
Design of a Multi-Epitope Vaccine Candidate Against Infectious Laryngotracheitis Virus Affecting Poultry by Computational Approaches

 , ,
, ,  ,
,
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Retrieval of ILTV’s Glycoprotein B and D Amino Acid Sequences
2.2. Physicochemical Parameters Evaluation
2.3. Homology Modeling
2.4. Cytotoxic T-Cell Lymphocytes (CTL) Epitopes Prediction
2.5. Antigenicity, Allergenicity, Toxicity Analysis and Conservancy Prediction of CTL Epitopes
2.6. Helper T Cell (HTL) Epitopes Prediction
2.7. Antigenicity, Allergenicity and Toxicity Analysis of HTL Epitopes
2.8. Interferon (IFN-γ) Epitopes Prediction
2.9. Linear B Cell Epitopes Prediction
2.10. Mapping of CTL and HTL Epitopes
2.11. Designing of Multi-Epitope Final Vaccine Construct
2.12. Physiochemical Parameters Evaluation of the Final Vaccine Design
2.13. Homology Modeling of the Final Vaccine Design
2.14. Molecular Docking
2.15. Binding Affinity Analysis
2.16. Immune Simulation
2.17. Codon Optimization
3. Results
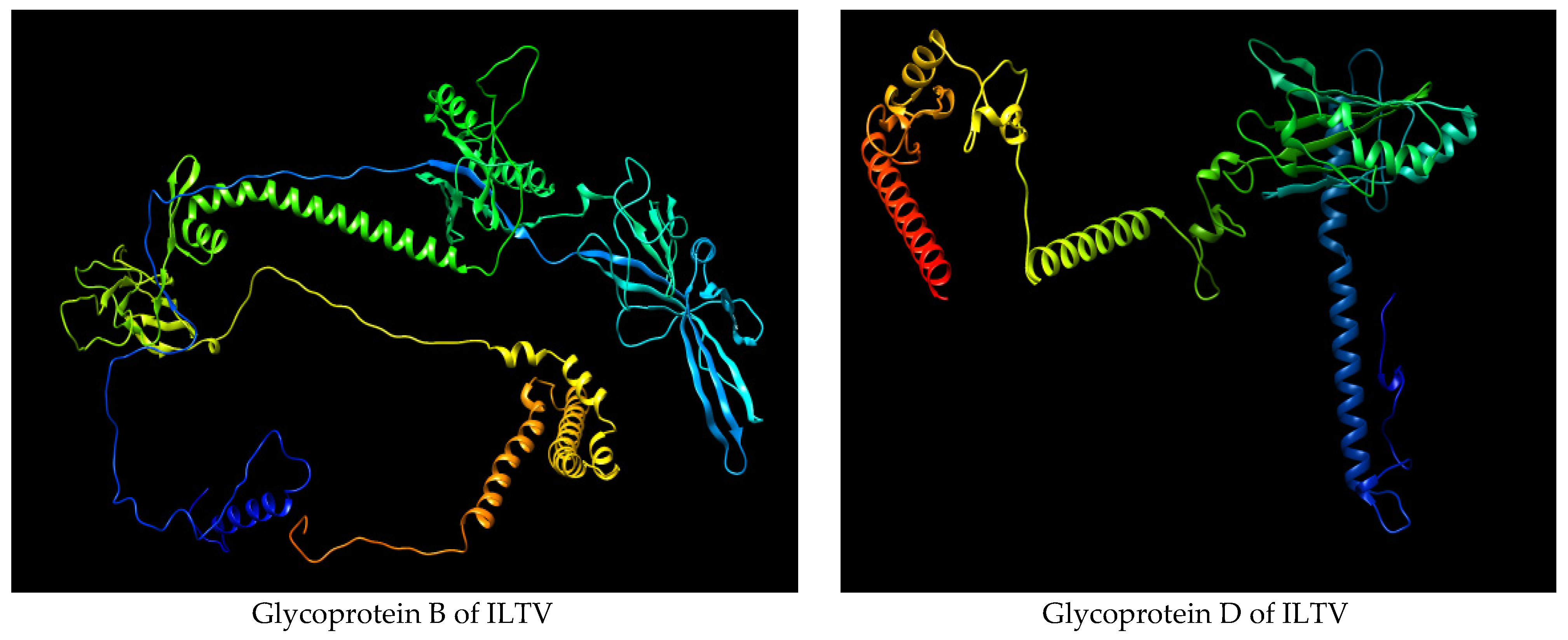
3.1. Retrieval of Glycoproteins B and D Sequences
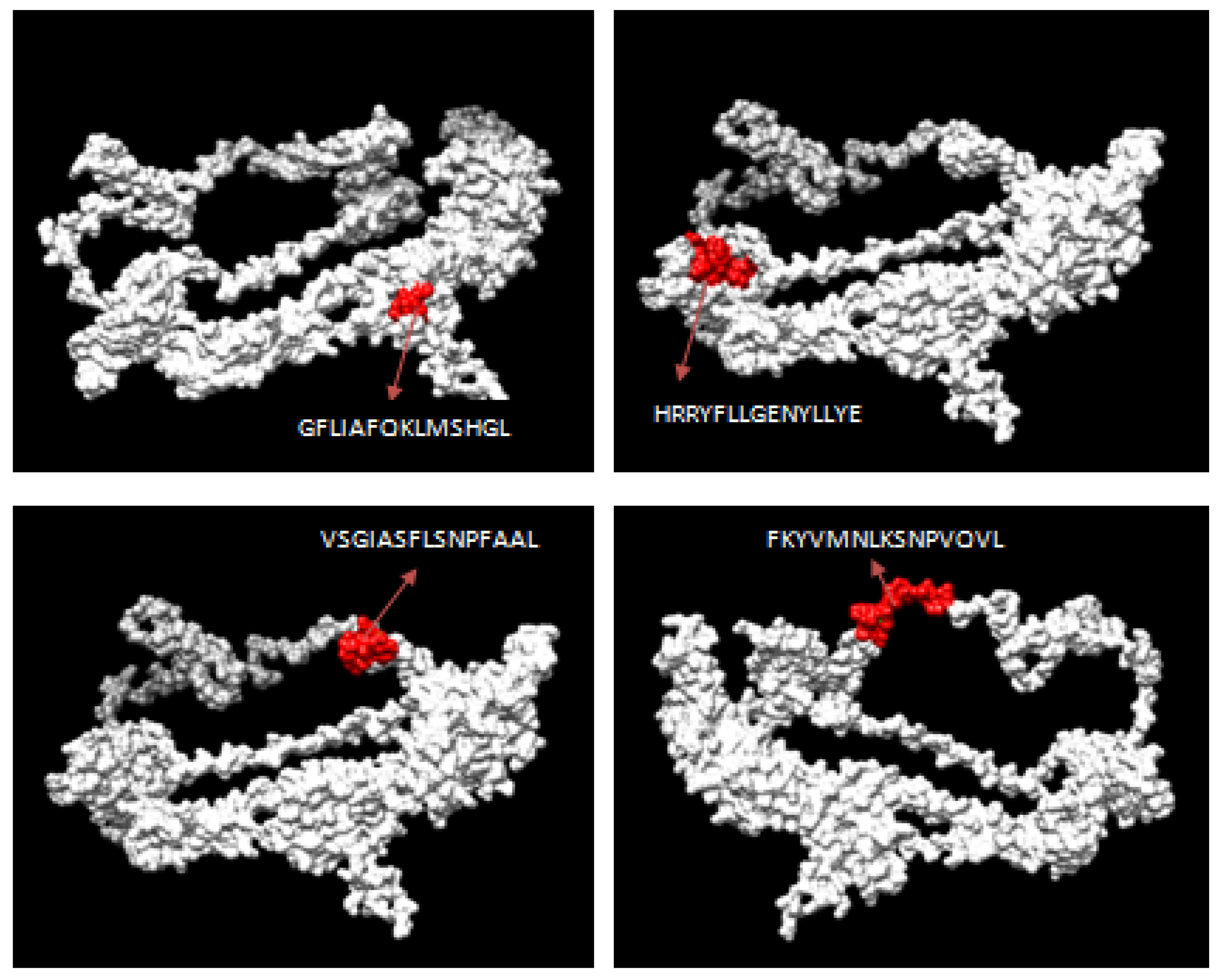
3.2. CTL Epitope Prediction
3.3. HTL Epitope Prediction
3.4. IFN-γ Inducing Epitopes Prediction
3.5. Designing the Final Multi-Epitope Vaccine Construct
3.6. Evaluation of Physicochemical Properties of Final Vaccine Design
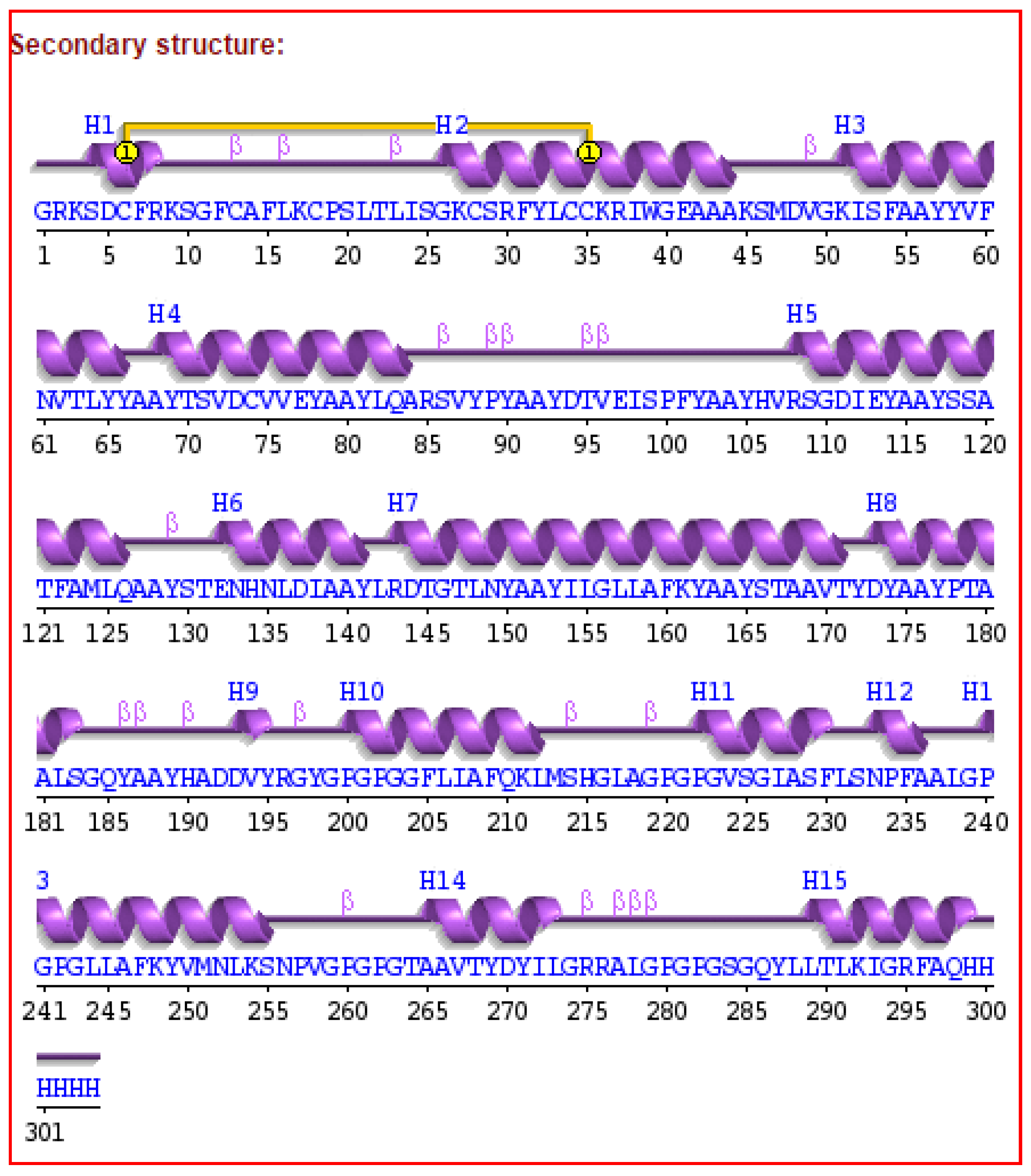
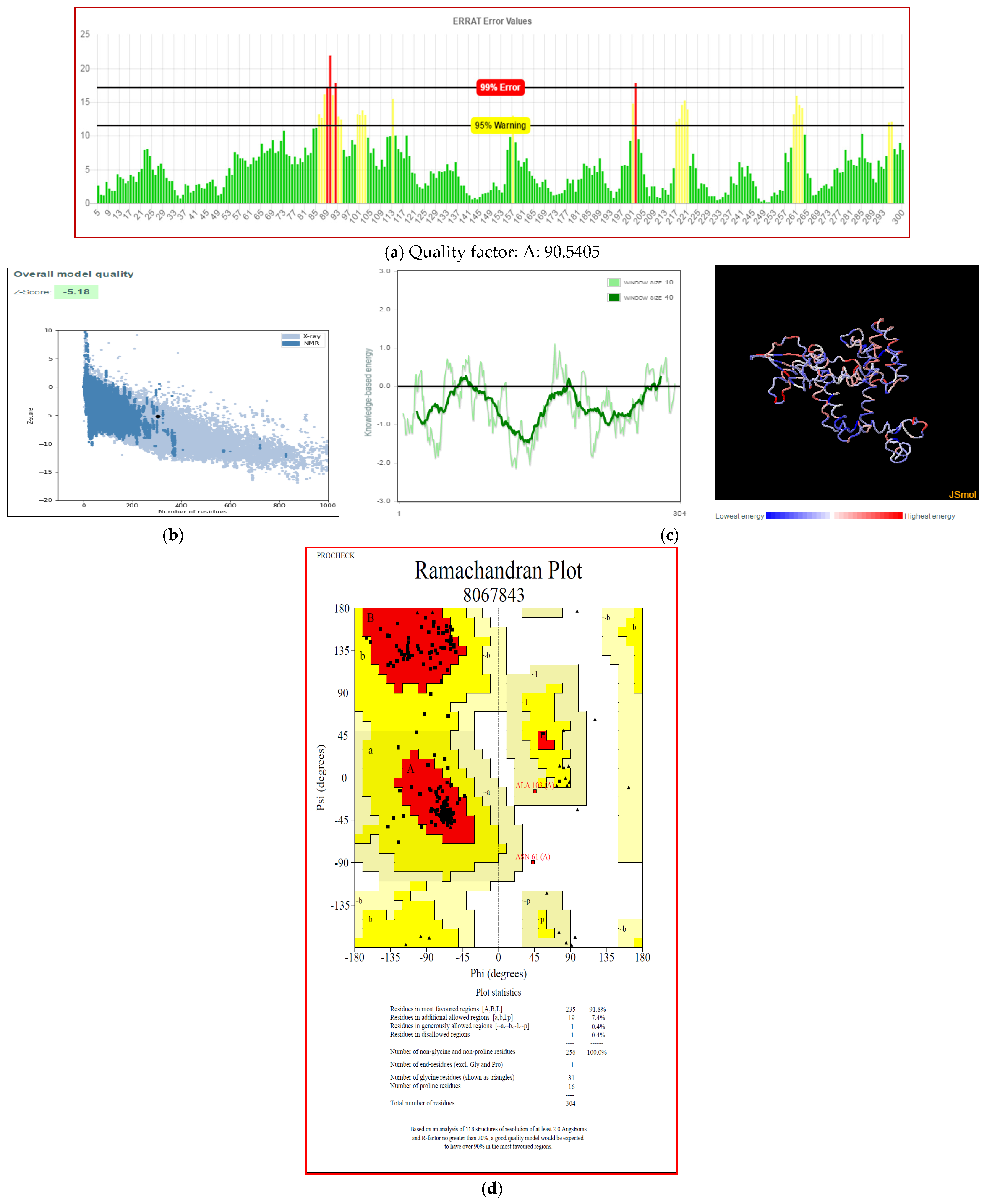
3.7. Secondary and Tertiary Structure Modeling
3.8. Molecular Docking with Immune Receptors
3.9. Binding Affinity Analysis
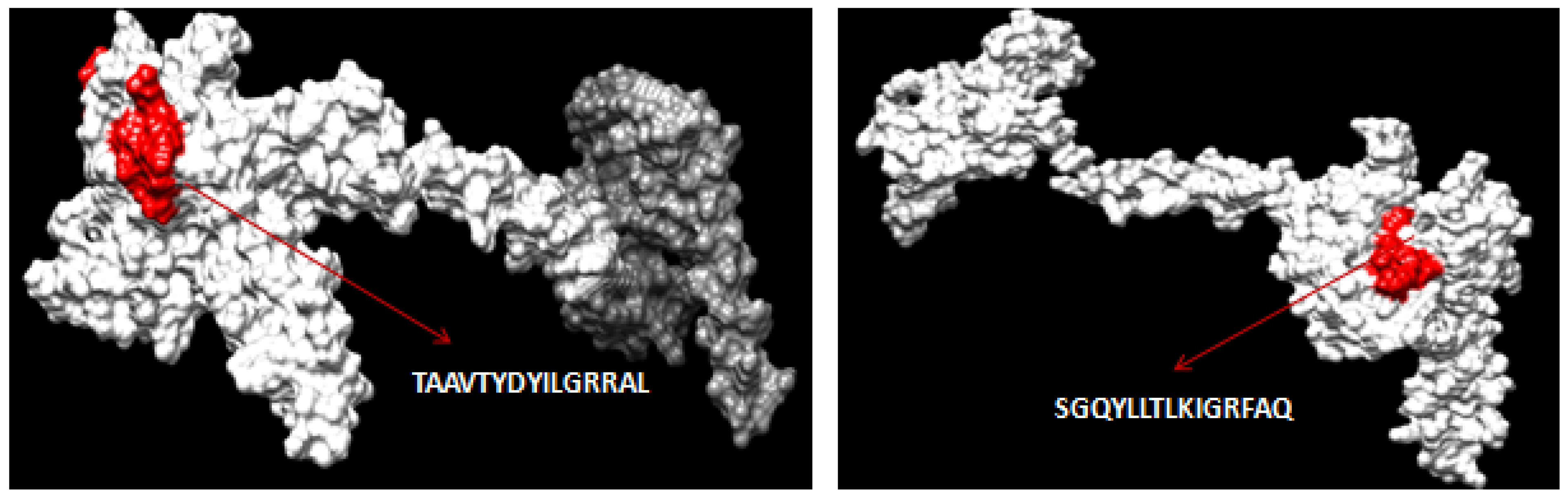
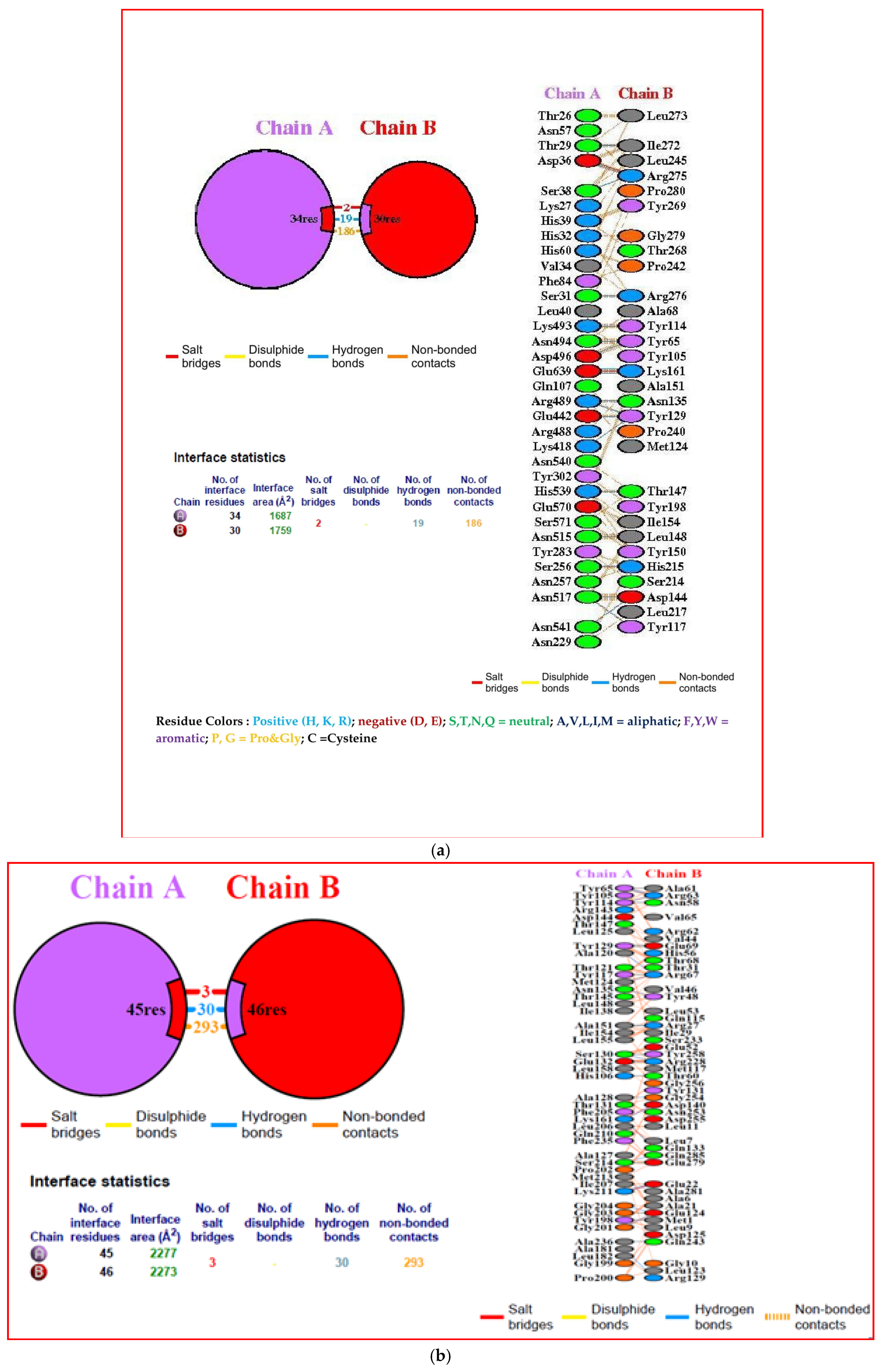
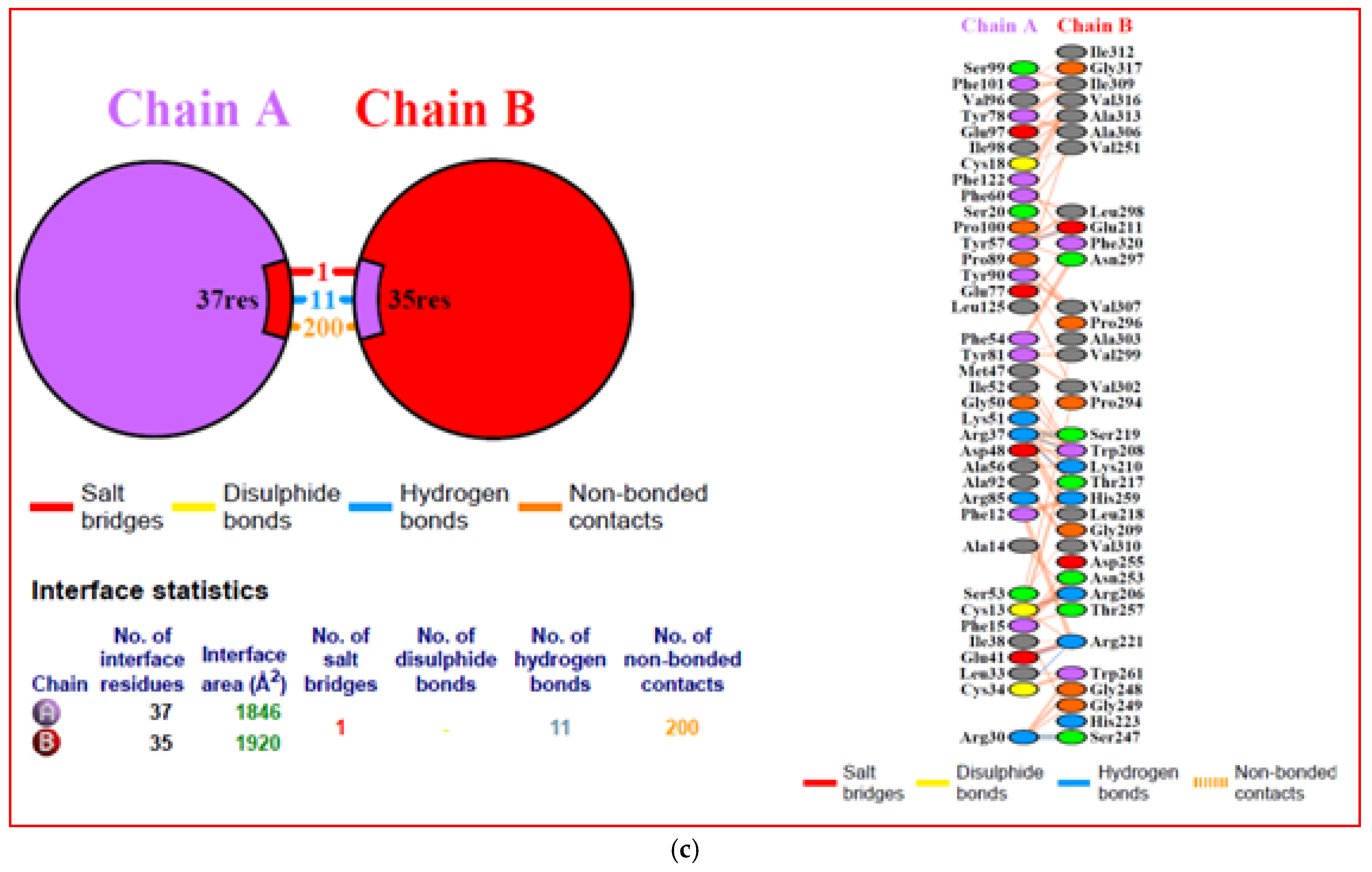
3.10. Interacting Residues
