
Anti-Inflammatory Pathways Mediating tDCS’s Effects on Neuropathic Pain

Simple Summary
Abstract
1. Introduction
2. Therapeutic Effects of tDCS on Neuropathic Pain Management

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studies | Design | Participants | Intervention | Outcomes | |||
|---|---|---|---|---|---|---|---|
| N (M/F) | Type of Injury | tDCS Group | Stimulated Area | Application Parameters | |||
| Ngernyam et al. [15] | Randomized controlled trial | 20 (15/5) | Spinal cord injury | 2 mA anodal tDCS | M1 | A single session of 20 min | Pain intensity (NRS): ↓ |
| Fregni et al. [16] | Randomized controlled trial | 17 (14/3) | Spinal cord injury | 2 mA anodal tDCS | M1 | 20 min for 5 consecutive days | Pain intensity (VAS): ↓ |
| Thibaut et al. [22] | Randomized controlled trial | 33 | Spinal cord injury | 2 mA anodal tDCS | M1 | Phase II: 10 sessions a day during weekday for 2 weeks | Pain intensity (VAS): ↓ |
| Bolognini et al. [18] | Randomized controlled trial | 8 (3/5) | Phantom limb pain | 2 mA anodal tDCS | M1 | A single session for 15 min | Pain intensity (VAS): ↓ |
| Bae et al. [21] | Randomized controlled trial | 14 (7/7) | Post-stroke pain | 2 mA anodal tDCS | M1 | 3 sessions per week for 20 min during 3 consecutive weeks | Pain intensity (VAS): ↓ |
| Kim et al. [25] | Randomized controlled trial | 20 | PDPN | 2 mA anodal tDCS | DLPFC | 20 min for 5 consecutive days | Pain intensity (VAS): ↓ |
| Young et al. [23] | Randomized controlled trial | 30 | Multiple sclerosis | 2 mA anodal tDCS | M1 | 20 min once a day during 5 consecutive days | Pain intensity (VAS): ↓ |
| Ayache et al. [27] | Randomized controlled trial | 16 | Multiple sclerosis | 2 mA anodal tDCS | DLPFC | 20 min for 3 consecutive daily session | Pain intensity (BPI and VAS): ↓ |
| Hagenacker et al. [24] | Randomized controlled trial | 10 | Trigeminal neuralgia | 1 mA anodal tDCS | M1 | 20 min for 2 weeks | Pain intensity (VRS): ↓ |
| Lewis et al. [17] | Randomized controlled trial | 30 (21/9) | Upper limb injury | 1 mA anodal tDCS | M1 | 20 min for 5 consecutive days | No significant improvement in pain intensity (BPI). |
| González-Zamorano et al. [25] | Randomized controlled trial | 22 (10/12) | Parkinson’s disease-related pain | 2 mA anodal tDCS | M1 | 10 sessions of 20 min for 10 consecutive days | Pain intensity (KPPS): ↓ |
3. Inflammatory Mechanisms of tDCS for NP
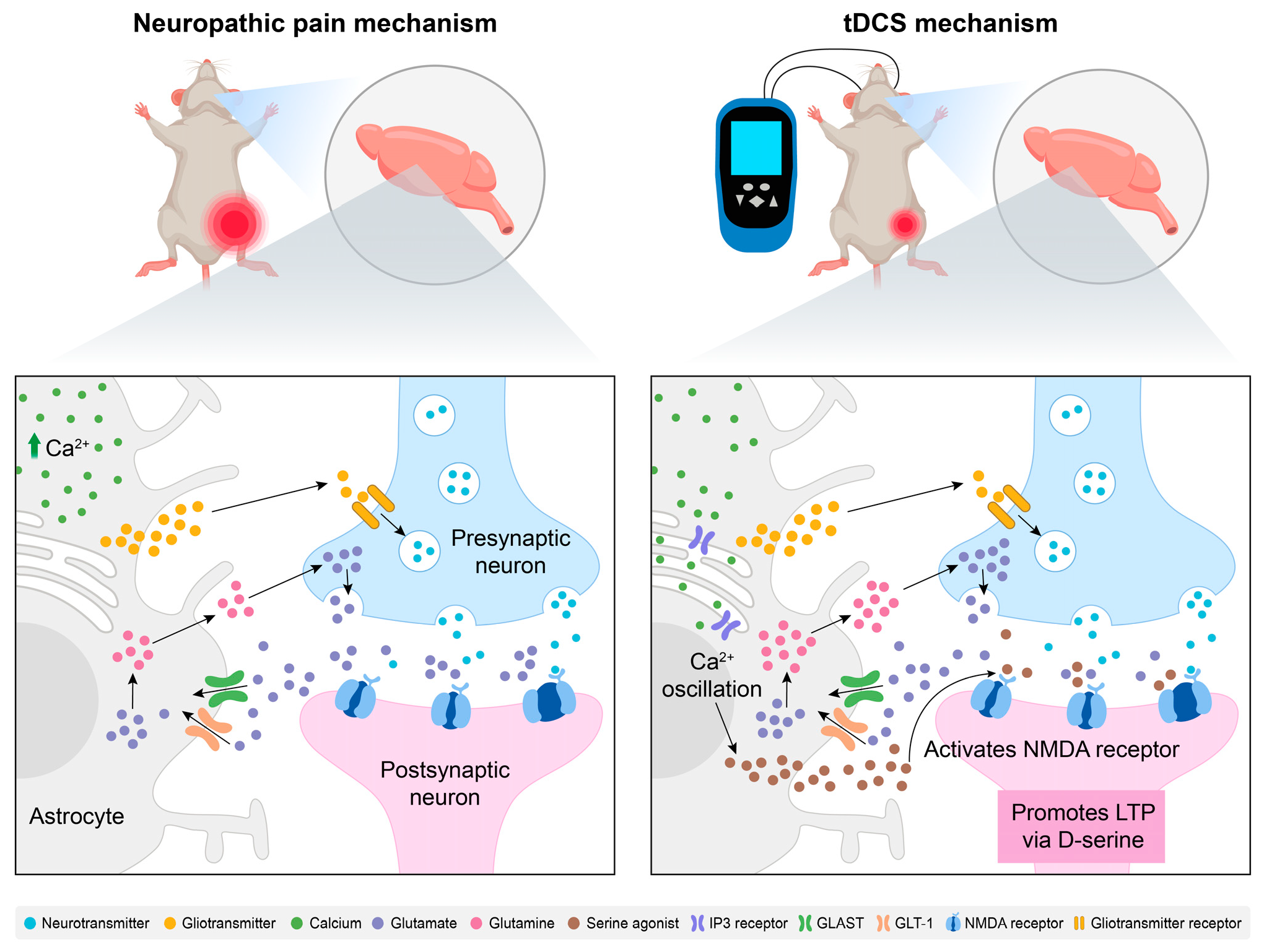
3.1. Brain Inflammatory Response
3.2. Spinal Cord Inflammatory Response
3.3. Dorsal Root Ganglion Inflammatory Response
3.4. Peripheral Nerve Inflammatory Reaction
4. Translational Limitations and Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jensen, T.S.; Baron, R.; Haanpää, M.; Kalso, E.; Loeser, J.D.; Rice, A.S.C.; Treede, R.D. A new definition of neuropathic pain. Pain 2011, 152, 2204–2205. [Google Scholar] [CrossRef] [PubMed]
- Bouhassira, D. Neuropathic pain: Definition, assessment and epidemiology. Rev. Neurol. 2019, 175, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.P.; Vase, L.; Hooten, W.M. Chronic pain: An update on burden, best practices, and new advances. Lancet 2021, 397, 2082–2097. [Google Scholar] [CrossRef] [PubMed]
- Lefaucheur, J.P. Cortical neurostimulation for neuropathic pain: State of the art and perspectives. Pain 2016, 157, S81–S89. [Google Scholar] [CrossRef] [PubMed]
- Antal, A.; Herrmann, C.S. Transcranial Alternating Current and Random Noise Stimulation: Possible Mechanisms. Neural Plast. 2016, 2016, 3616807. [Google Scholar] [CrossRef] [PubMed]
- Brunoni, A.R.; Amadera, J.; Berbel, B.; Volz, M.S.; Rizzerio, B.G.; Fregni, F. A systematic review on reporting and assessment of adverse effects associated with transcranial direct current stimulation. Int. J. Neuropsychopharmacol. 2011, 14, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, M.A.; Paulus, W. Excitability changes induced in the human motor cortex by weak transcranial direct current stimulation. J. Physiol. 2000, 527 Pt 3, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.Y.; Stohler, C.S.; Herr, D.R. Role of the Prefrontal Cortex in Pain Processing. Mol. Neurobiol. 2019, 56, 1137–1166. [Google Scholar] [CrossRef] [PubMed]
