
Heavy Metal-Associated (HMA) Domain-Containing Proteins: Insight into Their Features and Roles in Bread Wheat (Triticum aestivum L.)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of the HMA-Domain Containing Proteins (TaHMAs) in T. aestivum
2.2. Elucidation of HMA Characteristic Parameters
2.3. Gene Structure Identification and Cis-Regulatory Element Analysis in TaHMAs
2.4. Multiple Sequence Alignment and Phylogenetic Analysis
2.5. Domain Architecture, Sub-Cellular Localization, and Motif Analyses of TaHMAs
2.6. Identification of Duplicated Gene Pairs, Calculation for Ks/Ka, and Synteny Analysis
2.7. Expression Profiling
3. Results
3.1. Identification, Chromosomal Distribution, and Localisation of TaHMAs
3.2. Physiochemical Properties and Sub-Cellular Localization Analyses of TaHMAs
3.3. Gene Structure Analysis
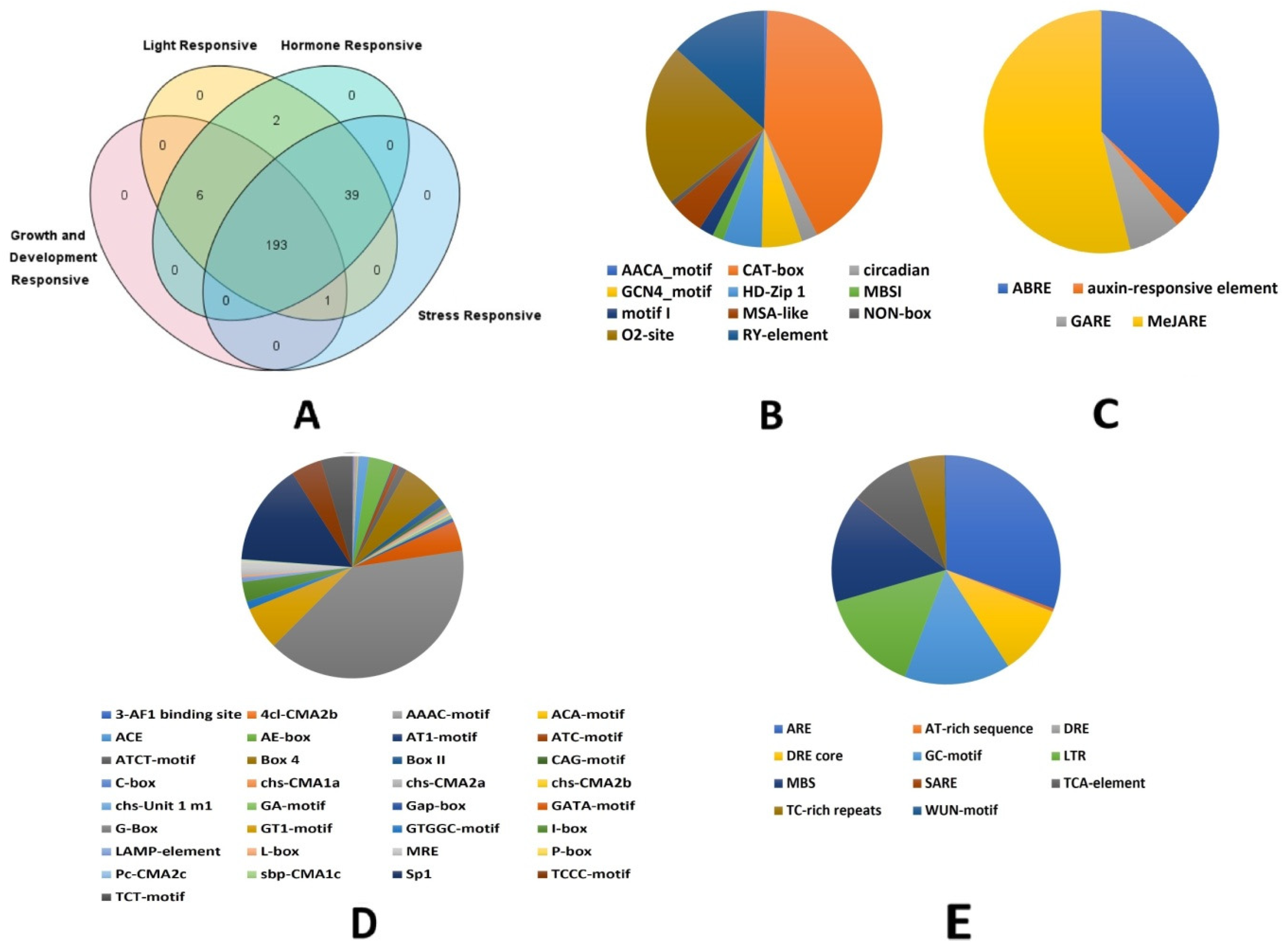
3.4. Cis-Regulatory Element Analysis
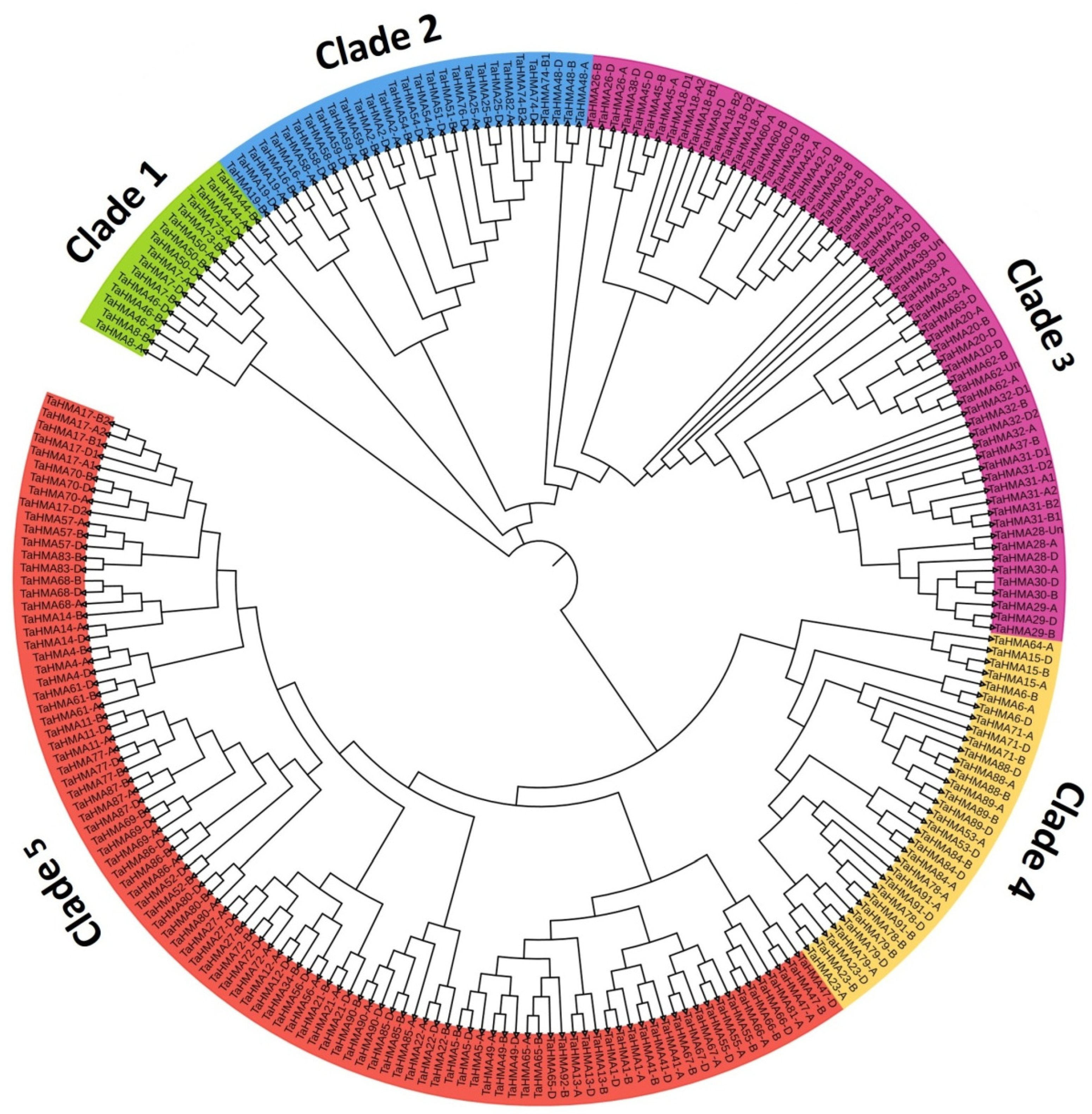
3.5. Phylogeny Analysis
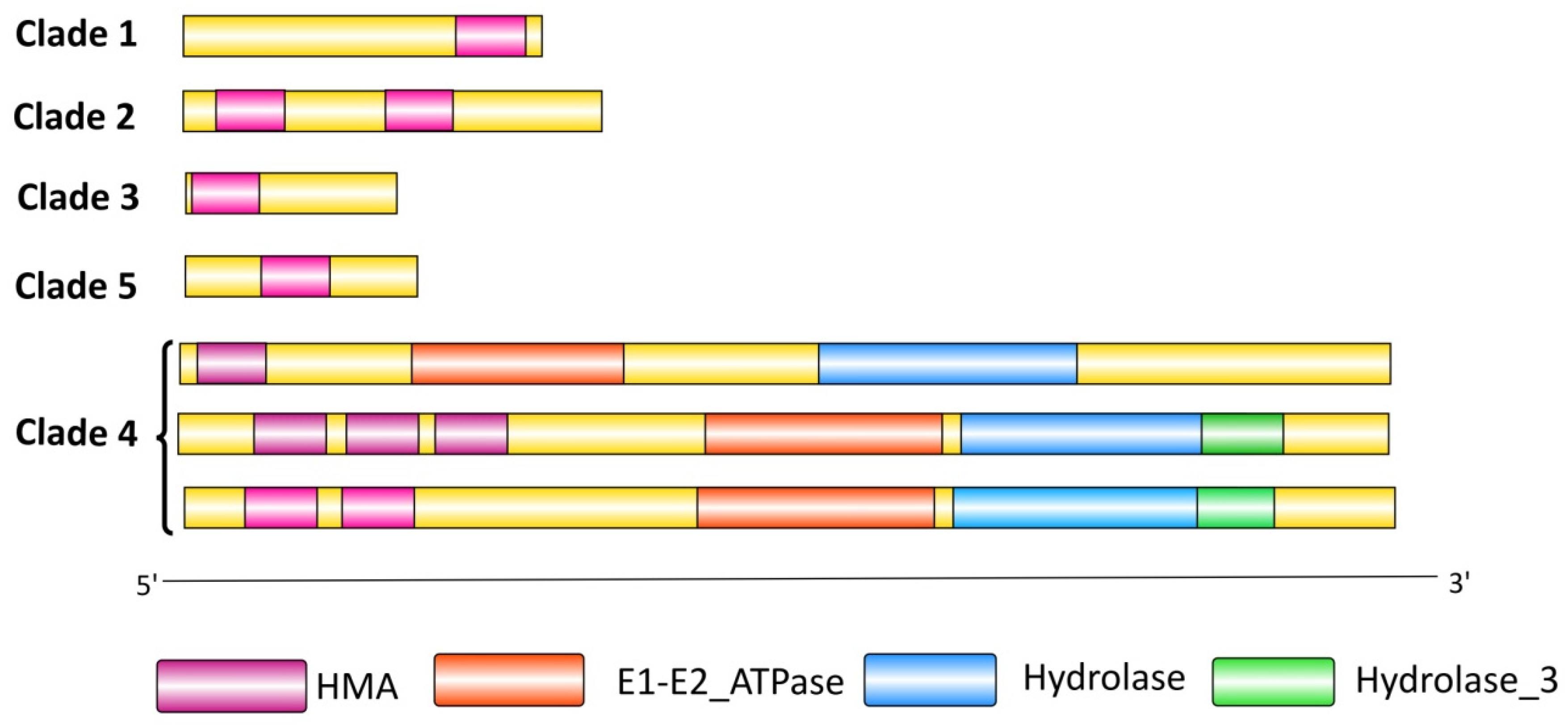
3.6. Domain Composition and Distribution Analysis
3.7. Analyses of Motif Diversity
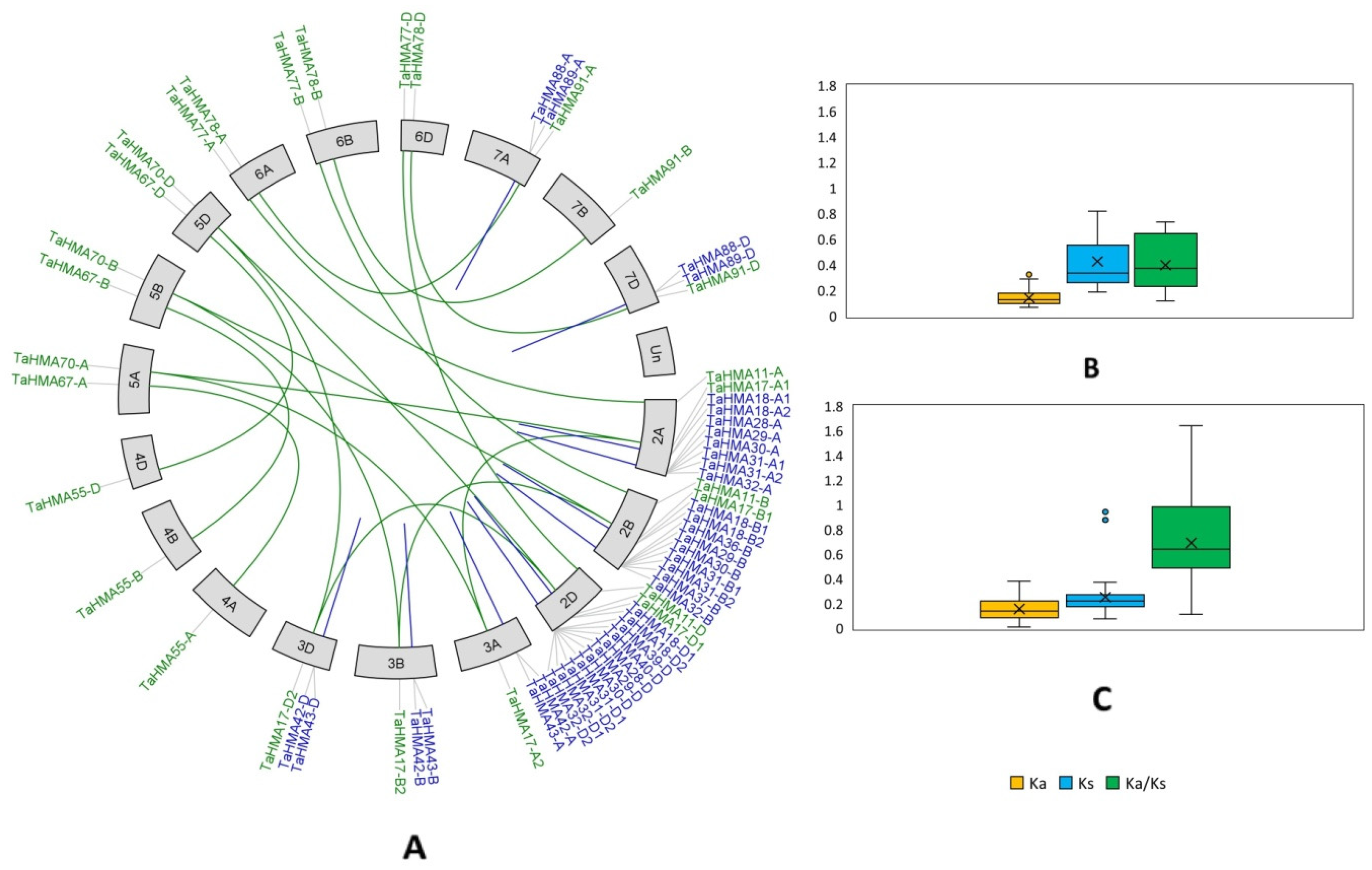
3.8. Gene Duplication Events and Ka/Ks Analyses
3.9. Synteny Analysis
3.10. Expression Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cao, Y.; Zhao, X.; Liu, Y.; Wang, Y.; Wu, W.; Jiang, Y.; Liao, C.; Xu, X.; Gao, S.; Shen, Y.; et al. Genome-Wide Identification of ZmHMAs and Association of Natural Variation in ZmHMA2 and ZmHMA3 with Leaf Cadmium Accumulation in Maize. PeerJ 2019, 7, e7877. [Google Scholar] [CrossRef]
- Kumar, S.; Prasad, S.; Yadav, K.K.; Shrivastava, M.; Gupta, N.; Nagar, S.; Bach, Q.-V.; Kamyab, H.; Khan, S.A.; Yadav, S.; et al. Hazardous Heavy Metals Contamination of Vegetables and Food Chain: Role of Sustainable Remediation Approaches—A Review. Environ. Res. 2019, 179, 108792. [Google Scholar] [CrossRef] [PubMed]