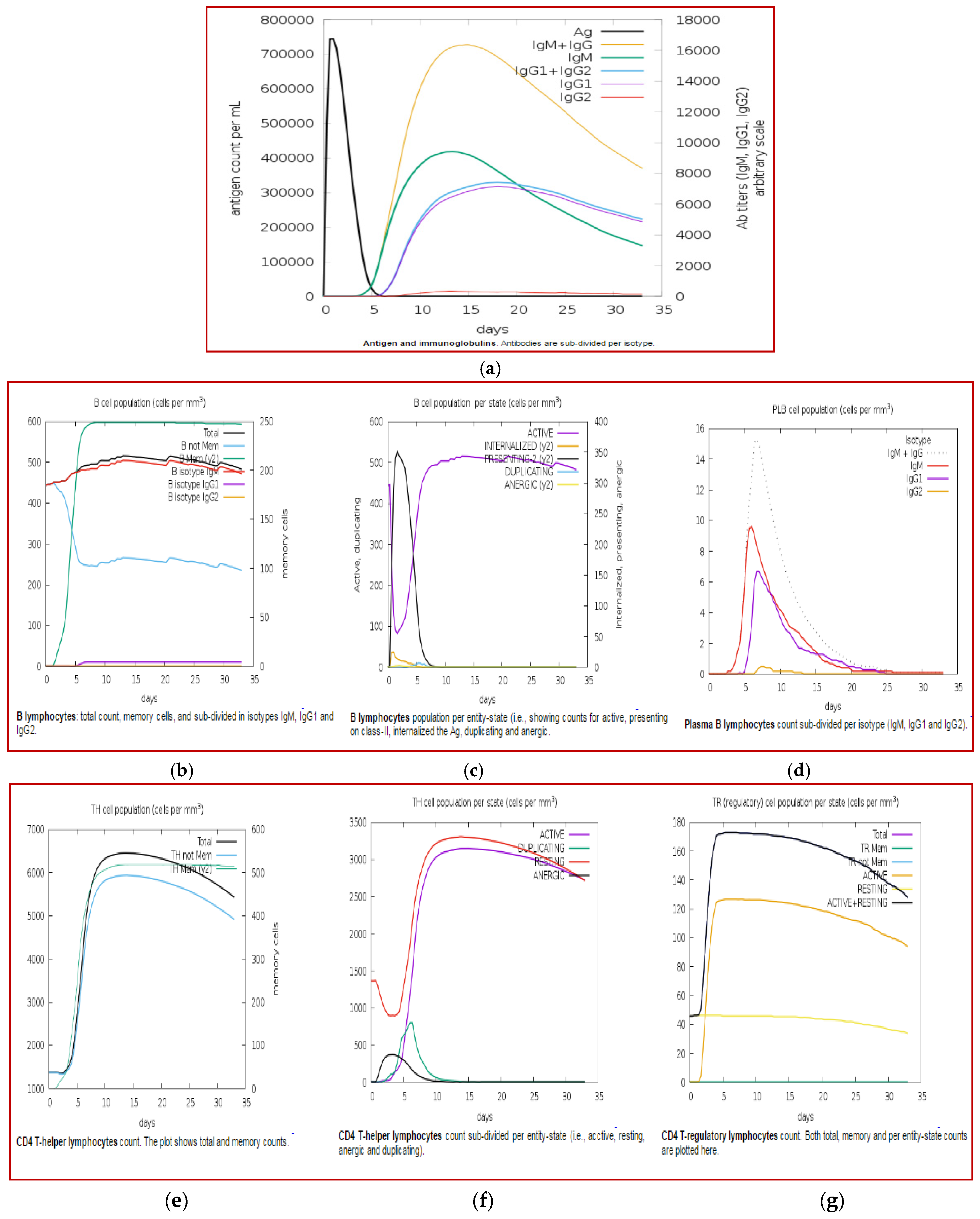
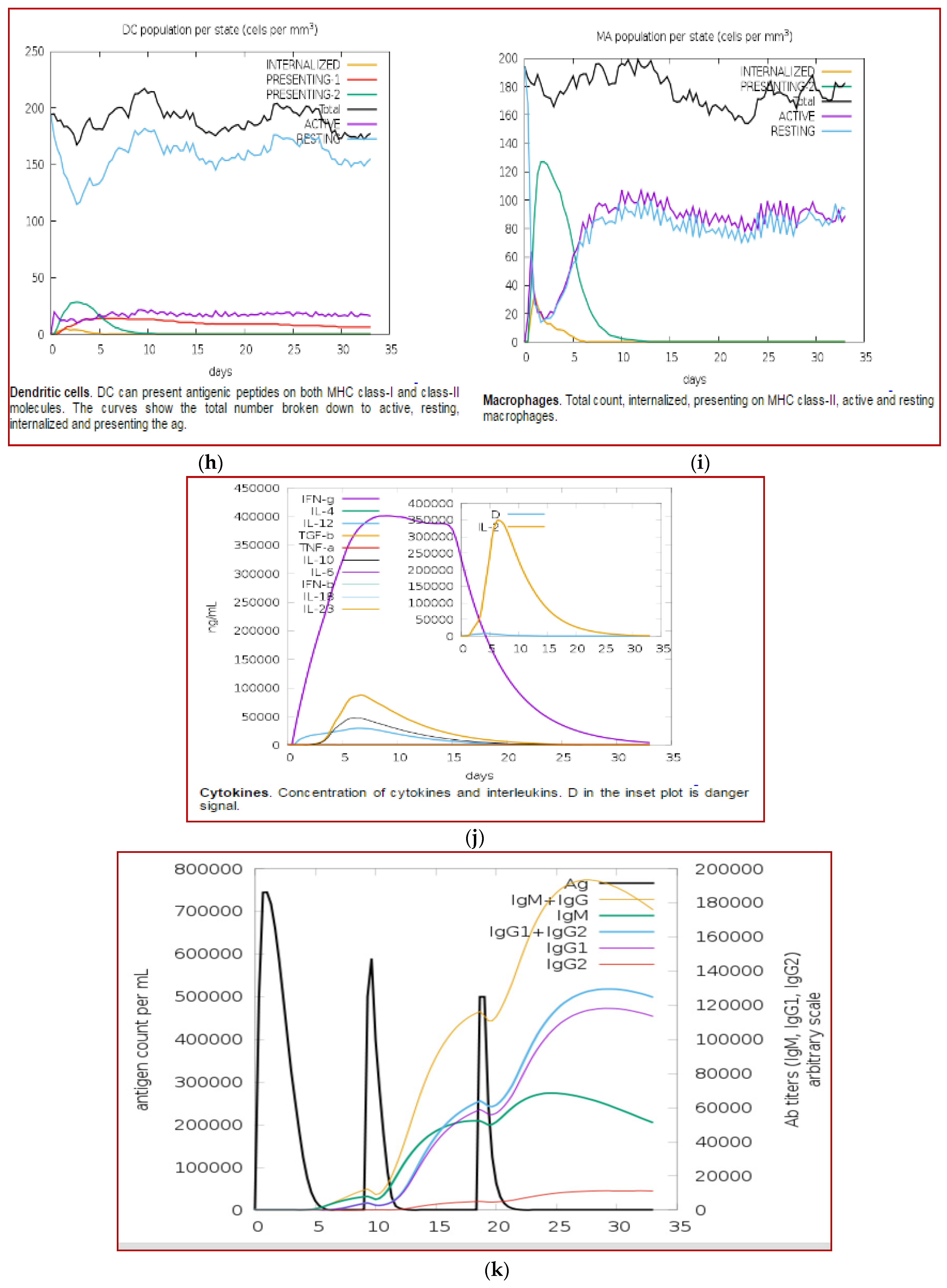
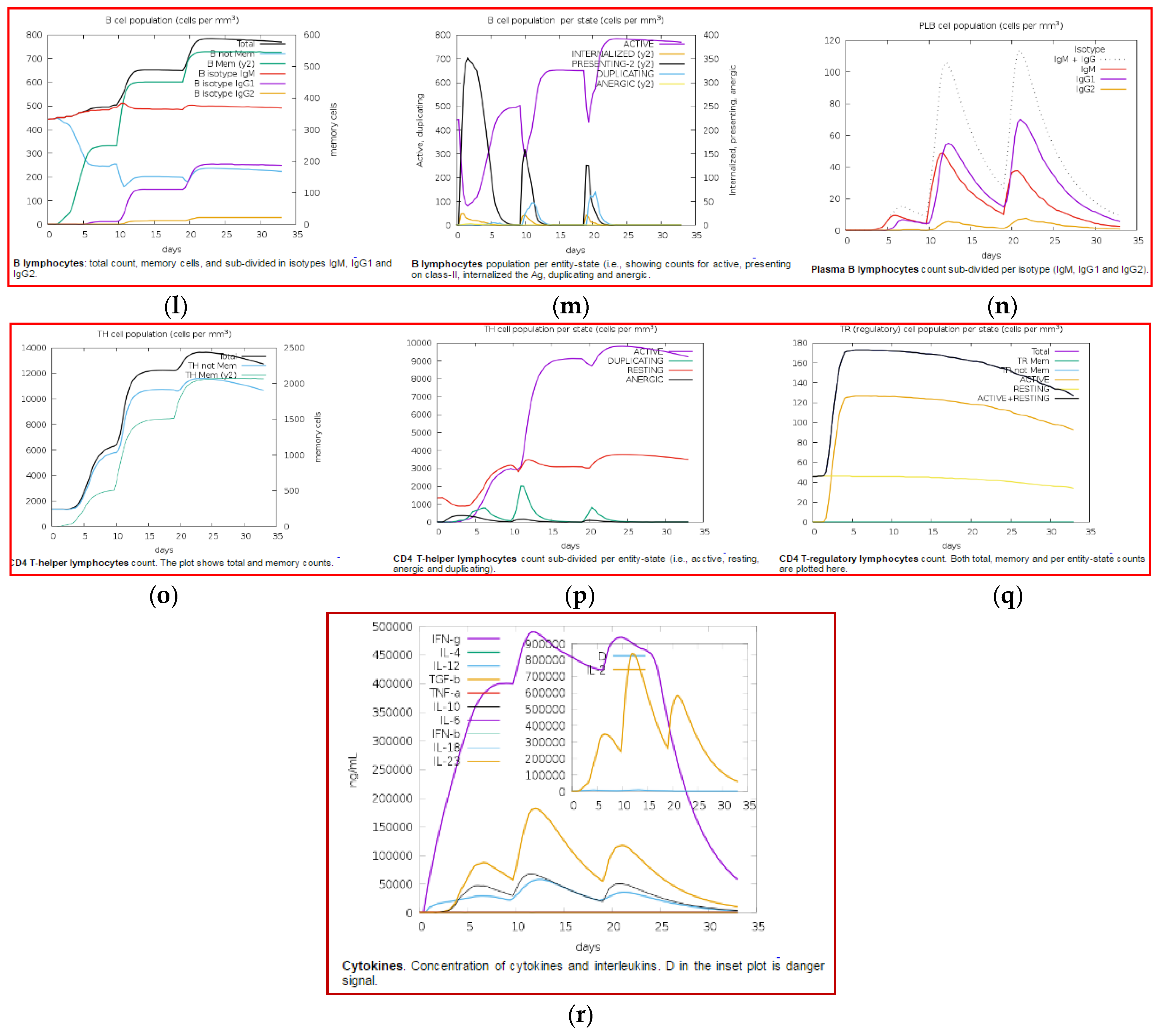
3.11. Immune Simulation
3.12. Codon Optimization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- García, M.; Spatz, S.J.; Guy, J.S. Infectious laryngotracheitis. In Diseases of Poultry, 13th ed.; Swayne, D.E., Glisson, J.R., McDougald, L.R., Nolan, L.K., Suarez, D.L., Nair, V., Eds.; Blackwell Publishing: Ames, IA, USA, 2013; pp. 161–179. [Google Scholar]
- Crawshaw, G.J.; Boycott, B.R. Infectious laryngotracheitis in peafowl and pheasants. Avian Dis. 1982, 26, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Portz, C.; Beltrão, N.; Furian, T.Q.; Júnior, A.B.; Macagnan, M.; Griebeler, J.; Lima Rosa, C.A.; Colodel, E.M.; Driemeier, D.; Back, A.; et al. Natural infection of turkeys by infectious laryngotracheitis virus. Vet. Microbiol. 2008, 131, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Eberle, R.; Hayward, G.S.; Mcgeoch, D.J.; Minson, A.C.; Pellet, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef]
- Granzow, H.; Klupp, B.G.; Fuchs, W.; Veits, J.; Osterrieder, N.; Mettenleiter, T.C. Egress of alphaherpes viruses: Comparative ultrastructural study. J. Virol. 2001, 75, 3675–3684. [Google Scholar] [CrossRef]
- Roziman, B.; Pellett, P.E. The family Herpesviridae: A brief introduction. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippinott Williams and Wilkins: Philadelphia, PA, USA, 2001; pp. 2381–2397. [Google Scholar]
- Meulemans, G.; Halen, P. Some physicochemical and biological properties of a Belgian strain (U76/1035) of infectious laryngotracheitis virus. Avian Pathol. 1978, 7, 311–325. [Google Scholar] [CrossRef]