- Sacerdote, P.; Franchi, S.; Moretti, S.; Castelli, M.; Procacci, P.; Magnaghi, V.; Panerai, A.E. Cytokine modulation is necessary for efficacious treatment of experimental neuropathic pain. J. Neuroimmune Pharmacol. 2013, 8, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Cioato, S.G.; Medeiros, L.F.; Marques Filho, P.R.; Vercelino, R.; de Souza, A.; Scarabelot, V.L.; de Oliveira, C.; Adachi, L.N.; Fregni, F.; Caumo, W.; et al. Long-Lasting Effect of Transcranial Direct Current Stimulation in the Reversal of Hyperalgesia and Cytokine Alterations Induced by the Neuropathic Pain Model. Brain Stimul. 2016, 9, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Lopes, B.C.; Medeiros, L.F.; Stein, D.J.; Cioato, S.G.; de Souza, V.S.; Medeiros, H.R.; Sanches, P.R.S.; Fregni, F.; Caumo, W.; Torres, I.L.S. tDCS and exercise improve anxiety-like behavior and locomotion in chronic pain rats via modulation of neurotrophins and inflammatory mediators. Behav. Brain Res. 2021, 404, 113173. [Google Scholar] [CrossRef] [PubMed]
- Fregni, F.; El-Hagrassy, M.M.; Pacheco-Barrios, K.; Carvalho, S.; Leite, J.; Simis, M.; Brunelin, J.; Nakamura-Palacios, E.M.; Marangolo, P.; Venkatasubramanian, G.; et al. Evidence-Based Guidelines and Secondary Meta-Analysis for the Use of Transcranial Direct Current Stimulation in Neurological and Psychiatric Disorders. Int. J. Neuropsychopharmacol. 2021, 24, 256–313. [Google Scholar] [CrossRef] [PubMed]
- Stagg, C.J.; Nitsche, M.A. Physiological basis of transcranial direct current stimulation. Neurosci. A Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2011, 17, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Lefaucheur, J.P.; Antal, A.; Ayache, S.S.; Benninger, D.H.; Brunelin, J.; Cogiamanian, F.; Cotelli, M.; De Ridder, D.; Ferrucci, R.; Langguth, B.; et al. Evidence-based guidelines on the therapeutic use of transcranial direct current stimulation (tDCS). Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2017, 128, 56–92. [Google Scholar] [CrossRef] [PubMed]
- Ngernyam, N.; Jensen, M.P.; Arayawichanon, P.; Auvichayapat, N.; Tiamkao, S.; Janjarasjitt, S.; Punjaruk, W.; Amatachaya, A.; Aree-uea, B.; Auvichayapat, P. The effects of transcranial direct current stimulation in patients with neuropathic pain from spinal cord injury. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2015, 126, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Fregni, F.; Boggio, P.S.; Lima, M.C.; Ferreira, M.J.; Wagner, T.; Rigonatti, S.P.; Castro, A.W.; Souza, D.R.; Riberto, M.; Freedman, S.D.; et al. A sham-controlled, phase II trial of transcranial direct current stimulation for the treatment of central pain in traumatic spinal cord injury. Pain 2006, 122, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.N.; Rice, D.A.; Kluger, M.; McNair, P.J. Transcranial direct current stimulation for upper limb neuropathic pain: A double-blind randomized controlled trial. Eur. J. Pain 2018, 22, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Bolognini, N.; Olgiati, E.; Maravita, A.; Ferraro, F.; Fregni, F. Motor and parietal cortex stimulation for phantom limb pain and sensations. Pain 2013, 154, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, Y.; Wan, R.; Feng, B.; Zhang, Z.; Lin, Y.; Wang, Y. The effect of high-definition transcranial direct current stimulation on pain processing in a healthy population: A single-blinded crossover controlled study. Neurosci. Lett. 2022, 767, 136304. [Google Scholar] [CrossRef] [PubMed]
