1. Introduction
Cancer is one of the diseases of modern times that is difficult to treat [
1]. Lifestyle, genetics, and environmental factors led to projections from demographics in the year 2022 that by 2030, cancer cases are expected to reach 35 million, with no signs of stopping [
2]. Chemotherapy is the most commonly used method to treat cancer, involving chemicals such as vincristine, vinblastine, and gemcitabine to induce apoptosis through cellular stress and suppress cell defense [
3]. However, problems such as fatigue, hair loss, nausea, vomiting, infertility, diarrhea, heart conditions, as well as memory and cognitive issues are just a few of the side effects commonly experienced by patients undergoing treatment [
4]. According to the log-kill hypothesis, chemotherapy drugs eliminate only a fixed percentage of cancer cells. This implies that cancer cells may develop chemoresistance and even evolve to advanced stages [
5]. This is part of the bigger issue of chemoresistance in cancer cells wherein cancer cells gain resistance to chemotherapeutic drugs. There are many proteins involved in chemoresistance in cancers—one protein of notable interest is clusterin (CLU), also known as lipoprotein J, a protein strongly associated with chemoresistance and considered one of the potential targets for treatments across various cancer cell lines [
6]. In normal functioning cells, it is responsible for neutralizing misfolded proteins, acting as a chaperone molecule. It is also responsible for numerous physio-pathological processes, including lipid transport, cell adhesion and aggregation, complement inhibition, sperm maturation, as well as anti-apoptosis [
7]. There are two forms of CLU, both of which are responsible for seemingly opposite functions. The nuclear form (nCLU), a 49 kDa protein, has been found to participate in pro-apoptotic pathways [