- Neighbour, N.K.; Newberry, L.A.; Bayyari, G.R.; Skeeles, J.K.; Beasley, J.N.; McNew, R.W. The effect of microaerosolized hydrogen peroxide on bacterial and viral pathogens. Poult. Sci. 1994, 73, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Khan, M.S.R.; Islam, M.A.; Hassan, J. Isolation and characterization of infectious laryngotracheitis virus in layer chickens. Bangladesh J. Vet. Med. 2010, 8, 123–130. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Dolan, A.; Ralph, A.C. Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J. Virol. 2000, 74, 10401–10406. [Google Scholar] [CrossRef]
- Morales Ruiz, S.; Bendezu Eguis, J.; Montesinos, R.; Tataje-Lavanda, L.; Fernández-Díaz, M. Full-genome sequence of infectious laryngotracheitis virus (GallidAlphaherpesvirus 1) strain VFAR-043, isolated in Peru. Genome Announc. 2018, 6, 10–1128. [Google Scholar] [CrossRef]
- Thureen, D.R.; Keeler, C.L. Psittacidherpesvirus 1 and infectious laryngotracheitis virus: Comparative genome sequence analysis of two avian alphaherpesviruses. J. Virol. 2006, 80, 7863–7872. [Google Scholar] [CrossRef]
- Lee, S.W.; Devlin, J.M.; Markham, J.F.; Noormohammadi, A.H.; Browning, G.F.; Ficorilli, N.P.; Hartley, C.A.; Markham, P.F. First complete genome sequence of infectious laryngotracheitis virus. BMC Genom. 2011, 12, 197. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, A.; Lavezzo, E.; Niero, G.; Moreno, A.; Massi, P.; Franchin, E.; Toppo, S.; Salata, C.; Palù, G. Full genome sequence based comparative study of wild type and vaccine strains of infectious laryngotracheitis virus from Italy. PLoS ONE 2016, 11, e0149529. