
The First Complete Mitogenome Characterization and Phylogenetic Implications of Elops machnata (Teleostei: Elopiformes: Elopidae)

 and
and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Sequencing and Assembly
2.3. Annotation and Sequence Analysis
2.4. Phylogenetic Tree Reconstruction
3. Results
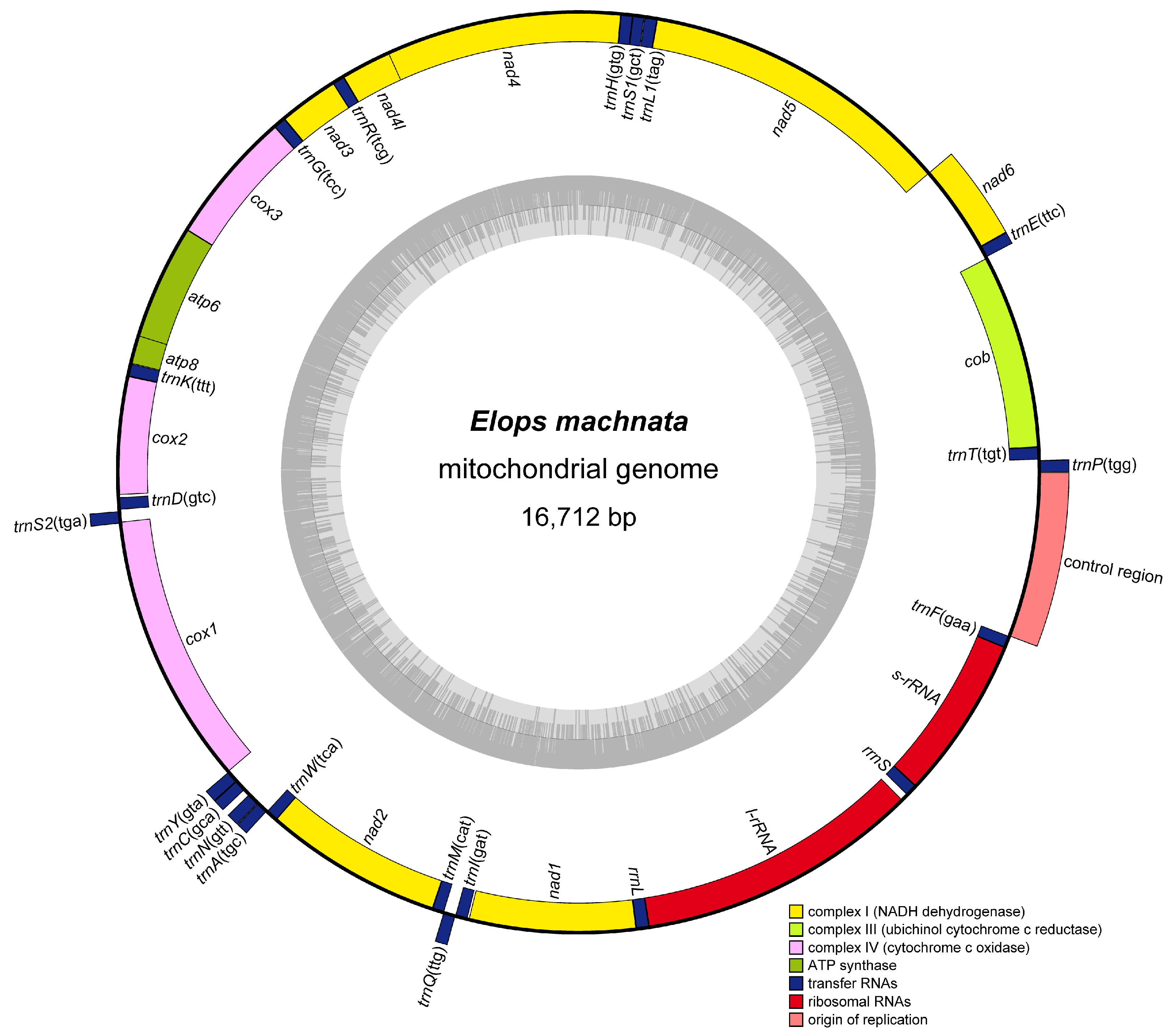
3.1. Genome Organization and Composition
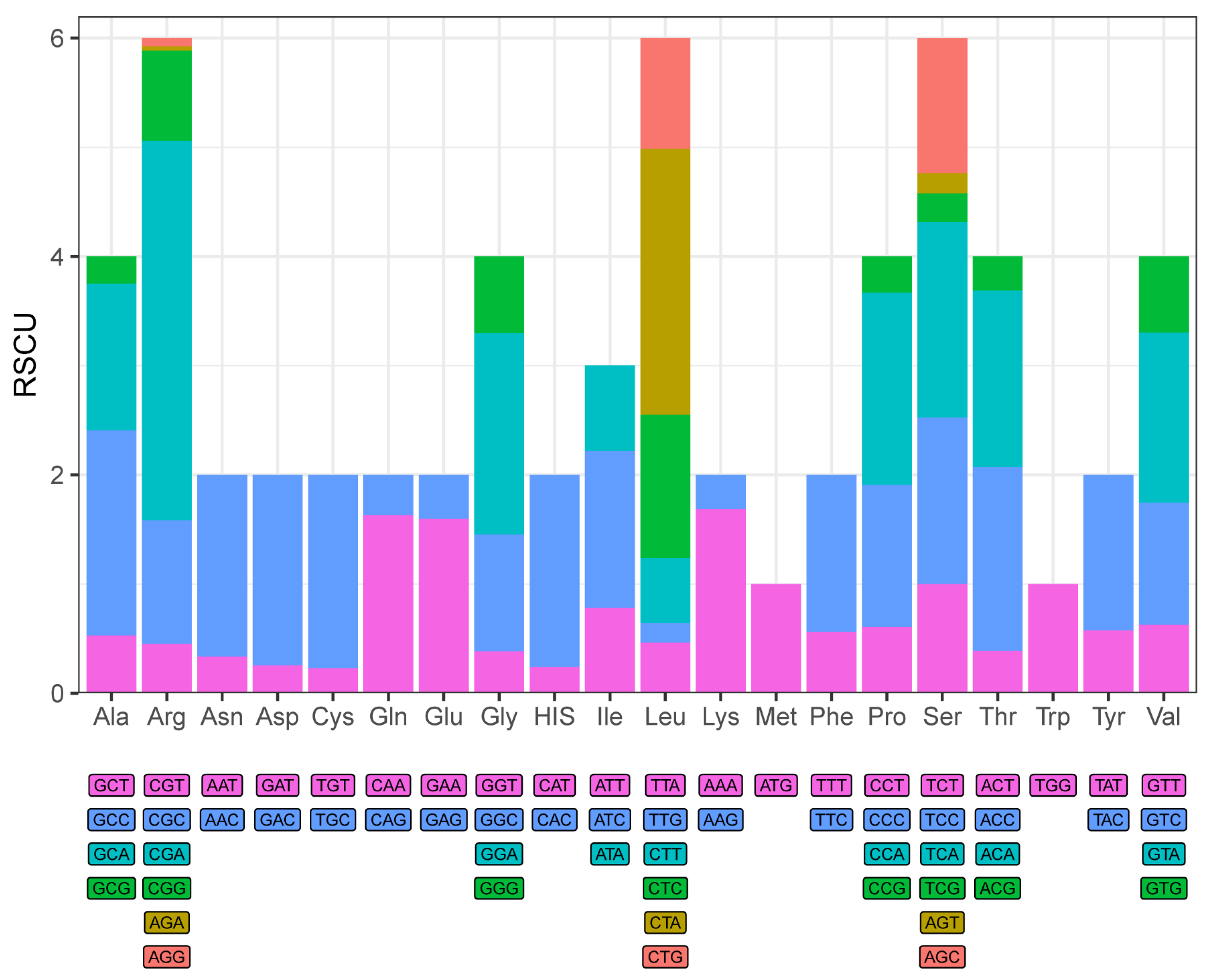
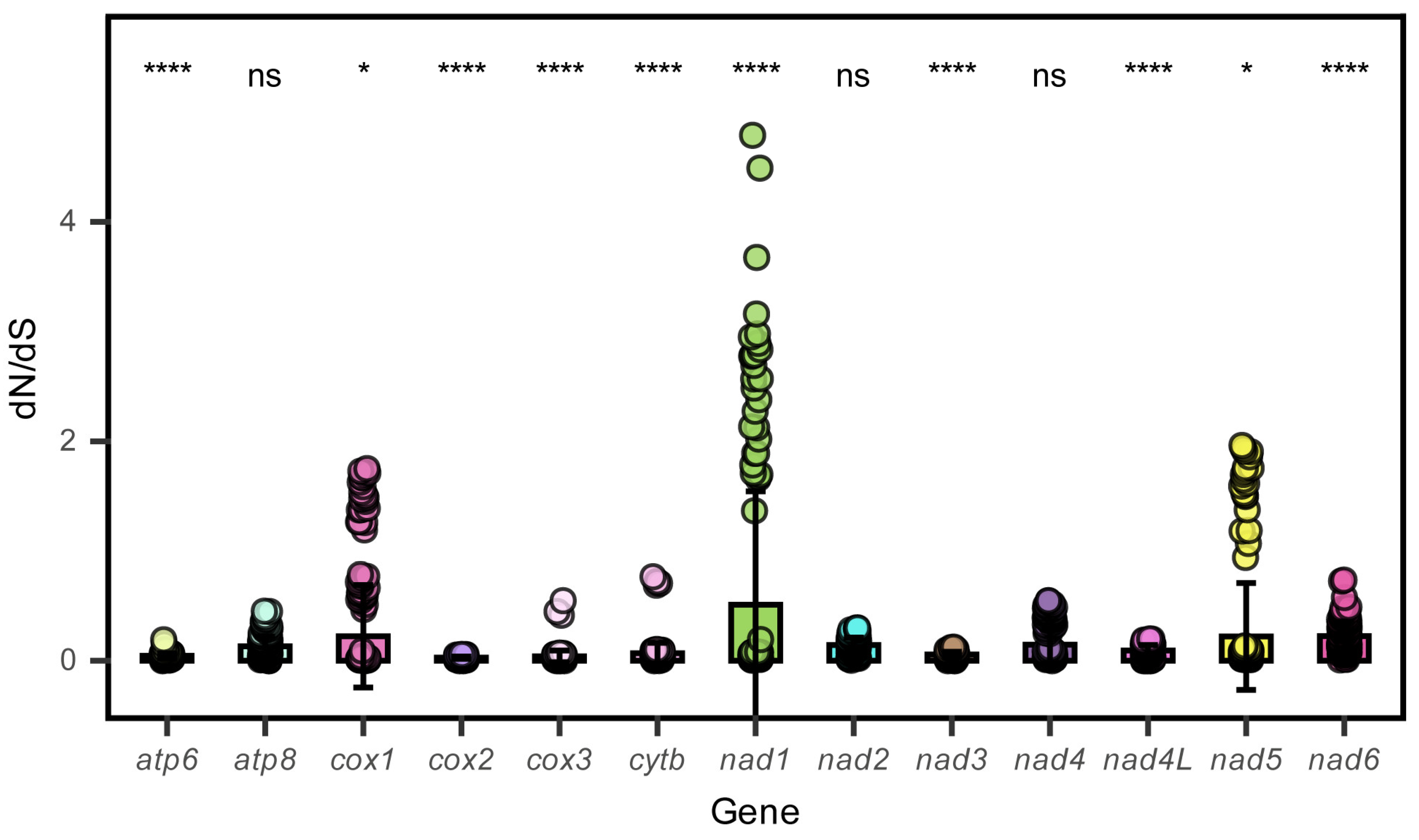
3.2. Protein-Coding Genes
3.3. Transfer and Ribosomal RNA Gene
3.4. Phylogenetic Analysis of Elopomorpha Species
4. Discussion
4.1. E. machnata Mitochondrial Genome Characteristics and Their Implications
4.2. Elopomorpha Phylogeny: Evolutionary Reconstruction and Relationships
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| QC | quality control |
| PCGs | protein-coding genes |
| RSCU | relative synonymous codon usage |
| CR | control region |
| DHU | dihydrouridine |
| DD | data deficient |
| BI | Bayesian inference |
| ML | maximum likelihood |
References
- Dornburg, A.; Friedman, M.; Near, T.J. Phylogenetic Analysis of Molecular and Morphological Data Highlights Uncertainty in the Relationships of Fossil and Living Species of Elopomorpha (Actinopterygii: Teleostei). Mol. Phylogenet. Evol. 2015, 89, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Alves, Y.M.; Alvarado-Ortega, J.; Brito, P.M. †Epaelops martinezi Gen. and Sp. Nov. from the Albian Limestone Deposits of the Tlayúa Quarry, Mexico—A New Late Mesozoic Record of Elopiformes of the Western Tethys. Cretac. Res. 2020, 110, 104260. [Google Scholar] [CrossRef]
- Adams, A.J.; Horodysky, A.Z.; McBride, R.S.; Guindon, K.; Shenker, J.; MacDonald, T.C.; Harwell, H.D.; Ward, R.; Carpenter, K. Global Conservation Status and Research Needs for Tarpons (Megalopidae), Ladyfishes (Elopidae) and Bonefishes (Albulidae). Fish Fish. 2014, 15, 280–311. [Google Scholar] [CrossRef]
