
The Influence of Parent Pairs with Different Genetic Distances on the Genetic Diversity of Offspring in Strongylocentrotus intermedius


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
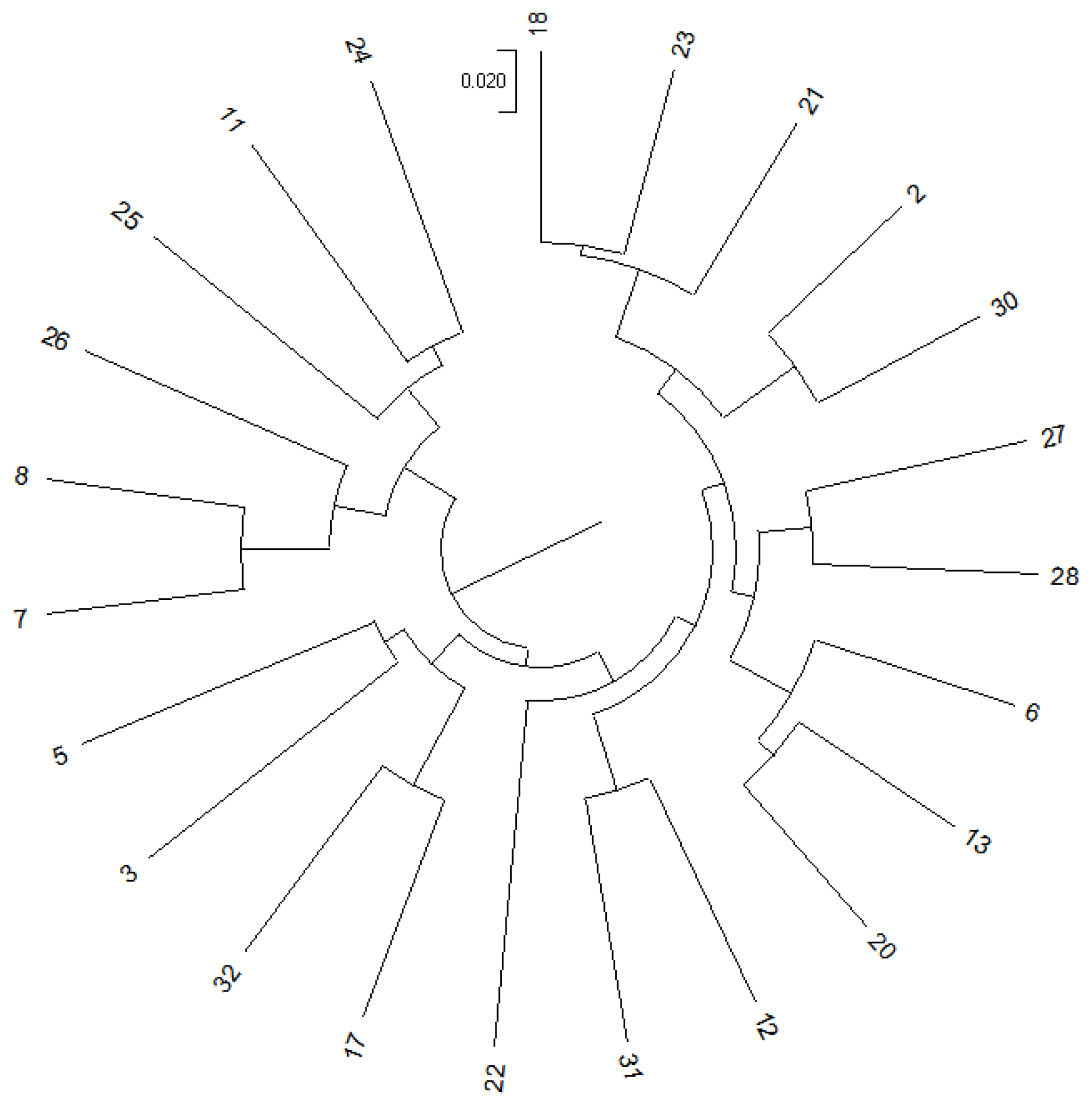
2.1. Experimental Material
2.2. Extraction of Genomic DNA
2.3. SSR-Seq Typing Process
2.4. Data Analysis
3. Results
3.1. Sequencing Results of the Offspring Population and SSR-Seq Genotyping Results
3.2. Genetic Diversity of Loci in the Offspring Population
3.3. Genetic Diversity of the Three Offspring Populations
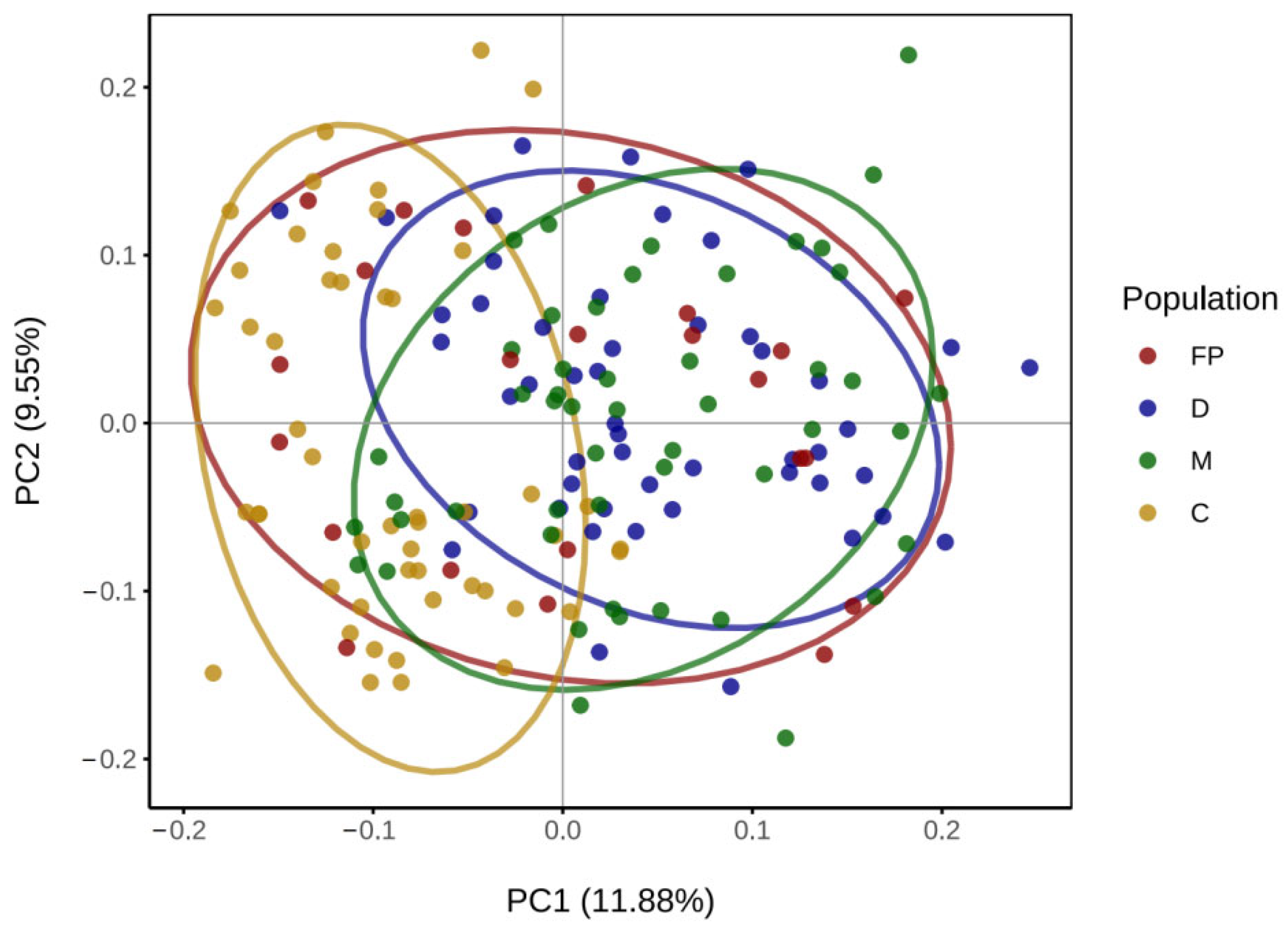
3.4. Analysis of Genetic Differentiation and Population Structure
4. Discussion
4.1. Genetic Diversity Analysis of 15 Loci in Offspring Populations
4.2. Genetic Diversity Analysis of the Three Offspring Populations
4.3. Genetic Differentiation and Genetic Structure of the Three Offspring Populations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andrew, N.L.; Agatsuma, Y.; Ballesteros, E.; Bazhin, A.G.; Creaser, E.P.; Barnes, D.K.A.; Botsford, L.W.; Bradbury, A.; Campbell, A.; Dixon, J.D.; et al. Status and management of world sea urchin fisheries. Oceanogr. Mar. Biol. 2002, 40, 343–425. [Google Scholar] [CrossRef]
- Lawrence, J.M.; Lawrence, A.L.; Watts, S.A. Ingestion, digestion, and digestibility of regular sea urchins. Dev. Aquac. Fish. Sci. 2020, 43, 165–190. [Google Scholar] [CrossRef]
- Wang, Z.; Chang, Y. Studies on hatching of Japanese sea urchin Strongylocentrotus intermedius. J. Fish. Sci. China 1997, 4, 60–67, (In Chinese with English abstract). [Google Scholar]
- Agatsuma, Y. Strongylocentrotus intermedius. Dev. Aquac. Fish. Sci. 2013, 38, 437–447. [Google Scholar] [CrossRef]
- Zhou, W.; Sun, J.; Wang, J.; Du, J. Current Status and Challenges of Sea Urchin Culture in China. J. Fish. Sci. 2008, 3, 151–153, (In Chinese with English abstract). [Google Scholar] [CrossRef]
- Yu, Y.; Hilsdorf, A.W.S.; Zhou, L.; Lin, Q.; Gao, Z.X. Genetics and molecular breeding in aquaculture animals. Front. Genet. 2022, 13, 1071303. [Google Scholar] [CrossRef] [PubMed]
- Gheyas, A.; Basavaraju, Y.; McAndrew, B.; Penman, D.J. Monitoring genetic diversity in a mass selection programme of common carp (Cyprinus carpio) using microsatellite markers. Aquaculture 2007, 272, S261–S262. [Google Scholar] [CrossRef]