8]. On the other hand, the secreted form of CLU (sCLU), approximately 80 kDa, have been discovered to be involved in anti-apoptotic activities [
9]. In this context, sCLU has been identified as one of the key factors responsible for the chemoresistance of various cancer cell lines. sCLU was found to be abnormally represented in various cancer cell lines, including skin, pancreatic, breast, colon, esophageal squamous, lung, and neuroblastoma [
10]. It was also found that the interplay between the expressions and functions of both forms of CLU changes depending on cancer progression [
11]. Additionally, clusterin is also involved in the progression and action of other conditions, particularly in neurological diseases, such as Alzheimer’s disease [
12] and Huntington’s disease [
13], among other conditions. The most common form of treatment for abnormal CLU expression involves the use of the drug Custirsen, also known as OGX-011 (where OGX is the acronym for the company OncoGenex Technologies and 011 is the compound’s code). This drug is a second-generation antisense oligonucleotide that inhibits CLU expression and is currently undergoing phase II clinical trials, as it has been found to resensitize cancer cells to chemotherapeutic agents [
14]. Despite CLU having an established treatment through OGX-011, it still has its drawbacks, namely, restricted drug delivery options, a lack of understanding of the required level of regulation adjustment to sufficiently affect the expression back to normal levels, genetic alterations leading to loss of adhesion to the target strand, and delayed onset of treatment efficacy, among other limitations [
15]. Such additional treatments for dealing with elevated CLU expression levels would prove very helpful. In this study, we present potential drug candidates targeting CLU using fragment-based drug discovery (FBDD) as potential effectors for resensitizing cancer cells to chemical treatments. Other sites of interest were