- De Sousa, R.P.C.; Silva-Oliveira, G.C.; Furo, I.O.; De Oliveira-Filho, A.B.; De Brito, C.D.B.; Rabelo, L.; Guimarães-Costa, A.; De Oliveira, E.H.C.; Vallinoto, M. The Role of the Chromosomal Rearrangements in the Evolution and Speciation of Elopiformes fishes (Teleostei; Elopomorpha). Zool. Anz. 2021, 290, 40–48. [Google Scholar] [CrossRef]
- Levesque, J.C. Age, Growth, and Recruitment Patterns of Juvenile Ladyfish (Elops sp.) from the East Coast of Florida (USA). PeerJ 2015, 3, e1392. [Google Scholar] [CrossRef]
- De Sousa, R.P.C.; Bessa-Brito, C.D.; Guimarães-Costa, A.; Evangelista-Gomes, G.; Sampaio, I.; De Oliveira, E.H.C.; Vallinoto, M. Exploring the Diversity of Elopidae (Teleostei; Elopiformes) Using DNA Barcoding Analysis. Diversity 2022, 14, 1008. [Google Scholar] [CrossRef]
- Ramanadevi, V.; Thangaraj, M. Comparative Phylogenetic Study of Four Genes of Mitochondrial Genome in Tenpounder Fishes (Order: Elopiformes). Not. Sci. Biol. 2013, 5, 282–289. [Google Scholar] [CrossRef]
- Ramanadevi, V.; Thangaraj, M. Genetic Diversity Analysis of Elops machnata (Forskal) Populations in South East and West Coasts of India Using RAPD Markers. Not. Sci. Biol. 2014, 6, 399–406. [Google Scholar] [CrossRef]
- Filleul, A.; Lavoué, S. Basal Teleosts and the Question of Elopomorph Monophyly. Morphological and Molecular Approaches. Comptes Rendus l’Académie Sci.—Ser. III—Sci. Vie 2001, 324, 393–399. [Google Scholar] [CrossRef]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. Mitogenomic Evidence for the Monophyly of Elopomorph Fishes (Teleostei) and the Evolutionary Origin of the Leptocephalus Larva. Mol. Phylogenet. Evol. 2004, 32, 274–286. [Google Scholar] [CrossRef]