- Kosakivska, I.V.; Babenko, L.M.; Romanenko, K.O.; Korotka, I.Y.; Potters, G. Molecular Mechanisms of Plant Adaptive Responses to Heavy Metals Stress. Cell Biol. Int. 2021, 45, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Batool, T.S.; Aslam, R.; Gul, A.; Paracha, R.Z.; Ilyas, M.; De Abreu, K.; Munir, F.; Amir, R.; Williams, L.E. Genome-Wide Analysis of Heavy Metal ATPases (HMAs) in Poaceae Species and Their Potential Role against Copper Stress in Triticum aestivum. Sci. Rep. 2023, 13, 7551. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.M.; Ali, A.; Wan, Y.; Zhou, G. Genome-Wide Identification of Heavy-Metal ATPases Genes in Areca catechu: Investigating Their Functionality under Heavy Metal Exposure. BMC Plant Biol. 2024, 24, 484. [Google Scholar] [CrossRef]
- Choppala, G.; Saifullah; Bolan, N.; Bibi, S.; Iqbal, M.; Rengel, Z.; Kunhikrishnan, A.; Ashwath, N.; Ok, Y.S. Cellular Mechanisms in Higher Plants Governing Tolerance to Cadmium Toxicity. Crit. Rev. Plant Sci. 2014, 33, 374–391. [Google Scholar] [CrossRef]
- Das, S.; Sultana, K.W.; Ndhlala, A.R.; Mondal, M.; Chandra, I. Heavy Metal Pollution in the Environment and Its Impact on Health: Exploring Green Technology for Remediation. Environ. Health Insights 2023, 17, 11786302231201259. [Google Scholar] [CrossRef]
- Li, J.; Zhang, M.; Sun, J.; Mao, X.; Wang, J.; Liu, H.; Zheng, H.; Li, X.; Zhao, H.; Zou, D. Heavy Metal Stress-Associated Proteins in Rice and Arabidopsis: Genome-Wide Identification, Phylogenetics, Duplication, and Expression Profiles Analysis. Front. Genet. 2020, 11, 477. [Google Scholar] [CrossRef]