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Spatz, S.; Zhang, Z.; Wen, G.; Garcia, M.; Zsak, L. Newcastle disease virus (NDV) recombinants expressing infectious laryngotracheitis virus (ILTV)glycoproteins gB and gD protect chickens against ILTV and NDV challenges. J. Virol. 2014, 88, 8397–8406. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.S.; Barnes, H.J.; Smith, L. Increased virulence of modified-live infectious laryngotracheitis vaccine virus following bird-to-bird passage. Avian Dis. 1991, 35, 348–355. [Google Scholar] [CrossRef]
- Oyarzun, P.; Ellis, J.J.; Gonzalez-Galarza, F.F.; Jones, A.R.; Middleton, D.; Boden, M.; Kobe, B. A bioinformatics tool for epitope based vaccine design that accounts for human ethnic diversity: Application to emerging infectious diseases. Vaccine 2015, 33, 1267–1273. [Google Scholar] [CrossRef]
- Chauhan, V.; Rungta, T.; Goyal, K.; Singh, M.P. Designing a multi-epitope based vaccine to combat Kaposi Sarcoma utilizing immunoinformatics approach. Sci. Rep. 2019, 9, 2517. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPAsy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar]
- Kallberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template based protein structure modelling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; Bakker, P.I.W.D.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cαgeometry: ϕ,ψ and Cβdeviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumor antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef]
- Pandey, R.K.; Ojha, R.; Mishra, A.; Prajapati, V.K. Designing B and T cell multiepitope based subunit vaccine using immunoinformatics approach to control Zika virus infection. J. Cell. Biochem. 2018, 119, 7631–7642. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon gamma inducing MHC class II binders. Biol. Direct. 