also determined in addition to the main binding site through hotspot searching. Qualitative structure-activity relationship (QSAR) analysis was also conducted to determine the structural properties relevant to designing effective drugs that target sCLU. Finally, molecular dynamics was investigated to determine the accurate binding energies as well as the structural stability of the complex, ligand, and protein.
4. Discussion
While clusterin has been identified for some time, its specific characteristics and functions are still being investigated. This includes the specific sections of the protein relevant to its interactions with ligands and residues with other proteins with respect to its main function as a chaperone protein [
59]. With the aid of a fragment-based methods approach, it is possible to elucidate the dynamic functions of clusterin along with potential drugs.
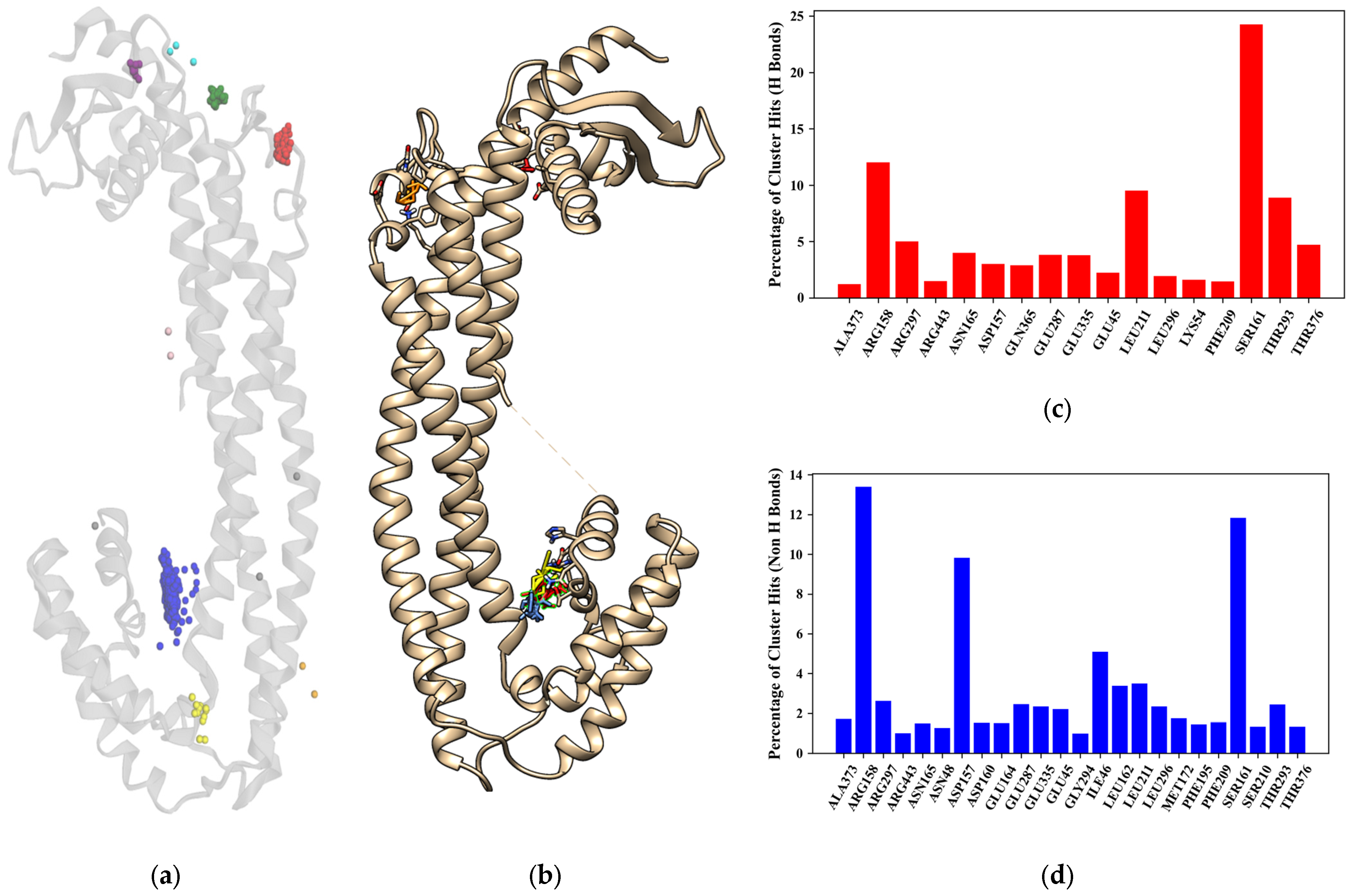
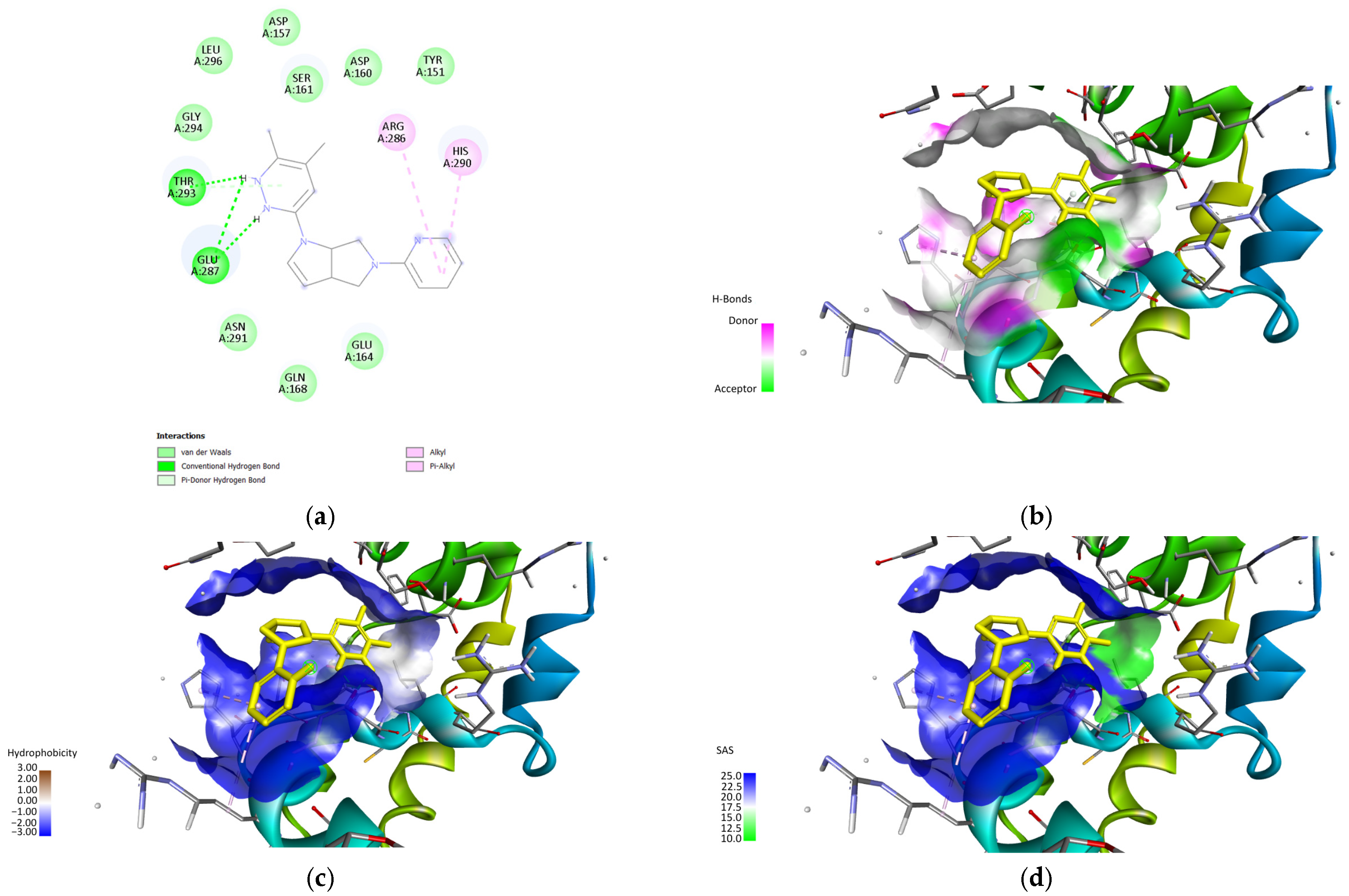
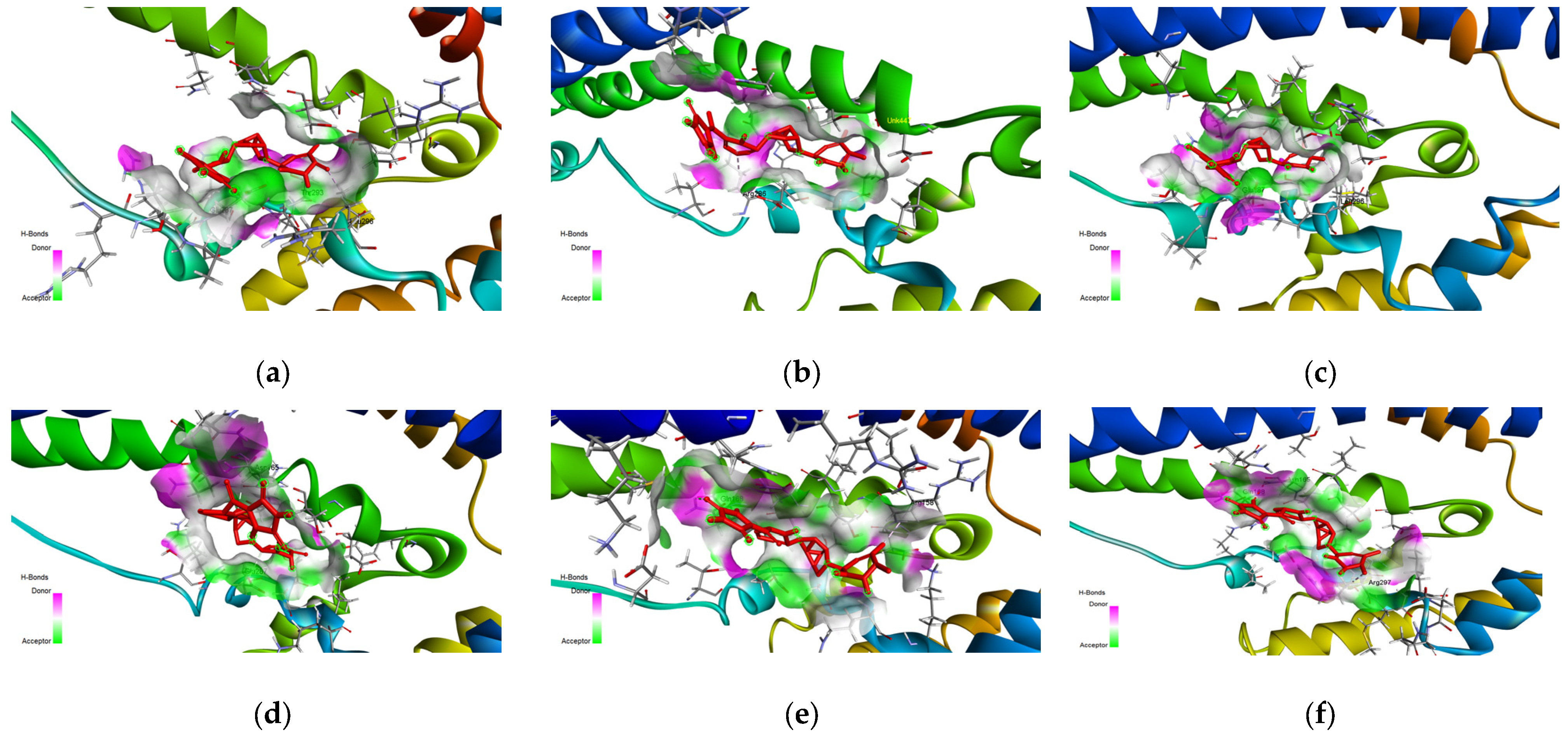
The main binding site as described in
Figure 3 is composed of the amino acids ARG158, ASP157, SER161, and THR293 and is of particular interest as the main ligand binding site as it can be treated as a helix-helix interface forming a pocket with an angle between the three main helix bundles and the bottom bundle. This formation has been cited in the literature as a motif with a high capability to host protein-ligand interactions, as shown in the leucine zipper nomenclature [
60].
Past studies on the dynamics of the structure indicate that this specific conformation is highly flexible, potentially accommodating a wide variety of ligands through allosteric or cooperative behavior [
61]. This is shown through docking studies conducted as well as the literature on chaperone proteins. Another aspect to take note of is the potential allosteric sites found using FTMap, as well as in the actual docking predictions.