- Betancur-R, R.; Broughton, R.E.; Wiley, E.O.; Carpenter, K.; López, J.A.; Li, C.; Holcroft, N.I.; Arcila, D.; Sanci angco, M.; Cureton, J.C., II; et al. The Tree of Life and a New Classification of Bony Fishes. PLoS Curr. 2013, 5, e1001550. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-N.; López, J.A.; Lavoué, S.; Miya, M.; Chen, W.-J. Phylogeny of the Elopomorpha (Teleostei): Evidence from Six Nuclear and Mitochondrial Markers. Mol. Phylogenet. Evol. 2014, 70, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Faircloth, B.C.; Sorenson, L.; Santini, F.; Alfaro, M.E. A Phylogenomic Perspective on the Radiation of Ray-Finned Fishes Based upon Targeted Sequencing of Ultraconserved Elements (UCEs). PLoS ONE 2013, 8, e65923. [Google Scholar] [CrossRef]
- Near, T.J.; Eytan, R.I.; Dornburg, A.; Kuhn, K.L.; Moore, J.A.; Davis, M.P.; Wainwright, P.C.; Friedman, M.; Smith, W.L. Resolution of Ray-Finned Fish Phylogeny and Timing of Diversification. Proc. Natl. Acad. Sci. USA 2012, 109, 13698–13703. [Google Scholar] [CrossRef] [PubMed]
- Sousa, R.P.C.D.; Sodré, D.; Costa, R.M.D.; Vallinoto, M.; Oliveira, E.H.C.; Silva-Oliveira, G.C.; Sampaio, I.; Guimarães-Costa, A. Range Distribution and Contributions to Taxonomy of Elops smithi (Elopiformes: Elopidae). An. Acad. Bras. Ciênc. 2019, 91, e20181240. [Google Scholar] [CrossRef]
- Andersson, G.E.; Karlberg, O.; Canbäck, B.; Kurland, C.G. On the Origin of Mitochondria: A Genomics Perspective. Philos. Trans. R. Soc. Lond. B 2003, 358, 165–179. [Google Scholar] [CrossRef]
- Hebert, S.L.; Lanza, I.R.; Nair, K.S. Mitochondrial DNA Alterations and Reduced Mitochondrial Function in Aging. Mech. Ageing Dev. 2010, 131, 451–462. [Google Scholar] [CrossRef]
- Cao, J.; Luo, Y.; Chen, Y.; Wu, Z.; Zhang, J.; Wu, Y.; Hu, W. Maternal Mitochondrial Function Affects Paternal Mitochondrial Inheritance in Drosophila. Genetics 2024, 226, iyae014. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Big Trees from Little Genomes: Mitochondrial Gene Order as a Phylogenetic Tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Song, Y.; Zhu, X.-Y.; Zhang, X.-M.; Xiong, H. Targeted Mitochondrial Epigenetics: A New Direction in Alzheimer’s Disease Treatment. Int. J. Mol. Sci. 2022, 23, 9703. [Google Scholar] [CrossRef]