2013, 8, 30. [Google Scholar] [CrossRef]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001, 14, 529–532. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. NucleicAcids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M. Contacts based prediction of binding affinity in protein-protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Munch, R.; Nortemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Zhang, L. Multi-epitope vaccines: A promising strategy against tumors and viral infections. Cell. Mol. Immunol. 2018, 15, 182–184. [Google Scholar] [CrossRef]
- KanabagatteBasavarajappa, M.; Song, H.; Lamichhane, C.; Samal, S.K. Glycoprotein based enzyme-linked immunosorbent assays for serodiagnosis of infectious laryngotracheitis. J. Clin. Microbiol. 2015, 53, 1727–1730. [Google Scholar] [CrossRef]
- Ali, S.A.; Almofti, Y.A.; Abd-Elrahman, K.A. Immunoinformatics approach for multiepitopes vaccine prediction against Glycoprotein B of Avian infectious laryngotracheitis virus. Adv. Bioinform. 2019, 2019, 1270485. [Google Scholar] [CrossRef]
- Hosseini, S.S.; Kolyani, K.A.; Tabatabaei, R.R.; Goudarzi, H.; Sepahi, A.A.; Salemi, M. In silico prediction of B and T cell epitopes based on NDV fusion protein for vaccine development against Newcastle disease virus. Vet. Res. Forum. 2021, 12, 157–165. [Google Scholar]
- Aziz, F.; Tufail, S.; Shah, M.A.; Salahuddin Shah, M.; Habib, M.; Mirza, O.; Iqbal, M.; Rahman, M. In Silico epitope prediction and immunogenic analysis for penton base epitope-focused vaccine against Hydropericardium Syndrome in Chicken. Virus Res. 2019, 273, 197750. [Google Scholar] [CrossRef]
- Fatoba, A.J.; Adeleke, V.T.; Maharaj, L.; Okpeku, M.; Adeniyi, A.A.; Adeleke, M.A. Design of a multiepitope vaccine against Chicken Anemia virus disease. Viruses 2022, 14, 1456. [Google Scholar] [CrossRef]