- Sui, J.; Luan, S.; Yang, G.; Chen, X.; Luo, K.; Gao, Q.; Wang, J.; Hu, H.; Kong, J. Genetic diversity and population structure of a giant freshwater prawn (Macrobrachium rosenbergii) breeding nucleus in China. Aquac. Res. 2018, 49, 2175–2183. [Google Scholar] [CrossRef]
- Ye, Y.; Ren, W.; Zhang, S.; Zhao, L.; Tang, J.; Hu, L.; Chen, X. Genetic diversity of fish in aquaculture and of common carp (Cyprinus carpio) in Traditional Rice–Fish Coculture. Agriculture 2022, 12, 997. [Google Scholar] [CrossRef]
- Robisalmi, A.; Gunadi, B.; Alipin, K.; Artati, D. Growth response and estimating heritability of synthetic base population (F0) of red tilapia (Oreochromis spp.) through family selection. IOP Conf. Ser. Earth Environ. Sci. 2023, 1260, 012005. [Google Scholar] [CrossRef]
- Liu, J.; Kong, J.; Dai, P.; Yu, Y.; Meng, X.; Luo, K.; Cao, B.; Chen, B.; Gao, H.; Luan, S. Development of SNP markers and verification analysis of relationship on family in Litopenaeus vannamei. Prog. Fish. Sci. 2021, 42, 108–116, (In Chinese with English abstract). [Google Scholar] [CrossRef]
- Yáñez, J.M.; Bassini, L.N.; Filp, M.; Lhorente, J.P.; Ponzoni, R.W.; Neira, R. Inbreeding and effective population size in a coho salmon (Oncorhynchus kisutch) breeding nucleus in Chile. Aquaculture 2014, 420, S15–S19. [Google Scholar] [CrossRef]
- Cheng, W.; Xia, R.; Wang, Q.; Zeng, K.; Song, W.; Wei, Q.; Deng, G.; Cheng, Y. Genetic diversity of Siniperca chuatsi in wild and cultivated populations. Freshw. Fish. 2020, 50, 31–37, (In Chinese with English abstract). [Google Scholar] [CrossRef]
- Fu, X.; Liu, J.; Liu, J. AFLP Analysis of Genetic Diversity in Five Selected Lines of abalone Haliotis diversicolor supertexta. J. Trop. Biol. 2016, 7, 422–426, (In Chinese with English abstract). [Google Scholar] [CrossRef]
- Willoughby, J.R.; Fernandez, N.B.; Lamb, M.C.; Ivy, J.A.; Lacy, R.C.; DeWoody, J.A. The impacts of inbreeding, drift and selection on genetic diversity in captive breeding populations. Mol. Ecol. 2015, 24, 98–110. [Google Scholar] [CrossRef]
- Howard, J.T.; Pryce, J.E.; Baes, C.; Maltecca, C. Invited review: Inbreeding in the genomics era: Inbreeding, inbreeding depression, and management of genomic variability. J. Dairy Sci. 2017, 100, 6009–6024. [Google Scholar] [CrossRef]
- D’ambrosio, J.; Phocas, F.; Haffray, P.; Bestin, A.; Brard-Fudulea, S.; Poncet, C.; Quillet, E.; Dechamp, N.; Fraslin, C.; Charles, M.; et al. Genome-wide estimates of genetic diversity, inbreeding and effective size of experimental and commercial Rainbow trout lines undergoing selective breeding. Genet. Sel. Evol. 2019, 51, 26. [Google Scholar] [CrossRef] [PubMed]