- Zhao, Y.; Peng, C.; Zhang, J.; Lai, R.; Zhang, X.; Guo, Z. Mitochondrial Displacement Loop Region SNPs Modify Sjögren’s Syndrome Development by Regulating Cytokines Expression in Female Patients. Front. Genet. 2022, 13, 847521. [Google Scholar] [CrossRef] [PubMed]
- Podsiadlowski, L.; Bartolomaeus, T. Major Rearrangements Characterize the Mitochondrial Genome of the Isopod Idotea baltica (Crustacea: Peracarida). Mol. Phylogenet. Evol. 2006, 40, 893–899. [Google Scholar] [CrossRef]
- Lin, B.; Chen, H.; Li, J.; Liao, J. Characteristics and Phylogenetic Implications of the Mitochondrial Genome of a Rare Species, Libellula melli. Gene Rep. 2024, 36, 101986. [Google Scholar] [CrossRef]
- Marnis, H.; Syahputra, K.; Kadarusman; Darmawan, J.; Cartealy, I.C.; Larashati, S.; Kusuma, W.E.; Hayuningtyas, E.P.; Iswanto, B.; Asaf, R.; et al. Insights into the Structural Features and Phylogenetic Implications of the Complete Mitochondrial Genome of Fasin Rainbow Fish (Melanotaenia fasinensis). BMC Genom. 2024, 25, 1066. [Google Scholar] [CrossRef]
- Sun, C.-H.; Huang, Q.; Zeng, X.-S.; Li, S.; Zhang, X.-L.; Zhang, Y.-N.; Liao, J.; Lu, C.-H.; Han, B.-P.; Zhang, Q. Comparative Analysis of the Mitogenomes of Two Corydoras (Siluriformes, Loricarioidei) with Nine Known Corydoras, and a Phylogenetic Analysis of Loricarioidei. ZooKeys 2022, 1083, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhou, L.; Yang, W.-T.; Miao, L.; Li, Z.; Zhang, X.-J.; Wang, Y.; Gui, J.-F. Comparative Mitogenome Analyses Uncover Mitogenome Features and Phylogenetic Implications of the Subfamily Cobitinae. BMC Genom. 2021, 22, 50. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de Novo Metazoan Mitochondrial Genome Annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved Detection and Functional Classification of Transfer RNA Genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) Version 1.3.1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Da tasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An Integrated and Scalable Desktop Platform for Streamlined Molecular Sequence Data Management and Evolutionary Phylogenetics Studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Z.; Gao, J.; Chen, X.; Li, Z.; Yu, Y.; Zhao, Y. The Complete Mitochondrial Genome of the Parkin son’s Rainbowfish, Melanotaenia parkinsoni (Atheriniformes: Melanotaeniidae). Mitochondrial DNA Part A 2016, 27, 2621–2622. [Google Scholar] [CrossRef]
- Ma, Z.; Yang, X.; Bercsenyi, M.; Wu, J.; Yu, Y.; Wei, K.; Fan, Q.; Yang, R. Comparative Mitogenomics of the Genus Odontobutis (Perciformes: Gobioidei: Odontobutidae) Revealed Conserved Gene Rearrangement and High Sequence Variations. Int. J. Mol. Sci. 2015, 16, 25031–25049. [Google Scholar] [CrossRef]
- Fonseca, M.M.; Harris, D.J.; Posada, D. The Inversion of the Control Region in Three Mitogenomes Provides Further Evidence for an Asymmetric Model of Vertebrate mtDNA Replication. PLoS ONE 2014, 9, e106654. [Google Scholar] [CrossRef]
- Das, P.J.; Kumar, S.; Choudhury, M.; Banik, S.; Pegu, S.R.; Kumar, S.; Deb, R.; Gupta, V.K. Characterization of the Complete Mitochondrial Genome and Identification of Signature Sequence of Indian Wild Pig. Gene 2024, 897, 148070. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal Mitochondrial Genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Patel, S.; Evans, C.W.; Stuckey, A.; Matzke, N.J.; Millar, C.D. A Unique Mitochondrial Gene Block Inversion in Antarctic Trematomin Fishes: A Cautionary Tale. J. Hered. 2022, 113, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Hardt, W.