- Burns, E.; Chipchase, L.S.; Schabrun, S.M. Primary sensory and motor cortex function in response to acute muscle pain: A systematic review and meta-analysis. Eur. J. Pain 2016, 20, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Kim, G.D.; Kim, K.Y. Analgesic effect of transcranial direct current stimulation on central post-stroke pain. Tohoku J. Exp. Med. 2014, 234, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Thibaut, A.; Carvalho, S.; Morse, L.R.; Zafonte, R.; Fregni, F. Delayed pain decrease following M1 tDCS in spinal cord injury: A randomized controlled clinical trial. Neurosci. Lett. 2017, 658, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Zoghi, M.; Khan, F.; Galea, M.P. The Effect of Transcranial Direct Current Stimulation on Chronic Neuropathic Pain in Patients with Multiple Sclerosis: Randomized Controlled Trial. Pain Med. 2020, 21, 3451–3457. [Google Scholar] [CrossRef] [PubMed]
- Hagenacker, T.; Bude, V.; Naegel, S.; Holle, D.; Katsarava, Z.; Diener, H.C.; Obermann, M. Patient-conducted anodal transcranial direct current stimulation of the motor cortex alleviates pain in trigeminal neuralgia. J. Headache Pain 2014, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- González-Zamorano, Y.; José Sánchez-Cuesta, F.; Moreno-Verdú, M.; Arroyo-Ferrer, A.; Fernández-Carnero, J.; Chaudhuri, K.R.; Fieldwalker, A.; Romero, J.P. TDCS for parkinson’s disease disease-related pain: A randomized trial. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2024, 161, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Ku, J.; Kim, H.J.; Im, D.J.; Lee, H.S.; Han, K.A.; Kang, Y.J. Randomized, sham controlled trial of transcranial direct current stimulation for painful diabetic polyneuropathy. Ann. Rehabil. Med. 2013, 37, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Ayache, S.S.; Palm, U.; Chalah, M.A.; Al-Ani, T.; Brignol, A.; Abdellaoui, M.; Dimitri, D.; Sorel, M.; Créange, A.; Lefaucheur, J.P. Prefrontal tDCS Decreases Pain in Patients with Multiple Sclerosis. Front. Neurosci. 2016, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.B.; Teixeira Costa, B.; Duarte, D.; Fregni, F. Transcranial Direct Current Stimulation as a Therapeutic Tool for Chronic Pain. J. ECT 2018, 34, e36–e50. [Google Scholar] [CrossRef] [PubMed]
- Kumru, H.; Soler, D.; Vidal, J.; Navarro, X.; Tormos, J.M.; Pascual-Leone, A.; Valls-Sole, J. The effects of transcranial direct current stimulation with visual illusion in neuropathic pain due to spinal cord injury: An evoked potentials and quantitative thermal testing study. Eur. J. Pain 2013, 17, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Boggio, P.S.; Amancio, E.J.; Correa, C.F.; Cecilio, S.; Valasek, C.; Bajwa, Z.; Freedman, S.D.; Pascual-Leone, A.; Edwards, D.J.; Fregni, F. Transcranial DC stimulation coupled with TENS for the treatment of chronic pain: A preliminary study. Clin. J. Pain 2009, 25, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.D.; Kumru, H.; Pelayo, R.; Vidal, J.; Tormos, J.M.; Fregni, F.; Navarro, X.; Pascual-Leone, A. Effectiveness of transcranial direct current stimulation and visual illusion on neuropathic pain in spinal cord injury. Brain 2010, 133, 2565–2577. [Google Scholar] [CrossRef] [PubMed]
- Flor, H.; Nikolajsen, L.; Staehelin Jensen, T. Phantom limb pain: A case of maladaptive CNS plasticity? Nat. Rev. Neurosci. 2006, 7, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Meacham, K.; Shepherd, A.; Mohapatra, D.P.; Haroutounian, S. Neuropathic Pain: Central vs. Peripheral Mechanisms. Curr. Pain Headache Rep. 2017, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Boggio, P.S.; Zaghi, S.; Fregni, F. Modulation of emotions associated with images of human pain using anodal transcranial direct current stimulation (tDCS). Neuropsychologia 2009, 47, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; McIntosh, J.M. Nicotinic acetylcholine receptors in neuropathic and inflammatory pain. FEBS Lett. 2018, 592, 1045–1062. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Xu, Y.; Cen, B.; Xia, Y.; Yao, L.; Zheng, Y.; Zhao, J.; He, S.; Chen, Y. Neuregulin1-ErbB4 Signaling in Spinal Cord Participates in Electroacupuncture Analgesia in Inflammatory Pain. Front. Neurosci. 2021, 15, 636348. [Google Scholar] [CrossRef] [PubMed]