- Würschum, T.; Zhu, X.; Zhao, Y.; Jiang, Y.; Reif, J.C.; Maurer, H.P. Maximization through optimization? On the relationship between hybrid performance and parental genetic distance. Theor. Appl. Genet. 2023, 136, 186. [Google Scholar] [CrossRef]
- Han, Z.; Han, Q.; Xia, Y.; Geng, X.; Du, K.; Yang, J.; Kang, X. Construction of a breeding parent population of Populus tomentosa based on SSR genetic distance analysis. Sci. Rep. 2020, 10, 18573. [Google Scholar] [CrossRef]
- Wei, J.; Zhao, L.; Wu, W.; Luo, K.; Ye, W.; Fu, Y.; Chen, C. Genetic characterization of Oreochromis niloticus (♀) × Sarotherodon melanotheron (♂) hybrid F2 and F3 by microsatellite analysis. S. China Fish. Sci. 2016, 12, 30–35, (In Chinese with English abstract). [Google Scholar]
- Chang, Y.; Zhang, W.; Zhao, C.; Song, J. Estimates of heritabilities and genetic correlations for growth and gonad traits in the sea urchin Strongylocentrotus intermedius. Aquac. Res. 2012, 43, 271–280. [Google Scholar] [CrossRef]
- Chang, Y.Q.; Zhang, W.J.; Leng, X.F.; Song, J. Strongylocentrotus intermedius “Dajin”. China Fish. 2015, 12, 58–59, (In Chinese with English abstract). [Google Scholar] [CrossRef]
- Wang, Z.Z.; Lu, B.Y.; Liu, L. Progress and prospects of scientific and technological innovation in China’s aquaculture seed industry during the 13th Five-Year Plan period. J. Dalian Ocean Univ. 2023, 38, 1–11, (In Chinese with English abstract). [Google Scholar]
- Jiang, H.; Zhang, W.; Chen, L.; Liu, J.; Lv, X.; Wang, Z.; Liu, L.; Chang, Y. Screening of disease-resistant families and analysis of growth performance in intermediate sea urchins. J. Dalian Ocean Univ. 2020, 35, 197–204, (In Chinese with English abstract). [Google Scholar] [CrossRef]
- Liu, L.; Zhang, W.; Liu, Y.; Leng, X.; Ou, F.; Zang, X.; Li, X.; Ding, J.; Chang, Y. Evaluation of the genetic diversity and genetic structure of multiple generation selection populations and unselected common population of sea urchin (Strongylocentrotus intermedius) using SSR-seq. J. Fish. China 2023, 47, 145–155, (In Chinese with English abstract). [Google Scholar] [CrossRef]
- Stoffel, M.A.; Esser, M.; Kardos, M.; Humble, E.; Nichols, H.; David, P.; Hoffman, J.I. inbreedR: An R package for the analysis of inbreeding based on genetic markers. Methods Ecol. Evol. 2016, 7, 1331–1339. [Google Scholar] [CrossRef]