- Mugunthan, S.P.; Mani Chandra, H. A computational reverse vaccinology approach for the design and development of multi-epitopic vaccine against avian pathogen Mycoplasma gallisepticum. Front. Vet. Sci. 2021, 8, 721061. [Google Scholar] [CrossRef]
- Elshafei, S.O.; Mahmoud, N.A.; Almofti, Y.A. Immunoinformatics, molecular docking and dynamics simulation approaches unveil a multi epitope-based potent peptide vaccine candidate against avian leukosis virus. Sci. Rep. 2024, 14, 2870. [Google Scholar] [CrossRef]
- Kar, T.; Narsaria, U.; Basak, S.; Deb, D.; Castiglione, F.; Mueller, D.M.; Srivastava, A.P. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020, 10, 10895. [Google Scholar] [CrossRef]
- Zhang, H.H.; Yang, X.M.; Xie, Q.M.; Ma, J.Y.; Luo, Y.N.; Cao, Y.C.; Chen, F.; Bi, Y.Z. The potent adjuvant effects of chicken beta-defensin-1 when genetically fused with infectious bursal disease virus VP2 gene. Vet. Immunol. Immunopathol. 2010, 136, 92–97. [Google Scholar] [CrossRef]
- Walker, J.M. (Ed.) The Proteomics Protocols Handbook; Springer Protocols Handbooks; Humana Press: Totowa, NJ, USA, 2005. [Google Scholar] [CrossRef]
- Ikai, A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar]
- Kyte, J.; Doolitttle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Foroutan, M.; Ghaffaifar, F.; Sharifi, Z.; Dalimi, A. Vaccination with a novel multi-epitope ROP8 DNA vaccine against acute Toxoplasma gondii infection induces strong B and T cell responses in mice. Comp. Immunol. Microbiol. Infect. Dis. 2020, 69, 101413. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Proteins (Consensus Sequence) | Mol. Wt. (Dalton) | Theoretical pI | Half-Life (in Mammalian Reticulocytes, In Vitro) | Antigeznecity (Threshold 0.4) | ||||
