
Integrative Single-Cell Transcriptomics and Network Modeling Reveal Modular Regulators of Sheep Zygotic Genome Activation

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Culture Media
2.2. Animals and Sample Collection
2.2.1. In Vivo Embryo Production
2.2.2. In Vitro Embryo Production
- Oocyte Collection and In Vitro Maturation (IVM)
- In Vitro Fertilization (IVF)
- In Vitro Culture (IVC)
2.3. RNA Extraction and High-Throughput Sequencing
2.4. RNA Data Analysis
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.6. Louvain Community Detection
2.7. Functional Annotation and Pathway Enrichment Analysis
3. Results
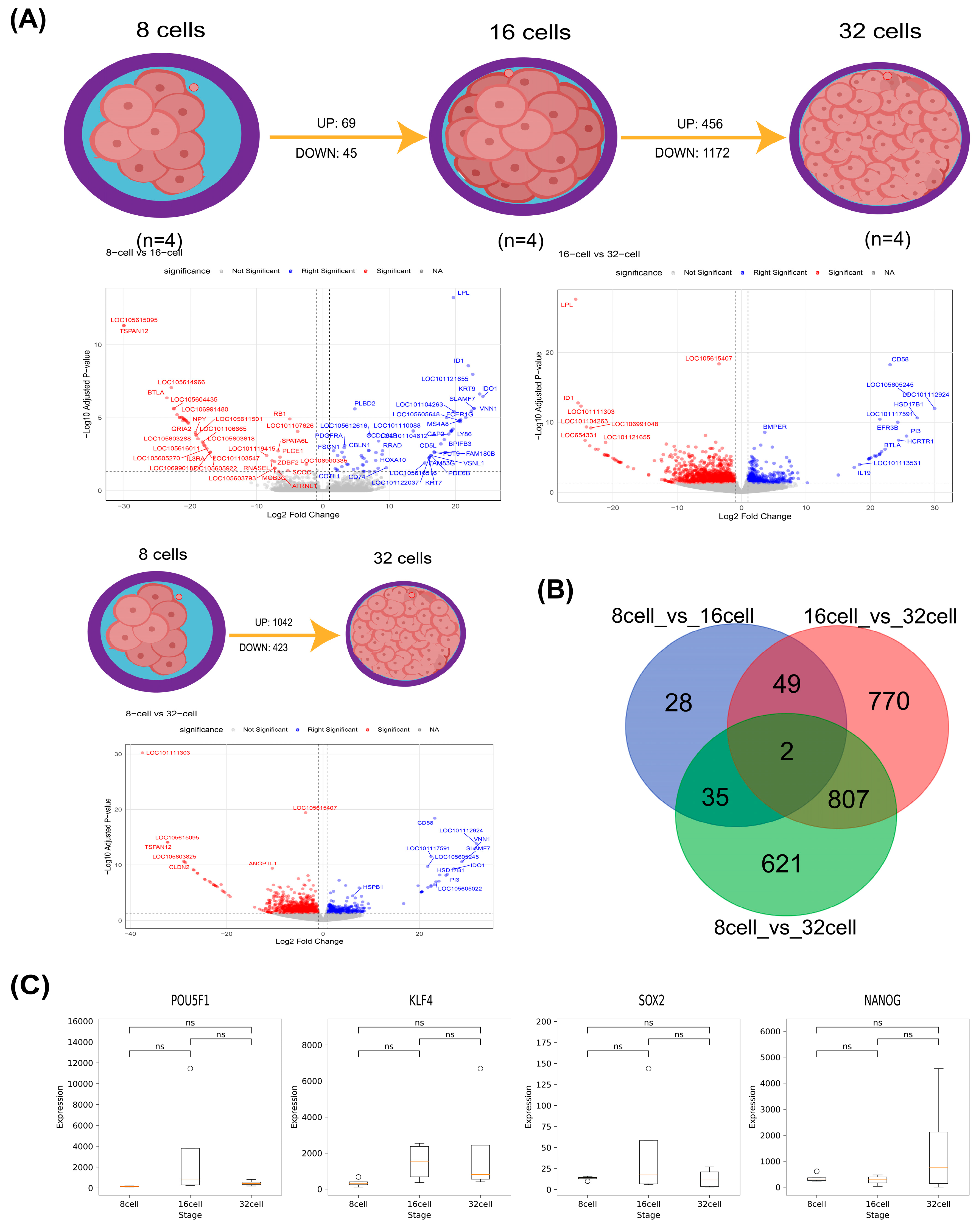
3.1. Summary of RNA-Seq Data
3.2. WGCNA Analysis Result
3.3. Louvain Community Detection Result
3.4. Results of Functional Annotation and Pathway Enrichment Analysis
4. Discussion
4.1. Two-Stage Regulatory Mechanisms of Sheep ZGA
4.2. Core Pluripotency Transcription Factors in Sheep Zygotic Genome Activation
4.3. Modular Logic of PPI–Louvain Integration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zou, Z.; Wang, Q.; Wu, X.; Schultz, R.M.; Xie, W. Kick-starting the zygotic genome: Licensors, specifiers, and beyond. EMBO Rep. 2024, 25, 4113–4130. [Google Scholar] [CrossRef] [PubMed]
- Phelps, W.A.; Hurton, M.D.; Ayers, T.N.; Carlson, A.E.; Rosenbaum, J.C.; Lee, M.T. Hybridization led to a rewired pluripotency network in the allotetraploid Xenopus laevis. eLife 2023, 12, e83952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, M.; Li, Y.; Zhang, Y. Profiling and functional characterization of maternal mRNA translation during mouse maternal-to-zygotic transition. Sci. Adv. 2022, 8, eabj3967. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Bonneau, A.R.; Giraldez, A.J. Zygotic genome activation during the maternal-to-zygotic transition. Annu. Rev. Cell Dev. Biol. 2014, 30, 581–613. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, W.; Zhu, J.; Kou, X.; Zhao, Y.; Wang, H.; Jiang, C.; Gao, S.; Kang, L. Pivotal role for long noncoding RNAs in zygotic genome activation in mice. Sci. China Life Sci. 2024, 67, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Yu, L.; Xu, Q.; Qiu, P.; Zhang, Y.; Dong, X.; Yan, G.; Sun, H.; Cao, G. GATAD2B is required for pre-implantation embryonic development by regulating zygotic genome activation. Cell Prolif. 2024, 57, e13647. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.H.; Zhan, C.L.; Li, X.H.; Lee, S.H.; Cui, X.S. Transcriptome analysis of the effects of high temperature on zygotic genome activation in porcine embryos. Sci. Rep. 2024, 14, 21849. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, Q.; Wang, Q.; Feng, S.; Lai, F.; Wang, P.; Zheng, F.; Xiang, Y.; Wu, J.; Nie, J.; et al. The landscape of RNA Pol II binding reveals a stepwise transition during ZGA. Nature 2020, 587, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.R.; Guo, S.M.; Shi, X.Y.; Ding, Y.W.; Li, H.B.; Li, L.; Xu, J.W.; He, X.; Ma, B.X.; Yin, Y.; et al. Deciphering transcription activity of mammalian early embryos unveils on/off of zygotic genome activation by protein translation/degradation. Cell Rep. 2025, 44, 115215. [Google Scholar] [CrossRef]
- Gentsch, G.E.; Owens, N.D.L.; Smith, J.C. The Spatiotemporal Control of Zygotic Genome Activation. iScience 2019, 16, 485–498. [Google Scholar] [CrossRef]
- Xu, T.; Liu, C.; Zhang, M.; Wang, X.; Yan, Y.; Liu, Q.; Ma, Y.; Yu, T.; Sathanawongs, A.; Jiao, J.; et al. Vitrification of Pronuclear Zygotes Perturbs Porcine Zygotic Genome Activation. Animals 2022, 12, 610. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Tiwari, M.; Goel, P.; Singh, M.K.; Selokar, N.L.; Palta, P. Comparative transcriptome profile of embryos at different developmental stages derived from somatic cell nuclear transfer (SCNT) and in-vitro fertilization (IVF) in riverine buffalo (Bubalus bubalis). Vet. Res. Commun. 2024, 48, 2457–2475. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, H.; Wang, L.; Chen, Y.; Yang, Y.; Chen, H.; Wang, F.; Zhang, Y.; Deng, M. H3K4me3 Genome-Wide Distribution and Transcriptional Regulation of Transposable Elements by RNA Pol2 Deposition. Int. J. Mol. Sci. 2024, 25, 13545. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Dai, L.; Xie, Y.; Zhang, W.; Zhong, X.; Wang, M.; Jiang, H.; He, Z.; Liu, X.; Zeng, H.; Wang, H. Weighted Gene Co-Expression Network Analysis Identifies ANGPTL4 as a Key Regulator in Diabetic Cardiomyopathy via FAK/SIRT3/ROS Pathway in Cardiomyocyte. Front. Endocrinol. 2021, 12, 705154. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Sanjeev, B.S. Over-representation analysis of angiogenic factors in immunosuppressive mechanisms in neoplasms and neurological conditions during COVID-19. Microb. Pathog. 2023, 185, 106386. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Hsaio, S.; Saglam, N.; Morrow, D.; Shain, D.H. Transcriptomic Profiling at the Maternal-to-Zygotic Transition in Leech, Helobdella austinensis. Genes 2024, 15, 283. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.; Gujar, G.; Shashank, C.G.; Sriranga, K.R.; Singh, R.J.; Singh, N. Omics strategies for unveiling male fertility-related biomarkers in livestock: A review. Gene Rep. 2024, 35, 101928. [Google Scholar] [CrossRef]
- Lee, M.T.; Bonneau, A.R.; Takacs, C.M.; Bazzini, A.A.; DiVito, K.R.; Fleming, E.S.; Giraldez, A.J. Nanog, Pou5f1 and SoxB1 activate zygotic gene expression during the maternal-to-zygotic transition. Nature 2013, 503, 360–364. [Google Scholar] [CrossRef]
