
Protective Effects of N-Acetylcysteine in Alleviating Cocaine-Mediated Microglial Activation and Neuroinflammation

 , , and
, , and
Simple Summary
Abstract
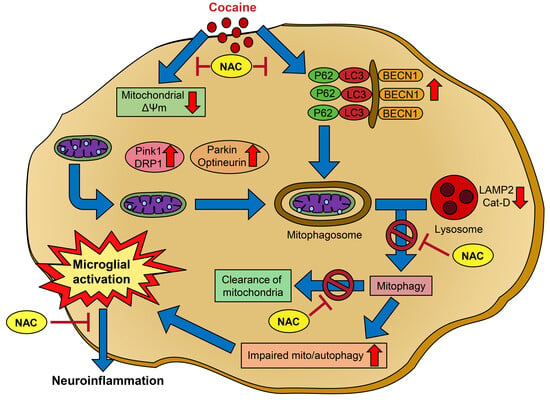
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture and Treatments
2.3. Animal Studies
2.4. Western Blotting
2.5. Measurement of Mitochondrial Membrane Potential
2.6. Measurement of Mitochondrial Superoxide Radicals
2.7. Seahorse XF96 Mitochondrial Stress Test
2.8. Lysosomal Membrane Permeability Assay
2.9. Lysosomal pH Detection
2.10. Behavioral Assessments
2.11. Statistical Analysis
3. Results
3.1. NAC Attenuates Cocaine-Mediated Mito/Autophagy Dysregulation in MPMs
3.2. NAC Alleviates Cocaine-Mediated Mitochondrial Dysfunction
3.3. NAC Ameliorates Cocaine-Mediated Lysosomal Dysfunction
3.4. Protective Effect of NAC on Cocaine-Induced Molecular and Behavioral Alterations in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CNS | Central nervous system |
| CUD | Cocaine use disorder |
| ECAR | Extracellular acidification rate |
| IL | Interleukin |
| LAMP2 | Lysosomal associated membrane protein 2 |
| LMP | Lysosomal membrane permeability |
| MPM | Mouse primary microglia |
| NAC | N-acetylcysteine |
| NLRP3 | NLR family pyrin domain containing 3 |
| NOR | Novel object recognition rate |
| OCR | Oxygen consumption rate |
| OFT | Open field test |
| TNF-α | Tumor necrosis factor-alpha |
| TLR | Toll like receptor |
References
- Substance Abuse and Mental Health Services Administration. 2023 Companion Infographic Report: Results from the 2021, 2022, and 2023 National Surveys on Drug Use and Health (SAMHSA Publication No. PEP24-07-020). Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental Health Services Administration. 2024. Available online: https://www.samhsa.gov/data/sites/default/files/reports/rpt47096/2023-nsduh-companion-report.pdf (accessed on 28 May 2025).