-D.; Schlegl, J.; Erdmann, V.A.; Hartmann’, R.K. Role of the D Arm and the Anticodon Arm in tRNA Recognition by Eubacterial and Eukaryotic RNase P Enzymesf. Biochemistry 1993, 32, 13046–13053. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.P.; Kim, J.-O.; Yoo, S.H.; Shin, J.; Yang, J.-Y.; Kim, K.; Kim, G.-D. Complete Mitochondrial Genome of Niphon spinosus (Perciformes: Niphonidae): Genome Characterization and Phylogenetic Analysis. Biomolecules 2025, 15, 52. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Selection on Codon Bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef]
- Wu, X.; Xu, M.; Yang, J.-R.; Lu, J. Genome-Wide Impact of Codon Usage Bias on Translation Optimization in Drosophila Melanogaster. Nat. Commun. 2024, 15, 8329. [Google Scholar] [CrossRef]
- Liao, J.; Li, J.-Y.; Li, Y.-Y.; Zhang, S.-Y.; Yang, Y.-F.; Guo, Y.-S.; Wang, Z.-D. In-Depth Analysis of the First Complete Mitochondrial Genome of the Rare Shrimp Alpheus euphrosyne, with Comparative Phylogenetic Genomics Insights into the Alpheus Species. Mol. Biol. Rep. 2025, 52, 579. [Google Scholar] [CrossRef]
- Liao, J.; Lin, B.-Q.; Wang, H.-J.; Wu, Z.-Q. Genetic Structural Variation in Mitochondrial Genomes of Four Species of Gomphidae and Their Phylogenetic Implications. Gene Rep. 2023, 33, 101808. [Google Scholar] [CrossRef]
- Bañón, R.; Arronte, J.C.; Armesto, Á.; Barros-García, D.; Carlos, A.D. Halosaur Fishes (Notacanthiformes: Halo sauridae) from Atlantic Spanish Waters According to Integrative Taxonomy. Zootaxa 2016, 4184, 471–490. [Google Scholar] [CrossRef]
- Zhang, Z.-Q. Animal Biodiversity: An Introduction to Higher-Level Classification and Taxonomic Richness. Zootaxa 2011, 3148, 7–12. [Google Scholar] [CrossRef]
- Barros-García, D.; Gomes-dos-Santos, A.; Machado, A.M.; Castro, L.F.C.; De Carlos, A.; Bañón, R.; Bruno, I.; Ar ronte, J.C.; Froufe, E. Complete Mitogenome of the Oven’s Halosaur, Halosaurus ovenii (Elopomorpha; Notacanthiformes). Mitochondrial DNA Part B 2021, 6, 1571–1572. [Google Scholar] [CrossRef]