- Navarro, P.; Festuccia, N.; Colby, D.; Gagliardi, A.; Mullin, N.P.; Zhang, W.; Karwacki-Neisius, V.; Osorno, R.; Kelly, D.; Robertson, M.; et al. OCT4/SOX2-independent Nanog autorepression modulates heterogeneous Nanog gene expression in mouse ES cells. EMBO J. 2012, 31, 4547–4562. [Google Scholar] [CrossRef]
- Shukla, M.; Duangrat, R.; Nopparat, C.; Sotthibundhu, A.; Govitrapong, P. Melatonin Augments the Expression of Core Transcription Factors in Aged and Alzheimer’s Patient Skin Fibroblasts. Biology 2024, 13, 698. [Google Scholar] [CrossRef]
- Meng, X.Y.; Sun, H.F.; Zhang, Y.J.; Li, W.X.; Wang, D.D.; Liu, D.H.; Li, S.M.; Wu, Y.; Li, J.K.; Liu, X.H.; et al. Generation of an induced pluripotent stem cell line (CSBZZUi001-A) from a female Alzheimer’s patient carrying the PSEN1 709 T > C heterozygous mutation. Stem Cell Res. 2024, 79, 103486. [Google Scholar] [CrossRef]
- He, X.D.; Phillips, S.; Hioki, K.; Majhi, P.D.; Babbitt, C.; Tremblay, K.D.; Pobezinsky, L.A.; Mager, J. TATA-binding associated factors have distinct roles during early mammalian development. Dev. Biol. 2024, 511, 53–62. [Google Scholar] [CrossRef]
- Tiwari, M.; Rawat, N.; Sharma, A.; Bhardwaj, P.; Roshan, M.; Nagoorvali, D.; Singh, M.K.; Chauhan, M.S. Methylation status of imprinted gene IGF2/ H19 DMR3 region in Goat (Capra hircus) blastocysts produced through parthenogenesis and in vitro fertilization. Small Rumin. Res. 2022, 216, 106796. [Google Scholar] [CrossRef]
- Tiwari, M.; Gujar, G.; Shashank, C.G.; Ponsuksili, S. Selection signatures for high altitude adaptation in livestock: A review. Gene 2024, 927, 148757. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Blondel, V.; Guillaume, J.L.; Lambiotte, R. Fast unfolding of communities in large networks: 15 years later. J. Stat. Mech-Theory E 2024, 2024, 10R001. [Google Scholar] [CrossRef]
- Sarkar, S.; Dong, A. Community detection in graphs using singular value decomposition. Phys. Rev. E 2011, 83, 046114. [Google Scholar] [CrossRef]
- Lu, W.; Li, Y.; Dai, Y.; Chen, K. Dominant Myocardial Fibrosis and Complex Immune Microenvironment Jointly Shape the Pathogenesis of Arrhythmogenic Right Ventricular Cardiomyopathy. Front. Cardiovasc. Med. 2022, 9, 900810. [Google Scholar] [CrossRef] [PubMed]
- Hasankhani, A.; Bakherad, M.; Bahrami, A.; Shahrbabak, H.M.; Pecho, R.D.C.; Shahrbabak, M.M. Integrated analysis of inflammatory mRNAs, miRNAs, and lncRNAs elucidates the molecular interactome behind bovine mastitis. Sci. Rep. 2023, 13, 13826. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, C.; Peng, J.; Tang, C.; Zhang, C.; Zhang, M.; Zou, X.; Zou, Y. Gender-specific lncRNA-miRNA-mRNA regulatory network to reveal potential genes for primary open-angle glaucoma. Exp. Eye Res. 2023, 236, 109668. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Pu, X.; Zhang, Y.; Dai, E. Renal tubular gen e biomarkers identification based on immune infiltrates in focal segmental glomerulosclerosis. Ren. Fail. 2022, 44, 966–986. [Google Scholar] [CrossRef]