- Jerry, D.R.; Preston, N.P.; Crocos, P.J.; Keys, S.; Meadows, J.R.; Li, Y. Parentage determination of Kuruma shrimp Penaeus (Marsupenaeus) japonicus using microsatellite markers (Bate). Aquaculture 2004, 235, 237–247. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, W.; Wang, W.; Gui, J.F.; Mei, J. Parentage determination of yellow catfish (Pelteobagrus Fulvidraco) based on microsatellite DNA markers. Aquac. Int. 2016, 24, 567–576. [Google Scholar] [CrossRef]
- Simonsen, K.L.; Churchill, G.A.; Aquadro, C.F. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics 1995, 141, 413–429. [Google Scholar] [CrossRef]
- Masatoshi, N.; Fumio, T.; Yoshio, T. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 1983, 19, 153–170. [Google Scholar] [CrossRef]
- Ke, X.; Liu, J.; Gao, F.; Cao, J.; Liu, Z.; Lu, M. Analysis of genetic diversity among six dojo loach (Misgurnus anguillicaudatus) populations in the Pearl River Basin based on microsatellite and mitochondrial DNA markers. Aquac. Rep. 2022, 27, 101346. [Google Scholar] [CrossRef]
- Qin, Y.; Shi, G.; Sun, Y. Evaluation of genetic diversity in Pampus argenteus using ssr markers. Genet. Mol. Res. 2013, 12, 5833–5841. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Li, Q.; Zhang, X. Analysis of genetic diversity in mass selection lines of white-shell Pacific oyster (Crassostrea gigas) using microsatellite fluorescent multiplex PCR technique. J. Fish. China 2017, 41, 1838–1846, (In Chinese with English abstract). [Google Scholar] [CrossRef]
- Wong, J.; Sun, F.; Wang, L.; Yang, Z.T.; Wen, Y.F.; Pang, H.Y.; Lee, M.; Yeo, S.T.; Liang, B.; Chen, K.; et al. Changes in genetic diversity of A*sian seabass in a 20-year breeding program. Aquaculture 2023, 575, 739738. [Google Scholar] [CrossRef]
- Hans, E.; Nicolas, G. Determinants of genetic diversity. Nat. Rev. Genet. 2016, 17, 422–433. [Google Scholar] [CrossRef] [PubMed]
- David, J.C.; Margaret, B.; Craig, M. Genetic Diversity and Conservation Units: Dealing With the Species-Population Continuum in the Age of Genomics. Front. Ecol. Evol. 2018, 6, 165. [Google Scholar] [CrossRef]
- Slatkin, M.; Barton, N.H. A comparison of three indirect methods for estimating average levels of gene flow. Evolution 1989, 43, 1349–1368. [Google Scholar] [CrossRef]