- Williams, L.E.; Mills, R.F. P1B-ATPases—An Ancient Family of Transition Metal Pumps with Diverse Functions in Plants. Trends Plant Sci. 2005, 10, 491–502. [Google Scholar] [CrossRef]
- Hasan, M.N.; Islam, S.; Bhuiyan, F.H.; Arefin, S.; Hoque, H.; Jewel, N.A.; Ghosh, A.; Prodhan, S.H. Genome Wide Analysis of the Heavy-Metal-Associated (HMA) Gene Family in Tomato and Expression Profiles under Different Stresses. Gene 2022, 835, 146664. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Q.; Zhang, X.; Zhang, X.; Yu, T.; Wu, Y.; Fang, Y.; Xue, D. Genome-Wide Identification of the HMA Gene Family and Expression Analysis under Cd Stress in Barley. Plants 2021, 10, 1849. [Google Scholar] [CrossRef] [PubMed]
- Zhiguo, E.; Tingting, L.; Chen, C.; Lei, W. Genome-Wide Survey and Expression Analysis of P 1B -ATPases in Rice, Maize and Sorghum. Rice Sci. 2018, 25, 208–217. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Shi, G. Genome-Wide Identification and Expression Profiling of Heavy Metal ATPase (HMA) Genes in Peanut: Potential Roles in Heavy Metal Transport. IJMS 2024, 25, 613. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xu, X.; Hu, X.; Liu, Q.; Wang, Z.; Zhang, H.; Wang, H.; Wei, M.; Wang, H.; Liu, H.; et al. Genome-Wide Analysis and Heavy Metal-Induced Expression Profiling of the HMA Gene Family in Populus trichocarpa. Front. Plant Sci. 2015, 6, 1149. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, L.; Deng, X.; Wang, P.; Ma, Q.; Nian, H.; Wang, Y.; Yang, C. Genome-Wide Characterization of Soybean P 1B-ATPases Gene Family Provides Functional Implications in Cadmium Responses. BMC Genom. 2016, 17, 376. [Google Scholar] [CrossRef]