- Hou, A.L.; Xu, W.D. A model of neuropathic pain in brachial plexus avulsion injury and associated spinal glial cell activation. J. Pain Res. 2018, 11, 3171–3179. [Google Scholar] [CrossRef] [PubMed]
- Nasseri, B.; Zaringhalam, J.; Daniali, S.; Manaheji, H.; Abbasnejad, Z.; Nazemian, V. Thymulin treatment attenuates inflammatory pain by modulating spinal cellular and molecular signaling pathways. Int. Immunopharmacol. 2019, 70, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Király, K.; Kozsurek, M.; Lukácsi, E.; Barta, B.; Alpár, A.; Balázsa, T.; Fekete, C.; Szabon, J.; Helyes, Z.; Bölcskei, K.; et al. Glial cell type-specific changes in spinal dipeptidyl peptidase 4 expression and effects of its inhibitors in inflammatory and neuropatic pain. Sci. Rep. 2018, 8, 3490. [Google Scholar] [CrossRef] [PubMed]
- Song, G.J.; Rahman, M.H.; Jha, M.K.; Gupta, D.P.; Park, S.H.; Kim, J.H.; Lee, S.H.; Lee, I.K.; Sim, T.; Bae, Y.C.; et al. A Bcr-Abl Inhibitor GNF-2 Attenuates Inflammatory Activation of Glia and Chronic Pain. Front. Pharmacol. 2019, 10, 543. [Google Scholar] [CrossRef] [PubMed]
- Bartolucci, M.L.; Marini, I.; Bortolotti, F.; Impellizzeri, D.; Di Paola, R.; Bruschetta, G.; Crupi, R.; Portelli, M.; Militi, A.; Oteri, G.; et al. Micronized palmitoylethanolamide reduces joint pain and glial cell activation. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2018, 67, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Scheuren, P.S.; Calvo, M. Exploring neuroinflammation: A key driver in neuropathic pain disorders. Int. Rev. Neurobiol. 2024, 179, 311–338. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Song, E.M.; Do, Y.H.; Ahn, S.; Oh, J.Y.; Hwang, T.Y.; Ryu, Y.; Jeon, S.; Song, M.Y.; Park, H.J. Acupuncture alleviates chronic pain and comorbid conditions in a mouse model of neuropathic pain: The involvement of DNA methylation in the prefrontal cortex. Pain 2021, 162, 514–530. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.H.; Zhao, W.J.; Yin, J.B.; Chen, Y.B.; Li, J.N.; Feng, B.; Lu, Y.C.; Wang, J.; Dong, Y.L.; Li, Y.Q. A Neural Circuit from Thalamic Paraventricular Nucleus to Central Amygdala for the Facilitation of Neuropathic Pain. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 7837–7854. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gadotti, V.M.; Chen, L.; Souza, I.A.; Huang, S.; Wang, D.; Ramakrishnan, C.; Deisseroth, K.; Zhang, Z.; Zamponi, G.W. A neuronal circuit for activating descending modulation of neuropathic pain. Nat. Neurosci. 2019, 22, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, T.; Ren, Q.; Li, N.; Wang, F.; Bi, Y.; Hu, L. Transcutaneous Electrical Nerve Stimulation in Relieving Neuropathic Pain: Basic Mechanisms and Clinical Applications. Curr. Pain Headache Rep. 2020, 24, 14. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Huh, Y. Astrocytic Calcium Dynamics Along the Pain Pathway. Front. Cell. Neurosci. 2020, 14, 594216. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Gharbi, T.; Zhang, Z.; Yang, G.Y. The Function of Astrocyte Mediated Extracellular Vesicles in Central Nervous System Diseases. Front. Cell Dev. Biol. 2020, 8, 568889. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Chen, F.; Tang, Y.; Zhao, Y.; Zhu, L.; Xiang, L.; Ning, L.; Zhou, W.; Chen, Y.; Wang, L.; et al. mGluR5-mediated astrocytes hyperactivity in the anterior cingulate cortex contributes to neuropathic pain in male mice. Commun. Biol. 2025, 8, 266. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Verkhratsky, A. The astrocyte excitability brief: From receptors to gliotransmission. Neurochem. Int. 2012, 61, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed]
- Zorec, R.; Araque, A.; Carmignoto, G.; Haydon, P.G.; Verkhratsky, A.; Parpura, V. Astroglial excitability and gliotransmission: An appraisal of Ca2+ as a signalling route. ASN Neuro 2012, 4, AN20110061. [Google Scholar] [CrossRef] [PubMed]