- Butler, A.J.; Rehm, J.; Fischer, B. Health outcomes associated with crack-cocaine use: Systematic review and meta-analyses. Drug Alcohol Depend. 2017, 180, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Hedegaard, H.; Bastian, B.A.; Trinidad, J.P.; Spencer, M.; Warner, M. Drugs Most Frequently Involved in Drug Overdose Deaths: United States, 2011–2016. Natl. Vital Stat. Rep. 2018, 67, 1–14. [Google Scholar] [PubMed]
- Garnett, M.F.; Miniño, A.M. Drug Overdose Deaths in the United States, 2003–2023; NCHS Data Brief, no 522; National Center for Health Statistics: Hyattsville, MD, USA, 2024. [Google Scholar] [CrossRef]
- Horiuchi, Y.; Hunai, H.; Ichimura, K.; Iinuma, T.; Oyama, K. Superstructure of stapes: An analysis by high-resolution computed tomography. Nihon Jibiinkoka Gakkai Kaiho 1989, 92, 383–389. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sharma, H.S.; Muresanu, D.; Sharma, A.; Patnaik, R. Cocaine-induced breakdown of the blood-brain barrier and neurotoxicity. Int. Rev. Neurobiol. 2009, 88, 297–334. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pedrajas, R.; Ramirez-Lamelas, D.T.; Muriach, B.; Sanchez-Villarejo, M.V.; Almansa, I.; Vidal-Gil, L.; Romero, F.J.; Barcia, J.M.; Muriach, M. Cocaine promotes oxidative stress and microglial-macrophage activation in rat cerebellum. Front. Cell. Neurosci. 2015, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, saboteur, or something else? Science 2013, 339, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Lewitus, G.M.; Konefal, S.C.; Greenhalgh, A.D.; Pribiag, H.; Augereau, K.; Stellwagen, D. Microglial TNF-alpha Suppresses Cocaine-Induced Plasticity and Behavioral Sensitization. Neuron 2016, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Krasnova, I.N.; Cadet, J.L. Methamphetamine toxicity and messengers of death. Brain Res. Rev. 2009, 60, 379–407. [Google Scholar] [CrossRef] [PubMed]
- McNally, L.; Bhagwagar, Z.; Hannestad, J. Inflammation, glutamate, and glia in depression: A literature review. CNS Spectr. 2008, 13, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Piechota, M.; Korostynski, M.; Solecki, W.; Gieryk, A.; Slezak, M.; Bilecki, W.; Ziolkowska, B.; Kostrzewa, E.; Cymerman, I.; Swiech, L.; et al. The dissection of transcriptional modules regulated by various drugs of abuse in the mouse striatum. Genome Biol. 2010, 11, R48. [Google Scholar] [CrossRef] [PubMed]
- Melser, S.; Lavie, J.; Benard, G. Mitochondrial degradation and energy metabolism. Biochim. Biophys. Acta 2015, 1853, 2812–2821. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Silva, L.; Silva, A.M.; Moreno, A.J.; Oliveira, C.R.; Santos, M.S. Mitochondrial complex I dysfunction induced by cocaine and cocaine plus morphine in brain and liver mitochondria. Toxicol. Lett. 2013, 219, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Rego, A.C.; Cardoso, S.M.; Borges, F.; Swerdlow, R.H.; Macedo, T.; de Oliveira, C.R. Mitochondrial dysfunction and caspase activation in rat cortical neurons treated with cocaine or amphetamine. Brain Res. 2006, 1089, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Thangaraj, A.; Periyasamy, P.; Guo, M.L.; Chivero, E.T.; Callen, S.; Buch, S. Mitigation of cocaine-mediated mitochondrial damage, defective mitophagy and microglial activation by superoxide dismutase mimetics. Autophagy 2020, 16, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Chivero, E.T.; Thangaraj, A.; Tripathi, A.; Periyasamy, P.; Guo, M.L.; Buch, S. NLRP3 Inflammasome Blockade Reduces Cocaine-Induced Microglial Activation and Neuroinflammation. Mol. Neurobiol. 2021, 58, 2215–2230. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Thangaraj, A.; Chivero, E.T.; Periyasamy, P.; Burkovetskaya, M.E.; Niu, F.; Guo, M.L.; Buch, S. N-Acetylcysteine Reverses Antiretroviral-Mediated Microglial Activation by Attenuating Autophagy-Lysosomal Dysfunction. Front. Neurol. 2020, 11, 840. [Google Scholar] [CrossRef] [PubMed]