- Zahra, S.; Shaheen, T.; Qasim, M.; Mahmood-ur-Rahman; Hussain, M.; Zulfiqar, S.; Shaukat, K.; Mehboob-ur-Rahman. Genome-Wide Survey of HMA Gene Family and Its Characterization in Wheat (Triticum aestivum). PeerJ 2023, 11, e14920. [Google Scholar] [CrossRef]
- Bolser, D.; Staines, D.M.; Pritchard, E.; Kersey, P. Ensembl Plants: Integrating Tools for Visualizing, Mining, and Analyzing Plant Genomic Data. In Plant Genomics Databases: Methods and Protocols; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; Volume 1533, pp. 1–31. [Google Scholar] [CrossRef]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent Updates, New Developments and Status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The Conserved Domain Database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- Sigrist, C.J.A.; De Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and Continuing Developments at PROSITE. Nucleic Acids Res. 2012, 41, D344–D347. [Google Scholar] [CrossRef]
- Blum, M.; Andreeva, A.; Florentino, L.C.; Chuguransky, S.R.; Grego, T.; Hobbs, E.; Pinto, B.L.; Orr, A.; Paysan-Lafosse, T.; Ponamareva, I.; et al. InterPro: The Protein Sequence Classification Resource in 2025. Nucleic Acids Res. 2025, 53, D444–D456. [Google Scholar] [CrossRef]
- Madhu; Kaur, A.; Singh, K.; Upadhyay, S.K. Ascorbate Oxidases in Bread Wheat: Gene Regulatory Network, Transcripts Profiling, and Interaction Analyses Provide Insight into Their Role in Plant Development and Stress Response. Plant Growth Regul. 2024, 103, 209–224. [Google Scholar] [CrossRef]
- Sharma, Y.; Dixit, S.; Singh, K.; Upadhyay, S.K. Vascular Plant One-Zinc Finger Transcription Factors: Exploration of Characteristic Features, Expression, Coexpression and Interaction Suggested Their Diverse Role in Bread Wheat (Triticum aestivum L.). Plant Growth Regul. 2025, 105, 71–87. [Google Scholar] [CrossRef]