- Zhadan, P.M.; Vaschenko, M.A.; Permyakov, P.A. Quantitative study of the behavior of two broadcast spawners, the sea urchins Strongylocentrotus intermedius and Mesocentrotus nudus, during mass spawning events in situ. PeerJ 2021, 9, e11058. [Google Scholar] [CrossRef]
- Wang, J.; Xia, D. Studies on fish heterosis with DNA fingerprinting. Aquac. Res. 2002, 33, 942–947. [Google Scholar] [CrossRef]
- Vallejo, R.L.; Leeds, T.D.; Gao, G.; Parsons, J.E.; Martin, K.E.; Evenhuis, J.P.; Fragomeni, B.O.; Wiens, G.D.; Palti, Y. Genomic selection models double the accuracy of predicted breeding values for bacterial cold water disease resistance compared to a traditional pedigree-based model in rainbow trout aquaculture. Genet. Sel. Evol. 2017, 49, 17. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Offspring Population ID | Female Parent ID | Male Parent ID | Genetic Distance Between Parents |
|---|---|---|---|
| D1 | 7 | 17 | 0.34297 |
| D2 | 8 | 12 | 0.35118 |
| D3 | 32 | 31 | 0.34743 |
| D4 | 24 | 12 | 0.31827 |
| D5 | 25 | 17 | 0.32214 |
| Mean | — | — | 0.33640 |
| C1 | 23 | 18 | 0.12109 |
| C2 | 13 | 20 | 0.12401 |
| C3 | 21 | 18 | 0.12875 |
| C4 | 6 | 20 | 0.13200 |
| C5 | 27 | 28 | 0.14672 |
| Mean | — | — | 0.13051 |
| M | 2 | 3 | 0.11891~0.32214 |
| 6 | 5 | ||
| 25 | 11 | ||
| 27 | 12 | ||
| — | 17 | ||
| — | 20 | ||
| — | 22 | ||
| — | 26 | ||
| — | 30 | ||
| — | 31 | ||
| Mean | — | — | 0.29916 |
| No. | Locus | Repeat Motif | Number of Alleles | Primer Sequence (5′–3′) |
|---|---|---|---|---|
| 1 | SSR1 | (CT)12 | 196~226 | F: TCGTCATGAGATGGTCGCT R: CATTTTACCGTGGTGGGGTC |
| 2 | SSR2 | (AG)13 | 179~187 | F: CGCAGGATGCAGTGATACC R: ATTCCACCAGTATCCCAGCT |
| 3 | SSR3 | (CT)18 | 136~180 | F: GCGCTTAATCTTTGGATAATTG R: CTGTAGTCGCTCCGCATGT |
| 4 | SSR4 | (AG)12 | 181~219 | F: GGGAAGTTTTCCCCACTGAC R: TGTCCATAACGCCACATTCG |
| 5 | SSR7 | (AC)10 | 199~213 | F: TCCCATATGATTGCTCGTGC R: AGCATTCACCGCGAAACTG |
| 6 | SSR14 | (AG)10 | 165~179 | F: ATCCCAAACTACGTTCAACC R: GGCTGCCTAGTTGCATAAAT |
| 7 | SSR16 | (CT)16 | 146~246 | F: CCTTGGAATGAGAACTTGT R: ACCGATTTTACTTGACCTG |
| 8 | SSR17 | (AT)6 | 231~237 | F: CTGTTTGGATGAGTGGAAT R: TTTGAACGAGCTTGCCTT |
| 9 | SSR18 | (TTGACT)4 | 112~130 | F: GTCAGGTAGCTATATGTTC R: TGGTGATAAACATGTCAGAA |
| 10 | SSR19 | (GCA)8 | 93~102 | F: AGCTCCTAGGGTTCTTACC R: ACATGGGTGGAGAGGTG |
| 11 | SSR20 | (GAA)5 | 147~156 | F: CTAATAGCCCTATGCCGCGT R: ATACACCACACGATTCGCAC |