- Barros-García, D.; Froufe, E.; Bañón, R.; Carlos Arronte, J.; De Carlos, A. Phylogenetic Analysis Shows the General Diversification Pattern of Deep-Sea Notacanthiforms (Teleostei: Elopomorpha). Mol. Phylogenet. Evol. 2018, 124, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Wu, Z.; Wang, H.; Xiao, S.; Mo, P.; Cui, X. Projected Effects of Climate Change on Species Range of Pantala flavescens, a Wandering Glider Dragonfly. Biology 2023, 12, 226. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Start | Stop | Strand | Length | Interval | Start Coding | Stop Coding |
|---|---|---|---|---|---|---|---|
| trnP(tgg) | 1 | 70 | + | 70 | 3 | ||
| trnT(tgt) | 74 | 146 | − | 73 | 0 | ||
| cytb | 147 | 1287 | − | 1141 | 5 | ATG | ACT |
| trnE(ttc) | 1293 | 1361 | + | 69 | 0 | ||
| nad6 | 1362 | 1883 | + | 522 | −4 | ATG | TAA |
| nad5 | 1880 | 3721 | − | 1842 | 0 | ATG | TAA |
| trnL1(tag) | 3722 | 3794 | − | 73 | 1 | ||
| trnS1(gct) | 3796 | 3863 | − | 68 | 0 | ||
| trnH(gtg) | 3864 | 3932 | − | 69 | 0 | ||
| nad4 | 3933 | 5313 | − | 1381 | −7 | ATG | TCT |
| nad4l | 5307 | 5603 | − | 297 | 0 | ATG | TAA |
| trnR(tcg) | 5604 | 5673 | − | 70 | −2 | ||
| nad3 | 5672 | 6022 | − | 351 | 0 | ATG | TAG |
| trnG(tcc) | 6023 | 6094 | − | 72 | −1 | ||
| cox3 | 6094 | 6879 | − | 786 | −1 | ATG | TAA |
| atp6 | 6879 | 7562 | − | 684 | −10 | ATG | TAA |
| atp8 | 7553 | 7720 | − | 168 | 1 | ATG | TAA |
| trnK(ttt) | 7722 | 7795 | − | 74 | 0 | ||
| cox2 | 7796 | 8486 | − | 691 | 14 | ATG | CCT |
| trnD(gtc) | 8501 | 8572 | − | 72 | 8 | ||
| trnS2(tga) | 8581 | 8651 | + | 71 | −9 | ||
| cox1 | 8643 | 10,238 | − | 1596 | 1 | GTG | AGG |
| trnY(gta) | 10,240 | 10,310 | + | 71 | 0 | ||
| trnC(gca) | 10,311 | 10,378 | + | 68 | 37 | ||
| trnN(gtt) | 10,416 | 10,487 | + | 72 | 1 | ||
| trnA(tgc) | 10,489 | 10,557 | + | 69 | 1 | ||