- Salamon, S.; Kramar, B.; Marolt, T.P.; Poljsak, B.; Milisav, I. Medical and Dietary Uses of N-Acetylcysteine. Antioxidants 2019, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Zhang, X.; Manda, K.R.; Banks, W.A.; Ercal, N. HIV proteins (gp120 and Tat) and methamphetamine in oxidative stress-induced damage in the brain: Potential role of the thiol antioxidant N-acetylcysteine amide. Free Radic. Biol. Med. 2010, 48, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Kerksick, C.; Willoughby, D. The antioxidant role of glutathione and N-acetyl-cysteine supplements and exercise-induced oxidative stress. J. Int. Soc. Sports Nutr. 2005, 2, 38–44. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, S.C.; Zaretsky, M.D.; Dubs, J.G.; Roederer, M.; Anderson, M.; Green, A.; Mitra, D.; Watanabe, N.; Nakamura, H.; Tjioe, I.; et al. N-acetylcysteine replenishes glutathione in HIV infection. Eur. J. Clin. Investig. 2000, 30, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Nocito Echevarria, M.A.; Andrade Reis, T.; Ruffo Capatti, G.; Siciliano Soares, V.; da Silveira, D.X.; Fidalgo, T.M. N-acetylcysteine for treating cocaine addiction—A systematic review. Psychiatry Res. 2017, 251, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Luo, A.Y.; Zhou, Y.R.; Ren, J.H. N-acetylcysteine reduces oxidative stress, nuclear factor-kappaB activity and cardiomyocyte apoptosis in heart failure. Mol. Med. Rep. 2014, 10, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Chivero, E.T.; Sil, S.; Singh, S.; Thangaraj, A.; Gordon, L.; Evah-Nzoughe, G.B.; Ferguson, N.; Callen, S.; Buch, S. Protective Role of Lactobacillus rhamnosus Probiotic in Reversing Cocaine-Induced Oxidative Stress, Glial Activation and Locomotion in Mice. J. Neuroimmune Pharmacol. 2022, 17, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.T.; Hsieh, P.J.; Lee, H.C.; Lo, C.H.; Tam, K.W.; Loh, E.W. Effectiveness of N-acetylcysteine in Treating Clinical Symptoms of Substance Abuse and Dependence: A Meta-analysis of Randomized Controlled Trials. Clin. Psychopharmacol. Neurosci. 2021, 19, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.Y.; Patwardhan, A.R.; Ang, K.H.; Wang, A.L.; Alquezar, C.; Welch, M.; Nguyen, P.T.; Grabe, M.; Molofsky, A.V.; Arkin, M.R.; et al. Genetically Encoded, pH-Sensitive mTFP1 Biosensor for Probing Lysosomal pH. ACS Sens. 2021, 6, 2168–2180. [Google Scholar] [CrossRef] [PubMed]
- Jedema, H.P.; Song, X.; Aizenstein, H.J.; Bonner, A.R.; Stein, E.A.; Yang, Y.; Bradberry, C.W. Long-Term Cocaine Self-administration Produces Structural Brain Changes That Correlate With Altered Cognition. Biol. Psychiatry 2021, 89, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Kutlu, M.G.; Gould, T.J. Effects of drugs of abuse on hippocampal plasticity and hippocampus-dependent learning and memory: Contributions to development and maintenance of addiction. Learn. Mem. 2016, 23, 515–533. [Google Scholar] [CrossRef] [PubMed]
- Arcuri, C.; Mecca, C.; Bianchi, R.; Giambanco, I.; Donato, R. The Pathophysiological Role of Microglia in Dynamic Surveillance, Phagocytosis and Structural Remodeling of the Developing CNS. Front. Mol. Neurosci. 2017, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Heredia, F.; Salgado, N.; Munoz, P. Microglia Function in the Normal Brain. Adv. Exp. Med. Biol. 2016, 949, 67–92. [Google Scholar] [CrossRef] [PubMed]
- Michell-Robinson, M.A.; Touil, H.; Healy, L.M.; Owen, D.R.; Durafourt, B.A.; Bar-Or, A.; Antel, J.P.; Moore, C.S. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef] [PubMed]