- Wolfe, D.; Dudek, S.; Ritchie, M.D.; Pendergrass, S.A. Visualizing Genomic Information across Chromosomes with PhenoGram. BioData Min. 2013, 6, 18. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An Upgraded Gene Feature Visualization Server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M. PlantCARE, a Database of Plant Cis-Acting Regulatory Elements and a Portal to Tools for in Silico Analysis of Promoter Sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Xie, Y.; Li, H.; Luo, X.; Li, H.; Gao, Q.; Zhang, L.; Teng, Y.; Zhao, Q.; Zuo, Z.; Ren, J. IBS 2.0: An Upgraded Illustrator for the Visualization of Biological Sequences. Nucleic Acids Res. 2022, 50, W420–W426. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein Localization Predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and Analyzing DNA and Protein Sequence Motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef] [PubMed]
- Darzentas, N. Circoletto: Visualizing Sequence Similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef]
- Pingault, L.; Choulet, F.; Alberti, A.; Glover, N.; Wincker, P.; Feuillet, C.; Paux, E. Deep Transcriptome Sequencing Provides New Insights into the Structural and Functional Organization of the Wheat Genome. Genome Biol. 2015, 16, 29. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Liu, Z.; Xin, M.; Qin, J.; Peng, H.; Ni, Z.; Yao, Y.; Sun, Q. Temporal Transcriptome Profiling Reveals Expression Partitioning of Homeologous Genes Contributing to Heat and Drought Acclimation in Wheat (Triticum aestivum L.). BMC Plant Biol. 2015, 15, 152. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.; Khan, A.A.; Lin, Q.; Han, Y.; Mu, P.; Liu, Y.; Zhang, H.; Li, L.; Meng, X.; et al. Expression Partitioning of Homeologs and Tandem Duplications Contribute to Salt Tolerance in Wheat (Triticum aestivum L.). Sci. Rep. 2016, 6, 21476. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Gordish-Dressman, H.; Hoffman, E.P. An Interactive Power Analysis Tool for Microarray Hypothesis Testing and Generation. Bioinformatics 2006, 22, 808–814. [Google Scholar] [CrossRef]
- Restrepo-Montoya, D.; McClean, P.E.; Osorno, J.M. Orthology and Synteny Analysis of Receptor-like Kinases “RLK” and Receptor-like Proteins “RLP” in Legumes. BMC Genom. 2021, 22, 113. [Google Scholar] [CrossRef]
- Taneja, M.; Upadhyay, S.K. Molecular Characterization and Differential Expression Suggested Diverse Functions of P-Type II Ca2+ATPases in Triticum aestivum L. BMC Genom. 2018, 19, 389. [Google Scholar] [CrossRef]
- Kaur, A.; Madhu; Sharma, A.; Singh, K.; Upadhyay, S.K. Exploration of Piezo Channels in Bread Wheat (Triticum aestivum L.). Agriculture 2023, 13, 783. [Google Scholar] [CrossRef]
- Kaur, A.; Madhu; Sharma, A.; Singh, K.; Upadhyay, S.K. Investigation of Two-Pore K+ (TPK) Channels in Triticum aestivum L. Suggests Their Role in Stress Response. Heliyon 2024, 10, e27814. [Google Scholar] [CrossRef] [PubMed]
- Deepika; Madhu; Upadhyay, S.K. Deciphering the features and functions of serine/arginine protein kinases in bread wheat. Plant Gene 2024, 38, 100451. [Google Scholar] [CrossRef]
- Deepika; Shekhawat, J.; Madhu; Verma, P.C.; Upadhyay, S.K. Genome-Wide Analysis Unveiled the Characteristic Features and Functions of AFC Genes of Bread Wheat (Triticum aestivum L.). J. Plant Growth Regul. 2025, 44, 2466–2484. [Google Scholar] [CrossRef]