- Muller, S. Update: On selected ROP cell polarity mechanisms in plant cell morphogenesis. Plant Physiol. 2023, 193, 26–41. [Google Scholar] [CrossRef]
- Pal, M.; Altamirano-Pacheco, L.; Schauer, T.; Torres-Padilla, M.E. Reorganization of lamina-associated domains in early mouse embryos is regulated by RNA polymerase II activity. Genes. Dev. 2023, 37, 901–912. [Google Scholar] [CrossRef]
- Su, Z.; Dong, Y.; Sun, J.; Wu, Y.; Wei, Q.; Liang, Y.; Lin, Z.; Li, Y.; Shen, L.; Xi, C.; et al. RNA m(6)A modification regulates cell fate transition between pluripotent stem cells and 2 cell-like cells. Cell Prolif. 2024, 57, e13696. [Google Scholar] [CrossRef] [PubMed]
- Pines, A.R.; Larsen, B.; Cui, Z.; Sydnor, V.J.; Bertolero, M.A.; Adebimpe, A.; Alexander-Bloch, A.F.; Davatzikos, C.; Fair, D.A.; Gur, R.C.; et al. Dissociable multi-scale patterns of development in personalized brain networks. Nat. Commun. 2022, 13, 2647. [Google Scholar] [CrossRef] [PubMed]
- Duran-Nebreda, S.; Jackson, M.D.B.; Bassel, G.W. A quantitative morphospace of multicellular organ design in the plant Arabidopsis. Curr. Biol. 2023, 33, 4798–4806.e4793. [Google Scholar] [CrossRef] [PubMed]
- Di Gaetano, L.; Carugno, G.; Battiston, F.; Coghi, F. Dynamical Fluctuations of Random Walks in Higher-Order Networks. Phys. Rev. Lett. 2024, 133, 107401. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Niu, P.; Hu, K.; Wang, X.; Huang, F.; Song, P.; Gao, Q.; Fang, D. Integrative Single-Cell Transcriptomics and Network Modeling Reveal Modular Regulators of Sheep Zygotic Genome Activation. Biology 2025, 14, 676. https://doi.org/10.3390/biology14060676
Li X, Niu P, Hu K, Wang X, Huang F, Song P, Gao Q, Fang D. Integrative Single-Cell Transcriptomics and Network Modeling Reveal Modular Regulators of Sheep Zygotic Genome Activation. Biology. 2025; 14(6):676. https://doi.org/10.3390/biology14060676
Chicago/Turabian StyleLi, Xiaopeng, Peng Niu, Kai Hu, Xueyan Wang, Fei Huang, Pengyan Song, Qinghua Gao, and Di Fang. 2025. "Integrative Single-Cell Transcriptomics and Network Modeling Reveal Modular Regulators of Sheep Zygotic Genome Activation" Biology 14, no. 6: 676. https://doi.org/10.3390/biology14060676
APA StyleLi, X., Niu, P., Hu, K., Wang, X., Huang, F., Song, P., Gao, Q., & Fang, D. (2025). Integrative Single-Cell Transcriptomics and Network Modeling Reveal Modular Regulators of Sheep Zygotic Genome Activation. Biology, 14(6), 676. https://doi.org/10.3390/biology14060676