- Danjo, Y.; Shigetomi, E.; Hirayama, Y.J.; Kobayashi, K.; Ishikawa, T.; Fukazawa, Y.; Shibata, K.; Takanashi, K.; Parajuli, B.; Shinozaki, Y.; et al. Transient astrocytic mGluR5 expression drives synaptic plasticity and subsequent chronic pain in mice. J. Exp. Med. 2022, 219, e20210989. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Hayashi, H.; Ishikawa, T.; Shibata, K.; Shigetomi, E.; Shinozaki, Y.; Inada, H.; Roh, S.E.; Kim, S.J.; Lee, G.; et al. Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J. Clin. Investig. 2016, 126, 1983–1997. [Google Scholar] [CrossRef] [PubMed]
- 5Liu, M.G.; Chen, J. Roles of the hippocampal formation in pain information processing. Neurosci. Bull. 2009, 25, 237–266. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.M.; Neugebauer, V. Cortico-limbic pain mechanisms. Neurosci. Lett. 2019, 702, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, H.; Yan, X.; Wu, Y.; Wei, G.; Wu, X.; Tian, X.; Xiong, Y.; Wu, G.; Wen, H. Transcranial Direct Current Stimulation Alleviates Neurovascular Unit Dysfunction in Mice With Preclinical Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 857415. [Google Scholar] [CrossRef] [PubMed]
- Leffa, D.T.; Bellaver, B.; Salvi, A.A.; de Oliveira, C.; Caumo, W.; Grevet, E.H.; Fregni, F.; Quincozes-Santos, A.; Rohde, L.A.; Torres, I.L.S. Transcranial direct current stimulation improves long-term memory deficits in an animal model of attention-deficit/hyperactivity disorder and modulates oxidative and inflammatory parameters. Brain Stimul. 2018, 11, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Zin, L.E.F.; Vizuete, A.F.K.; Callai, E.M.M.; Catarina, L.S.; Fróes, F.; Moreira, A.P.; de Oliveira Marques, C.; Leal, M.B.; Ponzoni, D.; Puricelli, E.; et al. Astroglial Alterations in the Hippocampus of Rats Submitted to a Single Trans-Cranial Direct Current Stimulation Trial. Neurochem. Res. 2023, 48, 3447–3456. [Google Scholar] [CrossRef] [PubMed]
- Ngernyam, N.; Jensen, M.P.; Auvichayapat, N.; Punjaruk, W.; Auvichayapat, P. Transcranial Direct Current Stimulation in Neuropathic Pain. J. Pain Relief 2013, S3, 1. [Google Scholar] [CrossRef] [PubMed]
- Liebetanz, D.; Nitsche, M.A.; Tergau, F.; Paulus, W. Pharmacological approach to the mechanisms of transcranial DC-stimulation-induced after-effects of human motor cortex excitability. Brain A J. Neurol. 2002, 125, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, B.; Reis, J.; Martinowich, K.; Schambra, H.M.; Ji, Y.; Cohen, L.G.; Lu, B. Direct current stimulation promotes BDNF-dependent synaptic plasticity: Potential implications for motor learning. Neuron 2010, 66, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Papouin, T.; Oliet, S.H.; Rusakov, D.A. Long-term potentiation depends on release of D-serine from astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Takata, N.; Mishima, T.; Hisatsune, C.; Nagai, T.; Ebisui, E.; Mikoshiba, K.; Hirase, H. Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 18155–18165. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ge, W.; Chen, Y.; Zhang, Z.; Shen, W.; Wu, C.; Poo, M.; Duan, S. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc. Natl. Acad. Sci. USA 2003, 100, 15194–15199. [Google Scholar] [CrossRef] [PubMed]
- Agulhon, C.; Petravicz, J.; McMullen, A.B.; Sweger, E.J.; Minton, S.K.; Taves, S.R.; Casper, K.B.; Fiacco, T.A.; McCarthy, K.D. What is the role of astrocyte calcium in neurophysiology? Neuron 2008, 59, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.; Robitaille, R.; Volterra, A. Gliotransmitters travel in time and space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Monai, H.; Ohkura, M.; Tanaka, M.; Oe, Y.; Konno, A.; Hirai, H.; Mikoshiba, K.; Itohara, S.; Nakai, J.; Iwai, Y.; et al. Calcium imaging reveals glial involvement in transcranial direct current stimulation-induced plasticity in mouse brain. Nat. Commun. 2016, 7, 11100. [Google Scholar] [CrossRef] [PubMed]