- Deepika; Madhu; Shekhawat, J.; Dixit, S.; Upadhyay, S.K. Pre-mRNA Processing Factor 4 Kinases (PRP4Ks): Exploration of Molecular Features, Interaction Network and Expression Profiling in Bread Wheat. J. Plant Growth Regul. 2025, 44, 806–820. [Google Scholar] [CrossRef]
- Kim, Y.; Choi, H.; Segami, S.; Cho, H.; Martinoia, E.; Maeshima, M.; Lee, Y. AtHMA1 Contributes to the Detoxification of Excess Zn(II) in Arabidopsis. Plant J. 2009, 58, 737–753. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, X.; Jiao, Y.; Qin, Y.; Liu, X.; He, K.; Chen, C.; Ma, L.; Wang, J.; Xiong, L.; et al. Global Genome Expression Analysis of Rice in Response to Drought and High-Salinity Stresses in Shoot, Flag Leaf, and Panicle. Plant Mol. Biol. 2007, 63, 591–608. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taneja, M.; Upadhyay, S.K. Heavy Metal-Associated (HMA) Domain-Containing Proteins: Insight into Their Features and Roles in Bread Wheat (Triticum aestivum L.). Biology 2025, 14, 818. https://doi.org/10.3390/biology14070818
Taneja M, Upadhyay SK. Heavy Metal-Associated (HMA) Domain-Containing Proteins: Insight into Their Features and Roles in Bread Wheat (Triticum aestivum L.). Biology. 2025; 14(7):818. https://doi.org/10.3390/biology14070818
Chicago/Turabian StyleTaneja, Mehak, and Santosh Kumar Upadhyay. 2025. "Heavy Metal-Associated (HMA) Domain-Containing Proteins: Insight into Their Features and Roles in Bread Wheat (Triticum aestivum L.)" Biology 14, no. 7: 818. https://doi.org/10.3390/biology14070818
APA StyleTaneja, M., & Upadhyay, S. K. (2025). Heavy Metal-Associated (HMA) Domain-Containing Proteins: Insight into Their Features and Roles in Bread Wheat (Triticum aestivum L.). Biology, 14(7), 818. https://doi.org/10.3390/biology14070818