|---|---|---|---|---|---|---|---|---|
| Glycoprotein B | 100,174.88 | 6.38 | 30 h | 0.5010 | ||||
| Glycoprotein D | 48,472.79 | 5.34 | 30 h | 0.4948 | ||||
| Secondary structure properties | ||||||||
| Glycoprotein B | Glycoprotein D | |||||||
| Alpha helix | (Hh) | 337 is 38.17% | Alpha helix | (Hh) | 152 is 35.02% | |||
| 310helix | (Gg) | 0 is 0.00% | 310helix | (Gg) | 0 is 0.00% | |||
| Pi helix | (Ii) | 0 is 0.00% | Pi helix | (Ii) | 0 is 0.00% | |||
| Beta bridge | (Bb) | 0 is 0.00% | Beta bridge | (Bb) | 0 is 0.00% | |||
| Extended Strand | (Ee) | 150 is 16.99% | Extended Strand | (Ee) | 68 is 15.67% | |||
| Beta turn | (Tt) | 35 is 3.96% | Beta turn | (Tt) | 34 is 7.83% | |||
| Bend region | (Ss) | 0 is 0.00% | Bend region | (Ss) | 0 is 0.00% | |||
| Random coil | (Cc) | 361 is 40.88% | Random coil | (Cc) | 180 is 41.47% | |||
| Ambiguous states | (?) | 0 is 0.00% | Ambiguous states | (?) | 0 is 0.00% | |||
| Other states | 0 is 0.00% | Other states | 0 is 0.00% | |||||
| S.No | Particulars | Value |
|---|---|---|
| 1. | Number of amino acids | 304 |
| 2. | Molecular weight | 32,768.29 |
| 3. | Theoretical pI | 8.87 |
| 4. | Total number of negatively charged residues (Asp + Glu) | 16 |
| 5. | Total number of positively charged residues (Arg + Lys) | 23 |
| 6. | Formula | C1508H2233N383O418S11 |
| 7. | Total number of atoms | 4553 |
| 8. | The estimated half-life is Mammalian reticulocytes-in vitro Yeast-in vivo Escherichia coli-in vivo | 30 h >20 h >10 h |
| 9. | Instability index | 33.88 |
| 10. | Aliphatic index | 78.52 |
| 11. | Grand average of hydropathicity (GRAVY) | 0.096 |