- Takeda, I.; Yoshihara, K.; Cheung, D.L.; Kobayashi, T.; Agetsuma, M.; Tsuda, M.; Eto, K.; Koizumi, S.; Wake, H.; Moorhouse, A.J.; et al. Controlled activation of cortical astrocytes modulates neuropathic pain-like behaviour. Nat. Commun. 2022, 13, 4100. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Nackley, A.; Huh, Y.; Terrando, N.; Maixner, W. Neuroinflammation and Central Sensitization in Chronic and Widespread Pain. Anesthesiology 2018, 129, 343–366. [Google Scholar] [CrossRef] [PubMed]
- Grace, P.M.; Hutchinson, M.R.; Maier, S.F.; Watkins, L.R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014, 14, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Zhang, L.; Cheng, J.K.; Ji, R.R. Cytokine mechanisms of central sensitization: Distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 5189–5194. [Google Scholar] [CrossRef] [PubMed]
- Lanza, M.; Casili, G.; Campolo, M.; Paterniti, I.; Colarossi, C.; Mare, M.; Giuffrida, R.; Caffo, M.; Esposito, E.; Cuzzocrea, S. Immunomodulatory Effect of Microglia-Released Cytokines in Gliomas. Brain Sci. 2021, 11, 466. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, D.R.; Blanco-Luquin, I.; Mendioroz, M. The Participation of Microglia in Neurogenesis: A Review. Brain Sci. 2021, 11, 658. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Xu, G.; Zhang, X.; Xu, L.; Zhou, H.; Ma, Z.; Shen, X.; Zhu, J.; Shen, R. Paeoniflorin Attenuates Inflammatory Pain by Inhibiting Microglial Activation and Akt-NF-κB Signaling in the Central Nervous System. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 47, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Beyer, C. A Fatal Alliance between Microglia, Inflammasomes, and Central Pain. Int. J. Mol. Sci. 2020, 21, 3764. [Google Scholar] [CrossRef] [PubMed]
- Ward, H.; West, S.J. Microglia: Sculptors of neuropathic pain? R. Soc. Open Sci. 2020, 7, 200260. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Luo, X.; Qadri, M.Y.; Berta, T.; Ji, R.R. Sex-Dependent Glial Signaling in Pathological Pain: Distinct Roles of Spinal Microglia and Astrocytes. Neurosci. Bull. 2018, 34, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Ferro, A.; Auguste, Y.S.S.; Cheadle, L. Microglia, Cytokines, and Neural Activity: Unexpected Interactions in Brain Development and Function. Front. Immunol. 2021, 12, 703527. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shui, X.; Sun, R.; Wan, L.; Zhang, B.; Xiao, B.; Luo, Z. Microglial Phenotypic Transition: Signaling Pathways and Influencing Modulators Involved in Regulation in Central Nervous System Diseases. Front. Cell. Neurosci. 2021, 15, 736310. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Feng, Z.; Chen, H.; Min, L.; Wen, H.; Liu, H.; Hou, J. Transcranial direct current stimulation regulates phenotypic transformation of microglia to relieve neuropathic pain induced by spinal cord injury. Front. Behav. Neurosci. 2023, 17, 1147693. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.S.; Lopes, B.C.; Medeiros, L.F.; Assumpção, J.A.F.; de Souza, A.; Salvi, A.A.; da Silva, L.S.; Fregni, F.; Caumo, W.; Torres, I.L.S. Transcranial Direct Current Stimulation (tDCS) Induces Analgesia in Rats with Neuropathic Pain and Alcohol Abstinence. Neurochem. Res. 2020, 45, 2653–2663. [Google Scholar] [CrossRef] [PubMed]
- Jayathilake, N.J.; Phan, T.T.; Kim, J.; Lee, K.P.; Park, J.M. Modulating neuroplasticity for chronic pain relief: Noninvasive neuromodulation as a promising approach. Exp. Mol. Med. 2025, 57, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.P.; Chen, S.Y.; Pang, Q.M.; Zhang, M.; Wu, X.C.; Wan, X.; Wan, W.H.; Ao, J.; Zhang, T. Advances in the research of the role of macrophage/microglia polarization-mediated inflammatory response in spinal cord injury. Front. Immunol. 2022, 13, 1014013. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jirachaipitak, S.; Wrigley, P.; Xu, H.; Euasobhon, P. Transcranial direct current stimulation for spinal cord injury-associated neuropathic pain. Korean J. Pain 2021, 34, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Stemkowski, P.L.; Smith, P.A. Sensory neurons, ion channels, inflammation and the onset of neuropathic pain. Can. J. Neurol. Sci. Le J. Can. Des Sci. Neurol. 2012, 39, 416–435. [Google Scholar] [CrossRef] [PubMed]