| 12 | SSRA6 | (TGA)6 | 179~194 | F: AAGCAGCCATTAAGGAAATG R: CAAGCAGGTTATCCGTTTCA |
| 13 | SSRA9 | (TC)9 | 185~211 | F: AAGCGAGCTTATGTCTAGTA R: CTAGAACCTTCATCAACTCT |
| 14 | SSRA10 | (AC)6 | 148~168 | F: CACGTATTTCGGATGGTGAC R: CTTATTATTAGCGCACGTCAT |
| 15 | SSRA22 | (TCTG)6 | 186~202 | F: GAAGAACCATGGACTTACTACA R: TGTTGTGAGAAAGGTAGCG |
| Clean Reads | Raw Reads | Clean Reads Ratio | Q20 | Q30 | |
|---|---|---|---|---|---|
| Average of all sampled offspring | 45,295 | 53,518 | 0.85 | 100.00% | 99.98% |
| Locus | Na | Ne | Ho | He | F | PIC |
|---|---|---|---|---|---|---|
| SSR1 | 10 | 6.553 | 0.820 | 0.847 | 0.032 | 0.829 |
| SSR2 | 4 | 1.576 | 0.298 | 0.365 | 0.185 | 0.337 |
| SSR3 | 3 | 2.693 | 0.567 | 0.629 | 0.099 | 0.579 |
| SSR4 | 9 | 3.280 | 0.380 | 0.695 | 0.453 | 0.664 |
| SSR7 | 8 | 3.540 | 0.740 | 0.718 | −0.031 | 0.673 |
| SSR14 | 6 | 3.023 | 0.613 | 0.669 | 0.083 | 0.623 |
| SSR16 | 3 | 1.743 | 0.473 | 0.426 | −0.111 | 0.387 |
| SSR17 | 4 | 1.975 | 0.567 | 0.494 | −0.148 | 0.444 |
| SSR18 | 3 | 2.144 | 0.252 | 0.534 | 0.528 | 0.445 |
| SSR19 | 3 | 1.461 | 0.347 | 0.315 | −0.099 | 0.285 |
| SSR20 | 4 | 2.206 | 0.487 | 0.547 | 0.11 | 0.482 |
| SSRA6 | 6 | 2.797 | 0.240 | 0.642 | 0.626 | 0.604 |
| SSRA9 | 5 | 1.757 | 0.420 | 0.431 | 0.025 | 0.397 |
| SSRA10 | 4 | 2.663 | 0.520 | 0.625 | 0.167 | 0.566 |
| SSRA22 | 3 | 2.631 | 0.281 | 0.620 | 0.546 | 0.550 |
| Mean | 5.067 | 2.669 | 0.467 | 0.570 | 0.164 | 0.524 |
| Population | Locus | Na | Ne | Ho | He | PIC | HWE |
|---|---|---|---|---|---|---|---|
| D | SSR1 | 9 | 6.624 | 0.842 | 0.849 | 0.831 | ns |
| SSR2 | 3 | 2.486 | 0.333 | 0.598 | 0.526 | * | |
| SSR3 | 4 | 2.473 | 0.520 | 0.596 | 0.518 | ns | |
| SSR4 | 7 | 3.748 | 0.480 | 0.733 | 0.704 | ns | |
| SSR7 | 7 | 3.846 | 0.620 | 0.740 | 0.705 | ** | |
| SSR14 | 5 | 3.408 | 0.780 | 0.707 | 0.655 | ns | |
| SSR16 | 3 | 1.644 | 0.420 | 0.392 | 0.356 | ns | |
| SSR17 | 3 | 1.738 | 0.540 | 0.425 | 0.379 | ns | |
| SSR18 | 3 | 1.971 | 0.180 | 0.493 | 0.405 | ns | |
| SSR19 | 3 | 1.678 | 0.500 | 0.404 | 0.337 | * | |
| SSR20 | 3 | 2.684 | 0.720 | 0.627 | 0.556 | ** | |
| SSRA6 | 4 | 2.641 | 0.340 | 0.621 | 0.572 | ns | |
| SSRA9 | 3 | 1.691 | 0.400 | 0.409 | 0.361 | ns | |
| SSRA10 | 3 | 2.145 | 0.420 | 0.534 | 0.462 | ** | |
| SSRA22 | 3 | 2.955 | 0.340 | 0.662 | 0.587 | ns | |
| Mean | 4.200 | 2.782 | 0.496 | 0.586 | 0.530 | — | |
| C | SSR1 | 6 | 4.909 | 0.778 | 0.796 | 0.766 | ns |