| trnW(tca) | 10,559 | 10,630 | − | 72 | −2 | ||
| nad2 | 10,629 | 11,675 | − | 1047 | 0 | ATG | TAG |
| trnM(cat) | 11,676 | 11,744 | − | 69 | −1 | ||
| trnQ(ttg) | 11,744 | 11,814 | + | 71 | −1 | ||
| trnI(gat) | 11,814 | 11,885 | − | 72 | 8 | ||
| nad1 | 11,894 | 12,868 | − | 975 | 0 | ATG | TAA |
| trnL2(taa) | 12,869 | 12,943 | − | 75 | 0 | ||
| rrnL | 12,944 | 14,606 | − | 1663 | 39 | ||
| trnV(tac) | 14,646 | 14,717 | − | 72 | 0 | ||
| rrnS | 14,718 | 15,674 | − | 957 | 0 | ||
| trnF(gaa) | 15,675 | 15,744 | − | 70 | 0 | ||
| D-loop | 15,745 | 16,712 | 968 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.-Y.; Cui, X.-F.; Chen, S.-H.; Li, Y.; Zhang, S.-Y.; Yang, Y.-F.; Li, Y.-Y.; Guo, Y.-S.; Wang, Z.-D.; Liao, J. The First Complete Mitogenome Characterization and Phylogenetic Implications of Elops machnata (Teleostei: Elopiformes: Elopidae). Biology 2025, 14, 739. https://doi.org/10.3390/biology14070739
Li J-Y, Cui X-F, Chen S-H, Li Y, Zhang S-Y, Yang Y-F, Li Y-Y, Guo Y-S, Wang Z-D, Liao J. The First Complete Mitogenome Characterization and Phylogenetic Implications of Elops machnata (Teleostei: Elopiformes: Elopidae). Biology. 2025; 14(7):739. https://doi.org/10.3390/biology14070739
Chicago/Turabian StyleLi, Jia-Yu, Xue-Fan Cui, Shu-He Chen, Yang Li, Shui-Yuan Zhang, Yuan-Feng Yang, Yi-Yang Li, Yu-Song Guo, Zhong-Duo Wang, and Jian Liao. 2025. "The First Complete Mitogenome Characterization and Phylogenetic Implications of Elops machnata (Teleostei: Elopiformes: Elopidae)" Biology 14, no. 7: 739. https://doi.org/10.3390/biology14070739
APA StyleLi, J.-Y., Cui, X.-F., Chen, S.-H., Li, Y., Zhang, S.-Y., Yang, Y.-F., Li, Y.-Y., Guo, Y.-S., Wang, Z.-D., & Liao, J. (2025). The First Complete Mitogenome Characterization and Phylogenetic Implications of Elops machnata (Teleostei: Elopiformes: Elopidae). Biology, 14(7), 739. https://doi.org/10.3390/biology14070739