- Cotto, B.; Li, H.; Tuma, R.F.; Ward, S.J.; Langford, D. Cocaine-mediated activation of microglia and microglial MeCP2 and BDNF production. Neurobiol. Dis. 2018, 117, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, P.; Liao, K.; Kook, Y.H.; Niu, F.; Callen, S.E.; Guo, M.L.; Buch, S. Cocaine-Mediated Downregulation of miR-124 Activates Microglia by Targeting KLF4 and TLR4 Signaling. Mol. Neurobiol. 2018, 55, 3196–3210. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.L.; Periyasamy, P.; Liao, K.; Kook, Y.H.; Niu, F.; Callen, S.E.; Buch, S. Cocaine-mediated downregulation of microglial miR-124 expression involves promoter DNA methylation. Epigenetics 2016, 11, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.; Guo, M.; Niu, F.; Yang, L.; Callen, S.E.; Buch, S. Cocaine-mediated induction of microglial activation involves the ER stress-TLR2 axis. J. Neuroinflamm. 2016, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Northcutt, A.L.; Hutchinson, M.R.; Wang, X.; Baratta, M.V.; Hiranita, T.; Cochran, T.A.; Pomrenze, M.B.; Galer, E.L.; Kopajtic, T.A.; Li, C.M.; et al. DAT isn’t all that: Cocaine reward and reinforcement require Toll-like receptor 4 signaling. Mol. Psychiatry 2015, 20, 1525–1537. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 2015, 11, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Mitochondrial dysfunction and longevity in animals: Untangling the knot. Science 2015, 350, 1204–1207. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Autophagy machinery in the context of mammalian mitophagy. Biochim. Biophys. Acta 2015, 1853, 2797–2801. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.R.; Jardim, F.R. Cocaine and mitochondria-related signaling in the brain: A mechanistic view and future directions. Neurochem. Int. 2016, 92, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, P.; Pozos, R.S.; Somanathan, R.; Shangari, N.; O’Brien, P.J. Mechanism of mitochondrial uncouplers, inhibitors, and toxins: Focus on electron transfer, free radicals, and structure-activity relationships. Curr. Med. Chem. 2005, 12, 2601–2623. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, M.T.; Moro, E.; Erickson, R.P. The pathogenesis of lysosomal storage disorders: Beyond the engorgement of lysosomes to abnormal development and neuroinflammation. Hum. Mol. Genet. 2018, 27, R119–R129. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.E.; Kielian, T. Neuroinflammatory paradigms in lysosomal storage diseases. Front. Neurosci. 2015, 9, 417. [Google Scholar] [CrossRef] [PubMed]
- LaRowe, S.D.; Kalivas, P.W.; Nicholas, J.S.; Randall, P.K.; Mardikian, P.N.; Malcolm, R.J. A double-blind placebo-controlled trial of N-acetylcysteine in the treatment of cocaine dependence. Am. J. Addict. 2013, 22, 443–452. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deshetty, U.M.; Oladapo, A.; Mohankumar, Y.; Horanieh, E.; Buch, S.; Periyasamy, P. Protective Effects of N-Acetylcysteine in Alleviating Cocaine-Mediated Microglial Activation and Neuroinflammation. Biology 2025, 14, 893. https://doi.org/10.3390/biology14070893
Deshetty UM, Oladapo A, Mohankumar Y, Horanieh E, Buch S, Periyasamy P. Protective Effects of N-Acetylcysteine in Alleviating Cocaine-Mediated Microglial Activation and Neuroinflammation. Biology. 2025; 14(7):893. https://doi.org/10.3390/biology14070893
Chicago/Turabian StyleDeshetty, Uma Maheswari, Abiola Oladapo, Yazhini Mohankumar, Elias Horanieh, Shilpa Buch, and Palsamy Periyasamy. 2025. "Protective Effects of N-Acetylcysteine in Alleviating Cocaine-Mediated Microglial Activation and Neuroinflammation" Biology 14, no. 7: 893. https://doi.org/10.3390/biology14070893
APA StyleDeshetty, U. M., Oladapo, A., Mohankumar, Y., Horanieh, E., Buch, S., & Periyasamy, P. (2025). Protective Effects of N-Acetylcysteine in Alleviating Cocaine-Mediated Microglial Activation and Neuroinflammation. Biology, 14(7), 893. https://doi.org/10.3390/biology14070893