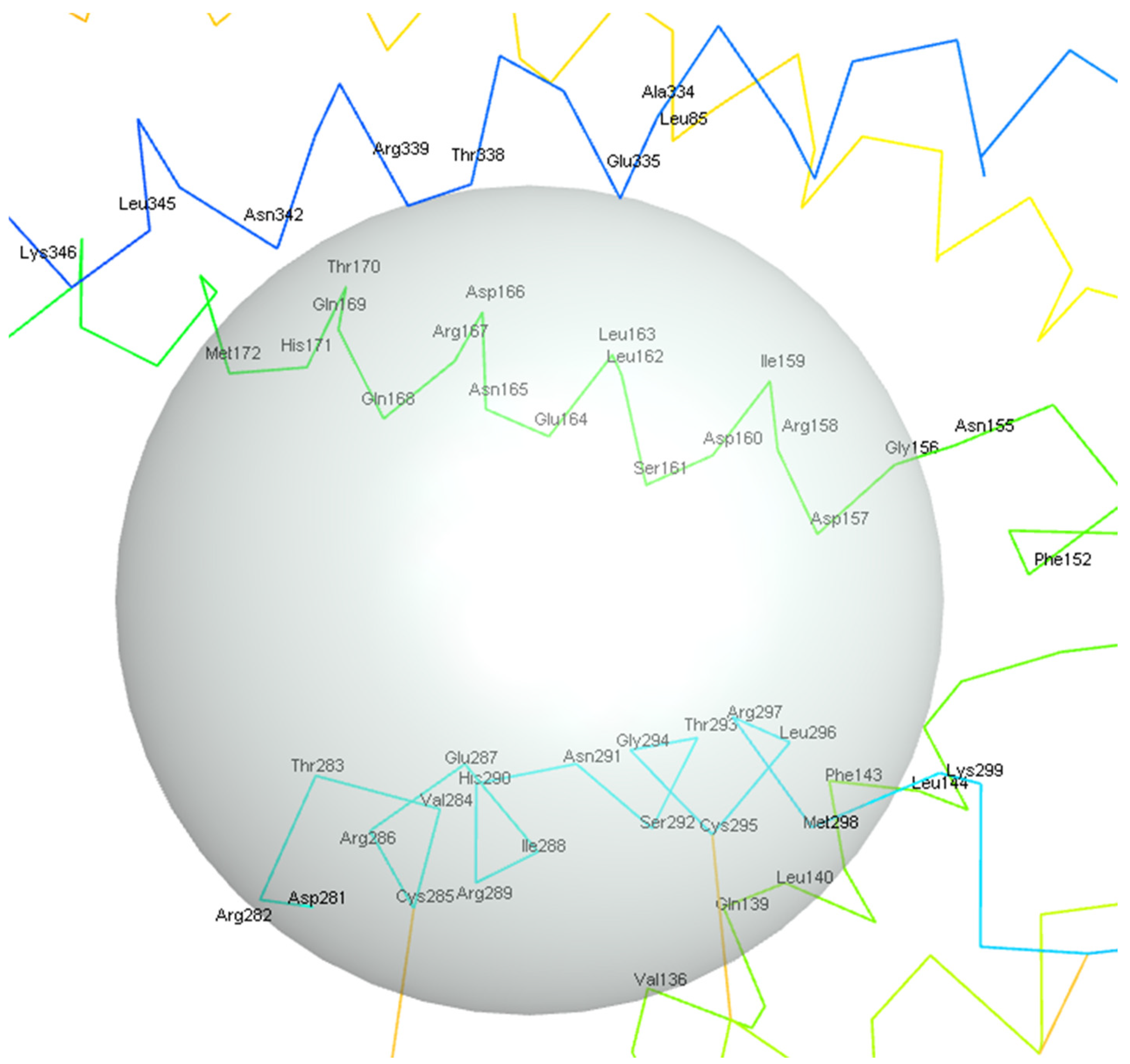
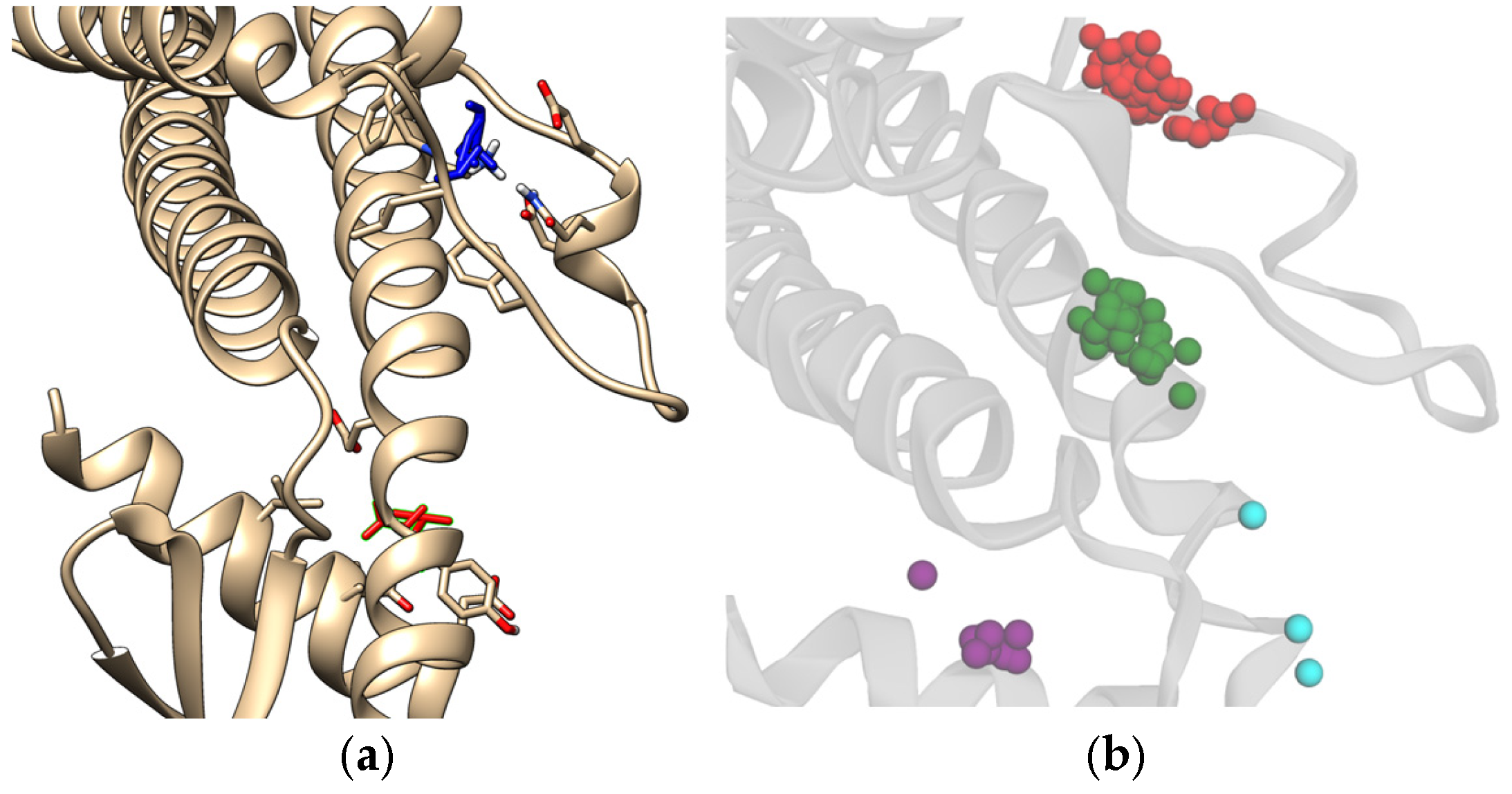
Figure 13 shows the potential allosteric binding sites, and based on
Table 2, the residues covered by the clusters are LEU211, ALA373, THR376, SER210, PRO212, PHE195, ARG194, GLN201, GLU199, LEU375, and GLY378 for the red, green, and purple clusters from the docking results. The residue interactions from FTMap also indicate LEU211, ALA373, THR376, SER210, and PHE195 residues as minor hotspots with respect to the main binding site previously described in this study. CLU has been established to display allosteric binding activity, namely, having three distinct classes of binding sites that cater to LRP-2, unstressed ligands, and stressed proteins [
46]. It is also established in the literature that the LRP-2 and stressed proteins are hypothesized to bind around the C-terminus of the alpha chain, which is also adjacent to the N-terminus of the beta chain, a region that would roughly coincide with the hotspots, specifically, near the green and purple clusters. This also suggests that the unstressed ligands do indeed bind to the specified binding site assigned for unstressed ligands. It should be noted that further analysis of these hotspots would be required to fully confirm the region in which the protein-protein interactions of sCLU take place.
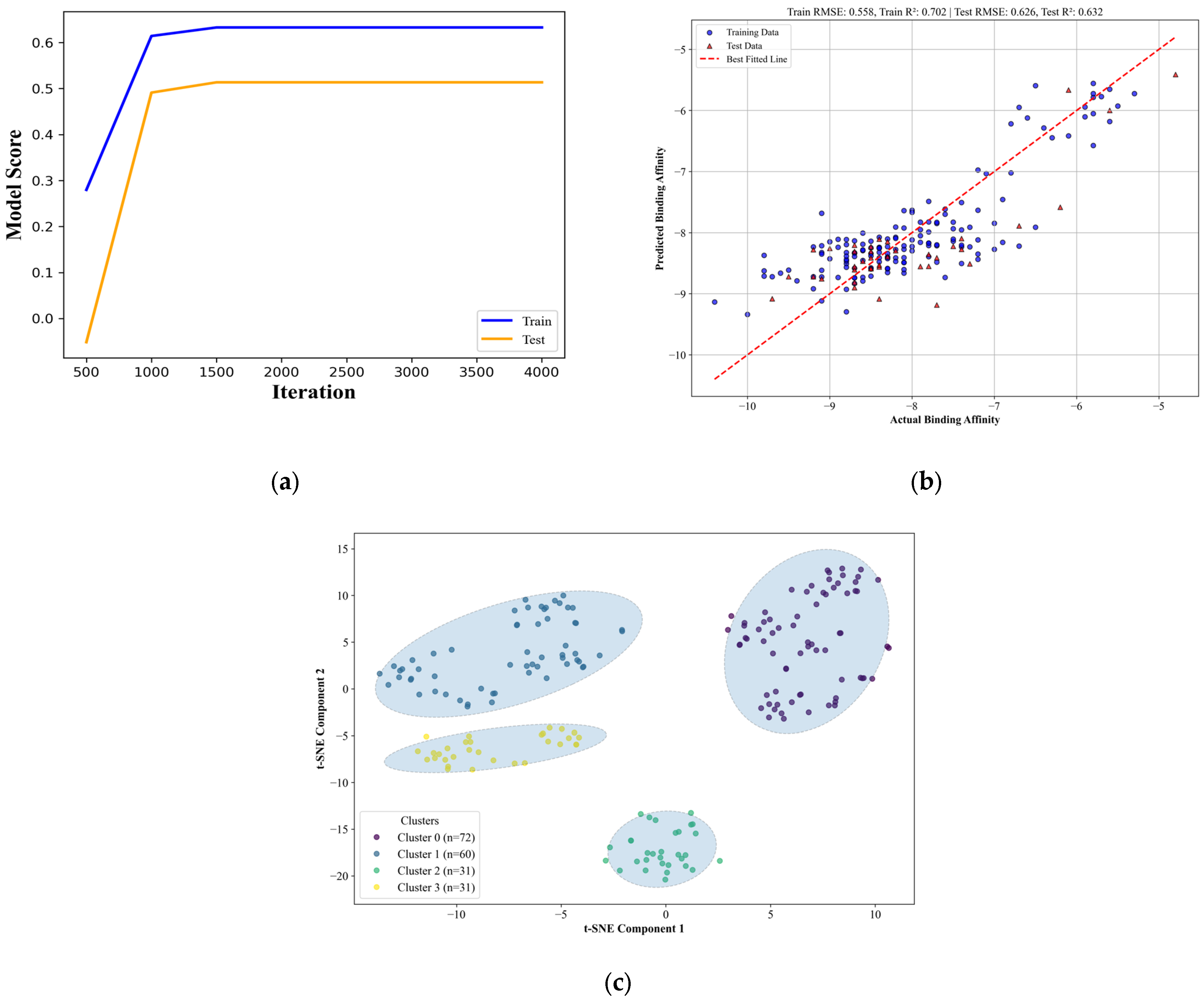