| SSR2 | 4 | 1.491 | 0.317 | 0.329 | 0.300 | * | |
| SSR3 | 4 | 2.643 | 0.640 | 0.622 | 0.554 | ns | |
| SSR4 | 4 | 2.859 | 0.340 | 0.650 | 0.583 | ns | |
| SSR7 | 4 | 2.767 | 0.860 | 0.639 | 0.568 | ns | |
| SSR14 | 3 | 2.256 | 0.440 | 0.557 | 0.494 | ** | |
| SSR16 | 3 | 1.744 | 0.500 | 0.427 | 0.383 | ns | |
| SSR17 | 4 | 1.986 | 0.540 | 0.497 | 0.437 | ns | |
| SSR18 | 2 | 1.777 | 0.229 | 0.437 | 0.342 | ** | |
| SSR20 | 2 | 1.173 | 0.160 | 0.147 | 0.136 | ns | |
| SSRA6 | 3 | 1.545 | 0.060 | 0.353 | 0.323 | ns | |
| SSRA9 | 4 | 1.660 | 0.420 | 0.398 | 0.369 | ns | |
| SSRA10 | 4 | 2.632 | 0.680 | 0.620 | 0.576 | ns | |
| SSRA22 | 3 | 1.525 | 0.300 | 0.344 | 0.300 | ns | |
| Mean | 3.571 | 2.212 | 0.447 | 0.487 | 0.438 | — | |
| M | SSR1 | 9 | 6.722 | 0.818 | 0.851 | 0.833 | ** |
| SSR2 | 3 | 1.348 | 0.263 | 0.258 | 0.242 | ns | |
| SSR3 | 4 | 2.234 | 0.540 | 0.552 | 0.494 | ns | |
| SSR4 | 9 | 2.194 | 0.320 | 0.544 | 0.526 | ns | |
| SSR7 | 7 | 3.802 | 0.740 | 0.737 | 0.695 | ** | |
| SSR14 | 5 | 2.921 | 0.620 | 0.658 | 0.608 | ns | |
| SSR16 | 3 | 1.823 | 0.500 | 0.451 | 0.406 | ns | |
| SSR17 | 3 | 2.121 | 0.620 | 0.529 | 0.468 | * | |
| SSR18 | 3 | 2.679 | 0.366 | 0.627 | 0.548 | ** | |
| SSR19 | 3 | 1.818 | 0.540 | 0.450 | 0.403 | ns | |
| SSR20 | 4 | 2.229 | 0.580 | 0.551 | 0.493 | ns | |
| SSRA6 | 6 | 3.544 | 0.320 | 0.718 | 0.674 | ns | |
| SSRA9 | 5 | 1.905 | 0.440 | 0.475 | 0.440 | ns | |
| SSRA10 | 4 | 2.677 | 0.460 | 0.626 | 0.550 | ns | |
| SSRA22 | 3 | 2.916 | 0.195 | 0.657 | 0.583 | ns | |
| Mean | 4.733 | 2.729 | 0.488 | 0.579 | 0.531 | — | |
| FP | SSR1 | 7 | 4.966 | 0.917 | 0.799 | 0.770 | * |
| SSR2 | 4 | 2.426 | 0.545 | 0.588 | 0.517 | ns | |
| SSR3 | 5 | 3.061 | 0.656 | 0.673 | 0.622 | ns | |
| SSR4 | 8 | 4.223 | 0.344 | 0.763 | 0.726 | ns | |
| SSR7 | 8 | 3.977 | 0.750 | 0.749 | 0.712 | * | |
| SSR14 | 6 | 3.606 | 0.781 | 0.723 | 0.681 | ns | |
| SSR16 | 4 | 1.750 | 0.469 | 0.429 | 0.394 | ns | |
| SSR17 | 4 | 2.181 | 0.656 | 0.542 | 0.487 | ns | |
| SSR18 | 3 | 2.217 | 0.258 | 0.549 | 0.463 | ns | |
| SSR19 | 3 | 1.724 | 0.344 | 0.420 | 0.354 | ns | |
| SSR20 | 4 | 2.325 | 0.594 | 0.570 | 0.496 | ** | |
| SSRA6 | 5 | 2.711 | 0.161 | 0.631 | 0.588 | ns | |
| SSRA9 | 5 | 1.438 | 0.313 | 0.305 | 0.292 | ns | |
| SSRA10 | 4 | 2.131 | 0.625 | 0.531 | 0.481 | ns | |
| SSRA22 | 3 | 2.186 | 0.296 | 0.543 | 0.478 | ** | |