- White, F.A.; Jung, H.; Miller, R.J. Chemokines and the pathophysiology of neuropathic pain. Proc. Natl. Acad. Sci. USA 2007, 104, 20151–20158. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.S.Y.; Kayani, K.; Whyte-Oshodi, D.; Whyte-Oshodi, A.; Nachiappan, N.; Gnanarajah, S.; Mohammed, R. Voltage gated sodium channels as therapeutic targets for chronic pain. J. Pain Res. 2019, 12, 2709–2722. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.A. K+ Channels in Primary Afferents and Their Role in Nerve Injury-Induced Pain. Front. Cell. Neurosci. 2020, 14, 566418. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.; Myers, R.R. Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. Neuroreport 1996, 7, 2897–2901. [Google Scholar] [CrossRef] [PubMed]
- Dubový, P.; Jancálek, R.; Klusáková, I.; Svízenská, I.; Pejchalová, K. Intra- and extraneuronal changes of immunofluorescence staining for TNF-alpha and TNFR1 in the dorsal root ganglia of rat peripheral neuropathic pain models. Cell. Mol. Neurobiol. 2006, 26, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pang, R.P.; Shen, K.F.; Zimmermann, M.; Xin, W.J.; Li, Y.Y.; Liu, X.G. TNF-α enhances the currents of voltage gated sodium channels in uninjured dorsal root ganglion neurons following motor nerve injury. Exp. Neurol. 2011, 227, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shen, Z.; Xu, H.; Zhang, K.; Guo, M.; Wang, F.; Li, J. BDNF Participates in Chronic Constriction Injury-Induced Neuropathic Pain via Transcriptionally Activating P2X7 in Primary Sensory Neurons. Mol. Neurobiol. 2021, 58, 4226–4236. [Google Scholar] [CrossRef] [PubMed]
- Michael, G.J.; Averill, S.; Shortland, P.J.; Yan, Q.; Priestley, J.V. Axotomy results in major changes in BDNF expression by dorsal root ganglion cells: BDNF expression in large trkB and trkC cells, in pericellular baskets, and in projections to deep dorsal horn and dorsal column nuclei. Eur. J. Neurosci. 1999, 11, 3539–3551. [Google Scholar] [CrossRef] [PubMed]
- Malcangio, M. Role of the immune system in neuropathic pain. Scand. J. Pain 2019, 20, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Muley, M.M.; Beggs, S.; Salter, M.W. Microglia-independent peripheral neuropathic pain in male and female mice. Pain 2022, 163, e1129–e1144. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Liu, H.; Hamel, K.A.; Morvan, M.G.; Yu, S.; Leff, J.; Guan, Z.; Braz, J.M.; Basbaum, A.I. Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain. Nat. Commun. 2020, 11, 264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; de Carvalho-Barbosa, M.; Kavelaars, A.; Heijnen, C.J.; Albrecht, P.J.; Dougherty, P.M. Dorsal Root Ganglion Infiltration by Macrophages Contributes to Paclitaxel Chemotherapy-Induced Peripheral Neuropathy. J. Pain 2016, 17, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.F.; Zhu, H.Q.; Wei, X.H.; Wang, J.; Li, Y.Y.; Pang, R.P.; Liu, X.G. Interleukin-10 down-regulates voltage gated sodium channels in rat dorsal root ganglion neurons. Exp. Neurol. 2013, 247, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Coull, J.A.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Pezet, S.; McMahon, S.B. Neurotrophins: Mediators and modulators of pain. Annu. Rev. Neurosci. 2006, 29, 507–538. [Google Scholar] [CrossRef] [PubMed]
- Lopes, B.C.; Medeiros, L.F.; Silva de Souza, V.; Cioato, S.G.; Medeiros, H.R.; Regner, G.G.; Lino de Oliveira, C.; Fregni, F.; Caumo, W.; Torres, I.L.S. Transcranial direct current stimulation combined with exercise modulates the inflammatory profile and hyperalgesic response in rats subjected to a neuropathic pain model: Long-term effects. Brain Stimul. 2020, 13, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Cocco, S.; Podda, M.V.; Grassi, C. Role of BDNF Signaling in Memory Enhancement Induced by Transcranial Direct Current Stimulation. Front. Neurosci. 2018, 12, 427. [Google Scholar] [CrossRef] [PubMed]