As a rule, chaperone proteins have a large range of targets they can bind to, much more so for chaperones which have complex roles in many signaling pathways, like sCLU, which is involved in pathways such as the NF-κB signaling pathway [
62], the PI3K/Akt pathway [
63], and the Reelin-signaling pathway [
64]. This can be seen in the results of the applicability domain of the QSAR model, where four distinct chemical spaces were found to be able to properly bind to the protein with a relatively good binding score. The chemical space covered by the candidate drugs showed what can be seen as an advantage and a disadvantage of the druggability of chaperone proteins [
65]. To address the concern of specificity, known anti-cancer drugs that have been approved by the FDA and EMA [
66] were docked for comparison; the results are available in
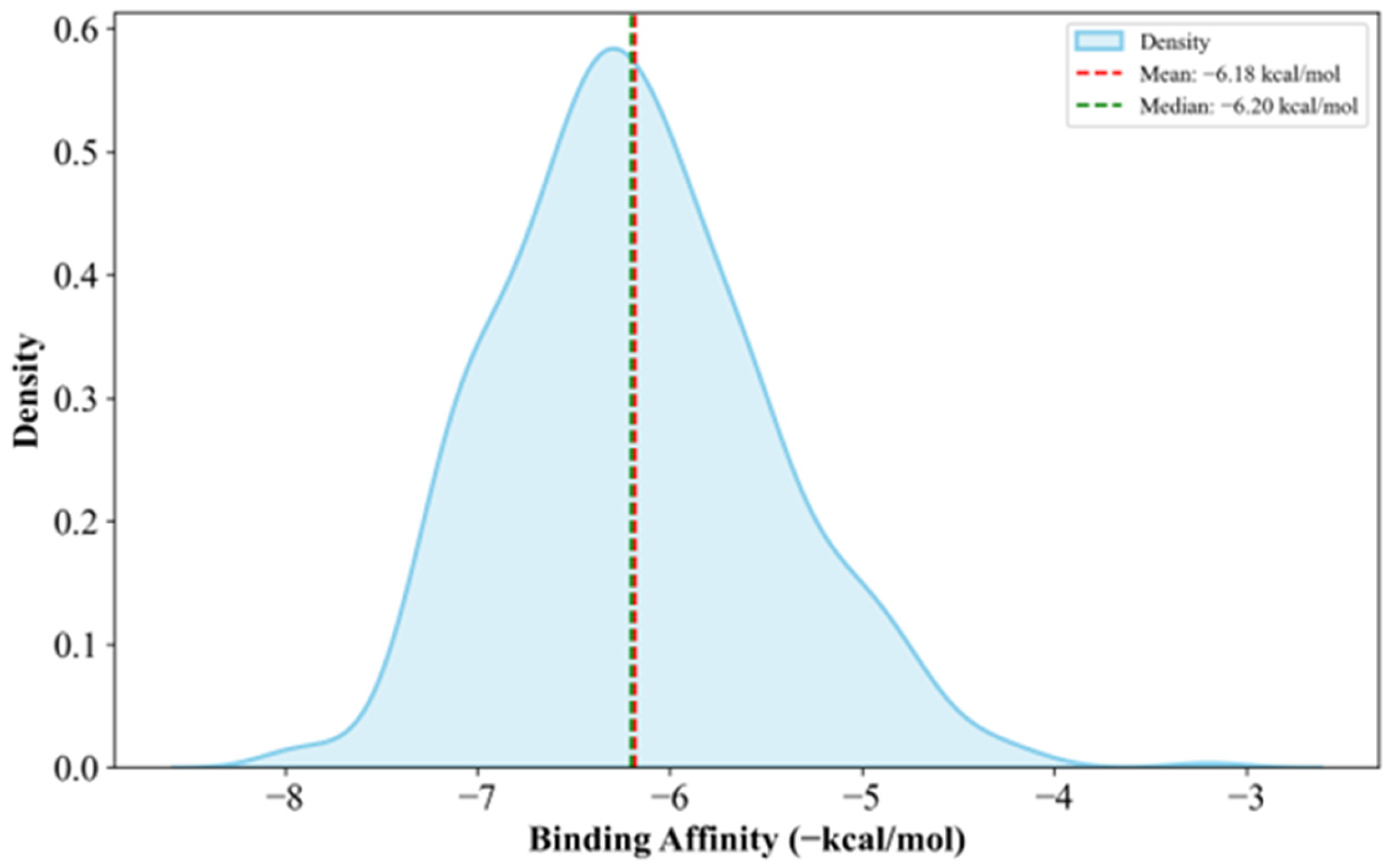
Supplementary Materials Table S16. Generally, the anti-cancer drugs show lower affinities to clusterin with a mean of −6.36 kcal/mol, which is 18.88% less potent than the candidate drugs, as shown in
Figure 7.
Another concern is the issue of the synthesizability of the resulting candidate drugs. A concern with candidate drugs produced for highly specific proteins is when they become so highly matched to the protein that it significantly reduces the synthesizability of the drug [
67]. Synthesizability analysis conducted using the DeepSA webserver (available from
https://bailab.siais.shanghaitech.edu.cn/services/deepsa/, accessed on 9 January 2024) [
68], shown in
Supplementary Materials Table S17, supports the idea that targeting less-specific proteins may potentially result in higher synthesizability results. The analysis showed that 148 compounds have higher easy-to-synthesize than hard-to-synthesize scores. This is also due to the algorithms used as the majority of the candidate drugs are from H-direct grow replacements and retrosynthesis-based replacements [
28]. This results in candidate drugs that can be easily synthesized, especially compared to compounds with alterations to the main substructures. On the other hand, the relatively low specificity of the ligands may present the possibility that the ligands may also bind to other proteins that are not the main target. This is not only potentially bad for the in vivo activity of the drug, as not only could it potentially disrupt the dosage required for appropriate inhibition of the receptor, but it may also have unintended toxic consequences on other parts of the cell [
69]. This can be seen with the toxicity results from T.E.S.T., as shown in
Table 4, whereby the candidate drugs produced a significant toxicity risk to certain benchmark strains—the nondescript nature of the compounds implicated them according to some structural qualities deemed toxic by the T.E.S.T. predictive models [
29].