| 12. | Solubility | 0.423 |
| 13. | Antigenicity | 0.6177 |
| 14. | Allergenicity | Non-allergen |
| S.No. | Complexes | Binding Affinity (ΔG) (kcal mol−1) | Dissociation Constant Kd(M) |
|---|---|---|---|
| 1. | TLR 3 + Vaccine construct | −18.7 | 1.9 × 10−14 |
| 2. | MHC I + Vaccine construct | −16.9 | 4.1 × 10−13 |
| 3. | MHC II + Vaccine construct | −11.6 | 2.9 × 10−9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponnusamy, P.; Sukumar, K.; Raja, A.; Saravanan, S.; Srinivasan, P.; Ramya, K.; Selvaraju, M.; Saravanan, R. Design of a Multi-Epitope Vaccine Candidate Against Infectious Laryngotracheitis Virus Affecting Poultry by Computational Approaches. Biology 2025, 14, 765. https://doi.org/10.3390/biology14070765
Ponnusamy P, Sukumar K, Raja A, Saravanan S, Srinivasan P, Ramya K, Selvaraju M, Saravanan R. Design of a Multi-Epitope Vaccine Candidate Against Infectious Laryngotracheitis Virus Affecting Poultry by Computational Approaches. Biology. 2025; 14(7):765. https://doi.org/10.3390/biology14070765
Chicago/Turabian StylePonnusamy, Periyasamy, Kuppannan Sukumar, Angamuthu Raja, Sellappan Saravanan, Palani Srinivasan, Kalaivanan Ramya, Mani Selvaraju, and Ramasamy Saravanan. 2025. "Design of a Multi-Epitope Vaccine Candidate Against Infectious Laryngotracheitis Virus Affecting Poultry by Computational Approaches" Biology 14, no. 7: 765. https://doi.org/10.3390/biology14070765
APA StylePonnusamy, P., Sukumar, K., Raja, A., Saravanan, S., Srinivasan, P., Ramya, K., Selvaraju, M., & Saravanan, R. (2025). Design of a Multi-Epitope Vaccine Candidate Against Infectious Laryngotracheitis Virus Affecting Poultry by Computational Approaches. Biology, 14(7), 765. https://doi.org/10.3390/biology14070765