco, G.; Coll, C.; Escrichs, A.; et al. Microbiota alterations in proline metabolism impact depression. Cell Metab. 2022, 34, 681–701.e10. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhao, X.; Liu, J.; Jin, M.; Liu, X.; Yan, J.; Yao, X.; Mao, X.; Li, N.; Liang, B.; et al. TNFα-induced IDH1 hyperacetylation reprograms redox homeostasis and promotes the chemotherapeutic sensitivity. Oncogene 2023, 42, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Ju, H.Q.; Yang, Z.; Ge, Q.; Zhang, Z.; Liu, F.; Yang, L.; Gong, H.; Shi, C.; Qu, L.; et al. Mitochondrial long non-coding RNA GAS5 tunes TCA metabolism in response to nutrient stress. Nat. Metab. 2021, 3, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhou, L.; Shi, Q.; Zhao, Y.; Lin, H.; Zhang, M.; Zhao, S.; Yang, Y.; Ling, Z.Q.; Guan, K.L.; et al. SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth. Embo J. 2015, 34, 1110–1125. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.Y.; Chen, M.H.; Tsao, C.M.; Hii, H.P.; Kuo, C.W.; Ka, S.M.; Wu, C.C.; Shih, C.C. Therapeutic potential of butyrate against heat Stress-Induced intestinal damage, systemic inflammation, and multiple organ Dysfunction: Insights from in vitro and in vivo experiments. Eur. J. Pharmacol. 2025, 999, 177710. [Google Scholar] [CrossRef] [PubMed]
- Michaudel, C.; Danne, C.; Agus, A.; Magniez, A.; Aucouturier, A.; Spatz, M.; Lefevre, A.; Kirchgesner, J.; Rolhion, N.; Wang, Y.; et al. Rewiring the altered tryptophan metabolism as a novel therapeutic strategy in inflammatory bowel diseases. Gut 2023, 72, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Duan, Y.; Luo, S.; Zhou, F.; Wu, Q.; Lu, Z. Short-chain fatty acids and cancer. Trends Cancer 2025, 11, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Kaiko, G.E.; Ryu, S.H.; Koues, O.I.; Collins, P.L.; Solnica-Krezel, L.; Pearce, E.J.; Pearce, E.L.; Oltz, E.M.; Stappenbeck, T.S. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 2016, 165, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, Z.; Lin, M.; Pan, H.; Liu, Y.; Liu, Y.; Xie, Y.; Zhang, J.; Guan, S.; Li, Y.; et al. Acetylation-induced degradation of ECHS1 enhances BCAA accumulation and proliferation in KRAS-mutant colorectal cancer. J. Exp. Clin. Cancer Res. 2025, 44, 164. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.; Sun, X.M.; Huang, F.Q.; Liu, J.F.; Cai, Y.Y.; Wu, X.; Alolga, R.N.; Li, P.; Liu, B.L.; Liu, Q.; et al. The mitochondrial β-oxidation enzyme HADHA restrains hepatic glucagon response by promoting β-hydroxybutyrate production. Nat. Commun. 2022, 13, 386. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Palmfeldt, J.; Gregersen, N.; Makhov, A.M.; Conway, J.F.; Wang, M.; McCalley, S.P.; Basu, S.; Alharbi, H.; Croix, C.S.; et al. Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J. Biol. Chem. 2019, 294, 12380–12391. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.; Chen, S.; Xu, T.; Zhen, W.; Yu, W.; Jiang, D.; Guo, X.; Wang, Z.; Zhang, K.; Li, M.; et al. Histone Deacetylase 3 Couples Mitochondria to Drive IL-1β-Dependent Inflammation by Configuring Fatty Acid Oxidation. Mol. Cell. 2020, 80, 43–58.e7. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ye, M.; Xia, A.; Jiang, H.; Huang, P.; Liu, H.; Hou, R.; Wang, Q.; Li, D.; Xu, J.R.; et al. The Fng3 ING protein regulates H3 acetylation and H4 deacetylation by interacting with two distinct histone-modifying complexes. N. Phytol. 2022, 235, 2350–2364. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.K.Y.; Hu, X.; Chosa, K.; Nguyen, C.; Lin, D.P.; Lai, K.K.; Kato, N.; Higuchi, Y.; Highlander, S.K.; Melendez, E.; et al. p300 Serine 89: A Critical Signaling Integrator and Its Effects on Intestinal Homeostasis and Repair. Cancers 2021, 13, 1288. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.C.; Lu, Y.X.; Ureña, E.; Meilenbrock, R.L.; Catterson, J.H.; Kißler, D.; Fröhlich, J.; Funk, E.; Partridge, L. Sexual identity of enterocytes regulates autophagy to determine intestinal health, lifespan and responses to rapamycin. Nat. Aging 2022, 2, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Cross, T.L.; Simpson, A.M.R.; Lin, C.Y.; Hottmann, N.M.; Bhatt, A.P.; Pellock, S.J.; Nelson, E.R.; Loman, B.R.; Wallig, M.A.; Vivas, E.I.; et al. Gut microbiome responds to alteration in female sex hormone status and exacerbates metabolic dysfunction. Gut Microbes 2024, 16, 2295429. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ding, J.; Ren, H.; Xin, Q.; Li, Z.; Han, L.; Liu, D.; Zhuo, Z.; Liu, C.; Ren, Z. Distinguishable Influence of the Delivery Mode, Feeding Pattern, and Infant Sex on Dynamic Alterations in the Intestinal Microbiota in the First Year of Life. Microb. Ecol. 2023, 86, 1799–1813. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Y.; Shen, J.; He, X.; Liu, F.; Wang, Y.; Wang, Y.; Qiao, Y.; Pei, P.; Wang, S. Gut Microbiota Dysbiosis Remodels the Lysine Acetylome of the Mouse Cecum in Early Life. Biology 2025, 14, 917. https://doi.org/10.3390/biology14080917
Zeng Y, Shen J, He X, Liu F, Wang Y, Wang Y, Qiao Y, Pei P, Wang S. Gut Microbiota Dysbiosis Remodels the Lysine Acetylome of the Mouse Cecum in Early Life. Biology. 2025; 14(8):917. https://doi.org/10.3390/biology14080917
Chicago/Turabian StyleZeng, Yubing, Jinying Shen, Xuejia He, Fan Liu, Yi Wang, Yi Wang, Yanan Qiao, Pei Pei, and Shan Wang. 2025. "Gut Microbiota Dysbiosis Remodels the Lysine Acetylome of the Mouse Cecum in Early Life" Biology 14, no. 8: 917. https://doi.org/10.3390/biology14080917
APA StyleZeng, Y., Shen, J., He, X., Liu, F., Wang, Y., Wang, Y., Qiao, Y., Pei, P., & Wang, S. (2025). Gut Microbiota Dysbiosis Remodels the Lysine Acetylome of the Mouse Cecum in Early Life. Biology, 14(8), 917. https://doi.org/10.3390/biology14080917