Currently, fragment-based drug candidates remain amenable to further optimization. Candidate drugs may be functionally modified to become more specific and thus obtain higher binding affinities through further optimization of the fragment growing process as well as by implementing other fragment optimization strategies, such as fragment reduction, to remove any unwanted structures in terms of toxicity and binding efficiency, as shown by other tools, such as ACFIS 2.0 [
70] and FragPELE [
71].
When comparing the designed drugs to other established anti-cancer drugs, it was found that the candidate drugs have low similarities to other known anti-cancer drugs.
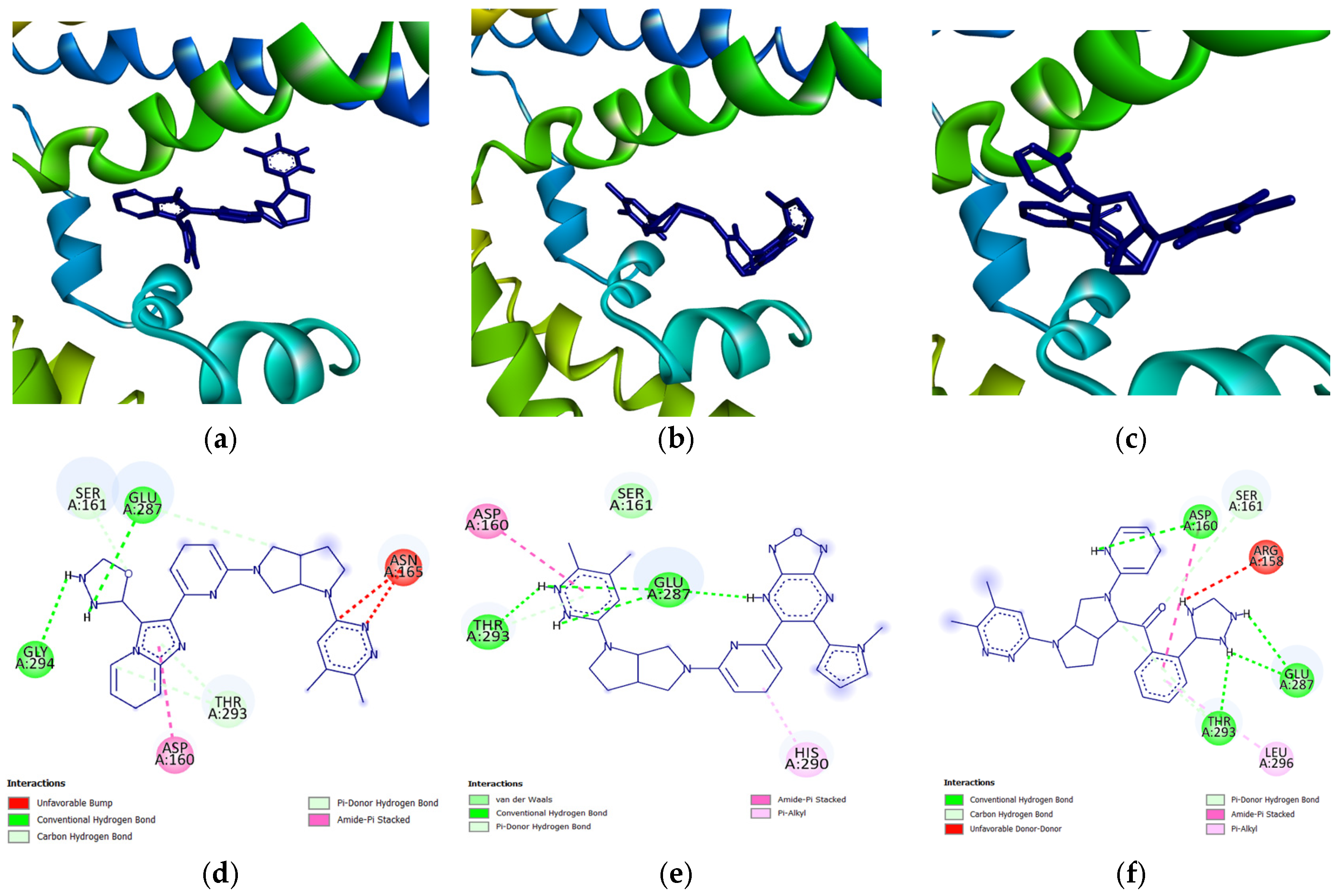
Figure 13 shows this (full results shown in
Supplementary Materials Table S18), with the highest Tanimoto similarity being only 0.2340, which indicates a generally low degree of overlap in the structures. This suggests that although the candidate drugs are meant for a chaperone protein which is inherently receptive to many types of ligands, it is still specific enough to possibly be able to target clusterin alone without interfering with other drug-protein interactions. Despite the weak overlapping of the molecular fingerprints, some structural features are still conserved among the candidate drugs and established anti-cancer drugs; this can be seen in
Supplementary Materials Figure S23.