- Hopf, A.; Schaefer, D.J.; Kalbermatten, D.F.; Guzman, R.; Madduri, S. Schwann Cell-Like Cells: Origin and Usability for Repair and Regeneration of the Peripheral and Central Nervous System. Cells 2020, 9, 1990. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Fan, B.; Ding, H.; Liu, Y.; Tang, H.; Pan, D.; Shi, J.; Zheng, P.; Shi, H.; Wu, H.; et al. Proteomics analysis of Schwann cell-derived exosomes: A novel therapeutic strategy for central nervous system injury. Mol. Cell. Biochem. 2019, 457, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. The Success and Failure of the Schwann Cell Response to Nerve Injury. Front. Cell. Neurosci. 2019, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Nocera, G.; Jacob, C. Mechanisms of Schwann cell plasticity involved in peripheral nerve repair after injury. Cell. Mol. Life Sci. CMLS 2020, 77, 3977–3989. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, M.; Kubo, A.; Hayashi, Y.; Iwata, K. Peripheral and Central Mechanisms of Persistent Orofacial Pain. Front. Neurosci. 2019, 13, 1227. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Ramadan, K.; Korczeniewska, O.; Anwer, M.M.; Benoliel, R.; Eliav, E. Interleukin-10 levels in rat models of nerve damage and neuropathic pain. Neurosci. Lett. 2015, 592, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Franchi, S.; Valsecchi, A.E.; Borsani, E.; Procacci, P.; Ferrari, D.; Zaffa, C.; Sartori, P.; Rodella, L.F.; Vescovi, A.; Maione, S.; et al. Intravenous neural stem cells abolish nociceptive hypersensitivity and trigger nerve regeneration in experimental neuropathy. Pain 2012, 153, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Austin, P.J.; Moalem-Taylor, G. The neuro-immune balance in neuropathic pain: Involvement of inflammatory immune cells, immune-like glial cells and cytokines. J. Neuroimmunol. 2010, 229, 26–50. [Google Scholar] [CrossRef] [PubMed]
- Antal, A.; Terney, D.; Kühnl, S.; Paulus, W. Anodal transcranial direct current stimulation of the motor cortex ameliorates chronic pain and reduces short intracortical inhibition. J. Pain Symptom Manag. 2010, 39, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.R.; Andriessen, A.S.; Chen, G.; Wang, K.; Jiang, C.; Maixner, W.; Ji, R.R. Central Nervous System Targets: Glial Cell Mechanisms in Chronic Pain. Neurother. J. Am. Soc. Exp. Neurother. 2020, 17, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Ethridge, V.T.; Gargas, N.M.; Sonner, M.J.; Moore, R.J.; Romer, S.H.; Hatcher-Solis, C.; Rohan, J.G. Effects of transcranial direct current stimulation on brain cytokine levels in rats. Front. Neurosci. 2022, 16, 1069484. [Google Scholar] [CrossRef] [PubMed]
- Bikson, M.; Grossman, P.; Thomas, C.; Zannou, A.L.; Jiang, J.; Adnan, T.; Mourdoukoutas, A.P.; Kronberg, G.; Truong, D.; Boggio, P.; et al. Safety of Transcranial Direct Current Stimulation: Evidence Based Update 2016. Brain Stimul. 2016, 9, 641–661. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Zheng, X.; Zhang, B. Anti-Inflammatory Pathways Mediating tDCS’s Effects on Neuropathic Pain. Biology 2025, 14, 892. https://doi.org/10.3390/biology14070892
Zhang H, Zheng X, Zhang B. Anti-Inflammatory Pathways Mediating tDCS’s Effects on Neuropathic Pain. Biology. 2025; 14(7):892. https://doi.org/10.3390/biology14070892
Chicago/Turabian StyleZhang, Haipeng, Xinyan Zheng, and Binn Zhang. 2025. "Anti-Inflammatory Pathways Mediating tDCS’s Effects on Neuropathic Pain" Biology 14, no. 7: 892. https://doi.org/10.3390/biology14070892
APA StyleZhang, H., Zheng, X., & Zhang, B. (2025). Anti-Inflammatory Pathways Mediating tDCS’s Effects on Neuropathic Pain. Biology, 14(7), 892. https://doi.org/10.3390/biology14070892