| mean | 5.077 | 2.816 | 0.522 | 0.595 | 0.546 | — |
| Locus | Fis | Fit | Fst | Nm |
|---|---|---|---|---|
| SSR1 | 0.023 | 0.034 | 0.011 | 23.532 |
| SSR2 | 0.229 | 0.292 | 0.082 | 2.795 |
| SSR3 | 0.039 | 0.099 | 0.062 | 3.796 |
| SSR4 | 0.409 | 0.453 | 0.076 | 3.053 |
| SSR7 | −0.049 | −0.031 | 0.017 | 14.320 |
| SSR14 | 0.042 | 0.083 | 0.043 | 5.554 |
| SSR16 | −0.118 | −0.111 | 0.007 | 37.201 |
| SSR17 | −0.173 | −0.148 | 0.021 | 11.717 |
| SSR18 | 0.502 | 0.522 | 0.041 | 5.911 |
| SSR19 | −0.218 | −0.099 | 0.097 | 2.317 |
| SSR20 | −0.101 | 0.110 | 0.191 | 1.056 |
| SSRA6 | 0.574 | 0.626 | 0.122 | 1.796 |
| SSRA9 | 0.017 | 0.025 | 0.009 | 28.947 |
| SSRA10 | 0.124 | 0.167 | 0.050 | 4.768 |
| SSRA22 | 0.497 | 0.547 | 0.099 | 2.275 |
| Mean | 0.120 | 0.171 | 0.062 | 9.936 |
| Source of Variation | df | Variance Components | Percentage of Variation | p-Value |
|---|---|---|---|---|
| Between population | 2 | 0.5537 | 7.984 | <0.01 |
| Between individuals within population | 147 | 0.7574 | 10.922 | <0.01 |
| Within individuals | 150 | 5.6233 | 81.093 | <0.01 |
| Total | 299 | 6.9344 | 100 | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, P.; Jiang, X.; Guo, H.; Jia, T.; Wu, S.; Ou, F.; Tian, W.; Liu, L.; Chang, Y.; Ding, J.; et al. The Influence of Parent Pairs with Different Genetic Distances on the Genetic Diversity of Offspring in Strongylocentrotus intermedius. Biology 2025, 14, 745. https://doi.org/10.3390/biology14070745
Liu P, Jiang X, Guo H, Jia T, Wu S, Ou F, Tian W, Liu L, Chang Y, Ding J, et al. The Influence of Parent Pairs with Different Genetic Distances on the Genetic Diversity of Offspring in Strongylocentrotus intermedius. Biology. 2025; 14(7):745. https://doi.org/10.3390/biology14070745
Chicago/Turabian StyleLiu, Peng, Xuechun Jiang, Hao Guo, Tongshan Jia, Shuaichen Wu, Fanjiang Ou, Wenzhuo Tian, Lei Liu, Yaqing Chang, Jun Ding, and et al. 2025. "The Influence of Parent Pairs with Different Genetic Distances on the Genetic Diversity of Offspring in Strongylocentrotus intermedius" Biology 14, no. 7: 745. https://doi.org/10.3390/biology14070745
APA StyleLiu, P., Jiang, X., Guo, H., Jia, T., Wu, S., Ou, F., Tian, W., Liu, L., Chang, Y., Ding, J., & Zhang, W. (2025). The Influence of Parent Pairs with Different Genetic Distances on the Genetic Diversity of Offspring in Strongylocentrotus intermedius. Biology, 14(7), 745. https://doi.org/10.3390/biology14070745