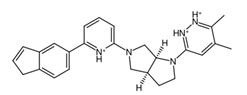
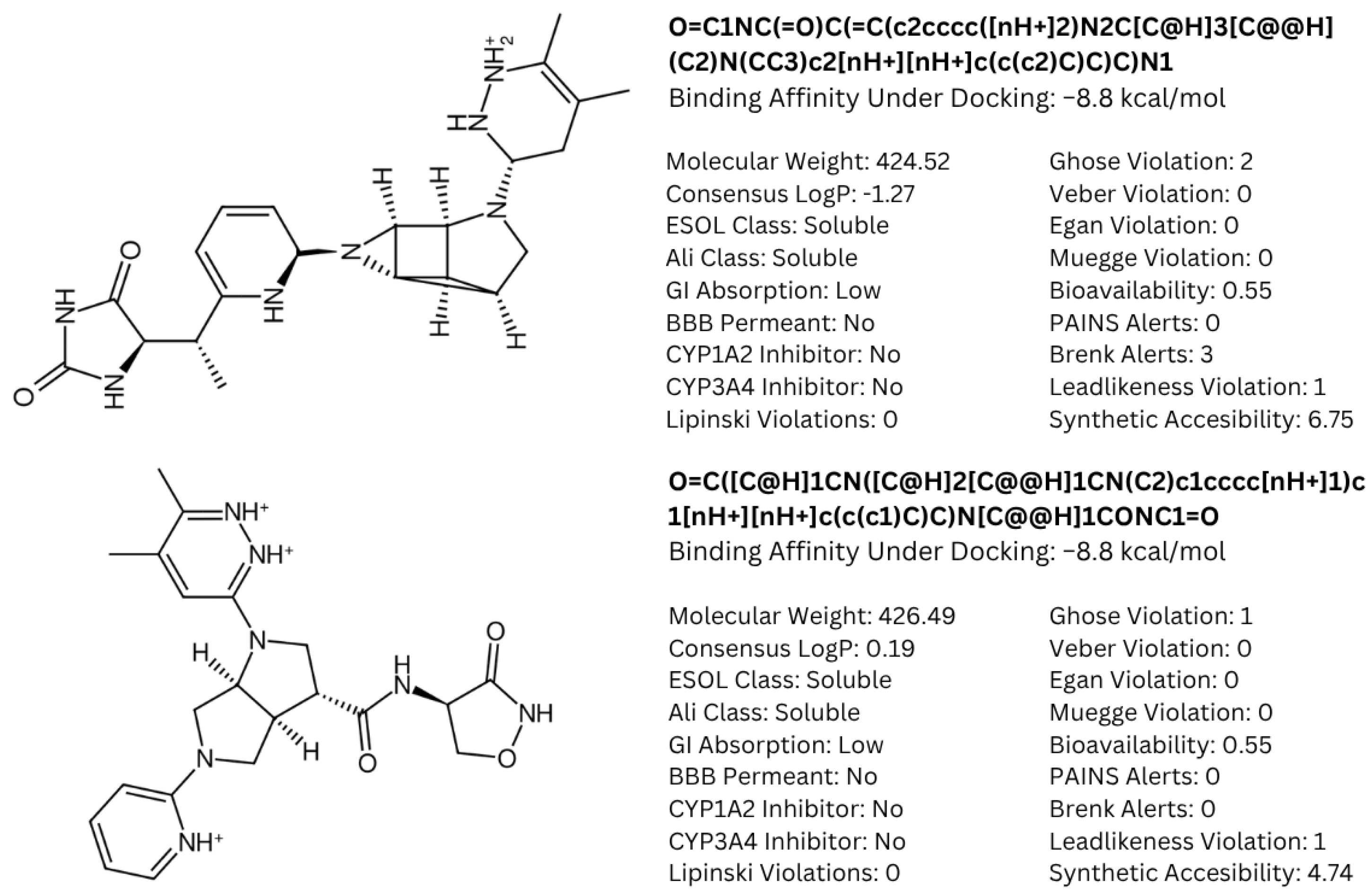
A common point of similarity across clusters 0, 1, and 3 is their structural overlaps with small molecule inhibitors. Such drugs are responsible for protein binding, targeting key signaling pathways, modulation of immune cell activity, and interrupting the tumor microenvironment. Among nitrogen-containing anti-cancer drugs, one of the most common structures is that of pyridine. It was found that this substructure is involved in drugs that function as anti-tumor and anti-proliferation agents, cell-cycle regulators, and cytotoxic agents through challenging binding with their target proteins [
72]. In addition, pyridazine is also implicated as a prominent contributor to the anti-cancer activity of certain drugs responsible for the inhibition of proteins involved in the onset and progression of cancer. Recent literature suggests that this substructure can act as either a core framework or as a warhead [
73]. Both substructures were found to be part of the main fragment backbone of the peripheral structures, as shown in
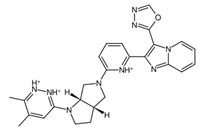
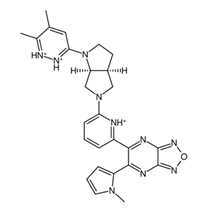
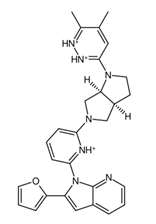
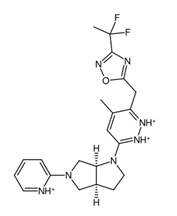
Figure 5a. In addition, the central structure is a fused pair of pyrrolidine rings. Although this substructure has been reported in some anti-cancer drugs and its derivatives can show anti-cancer activity, it is still undetermined if the pyrrolidine derivative found in the top 1 fragment, and consequently, the candidate drugs has any bearing on its potential anti-cancer activity. However, it should be noted that many anti-cancer drugs that feature pyrrolidine derivatives also prominently feature fused ring structures [
74]. This suggests that there is a good probability that the candidate drugs can perform the same anti-cancer activities; however, much experimental testing is required to substantiate this hypothesis. Additionally, it was also noted that the added structures from the fragment growing were primarily responsible for making the candidates fit better on the clusterin structure, as evidenced by their 3D conformations and prominent polar groups that can serve as hydrogen bond acceptors and donors [
75], as well as aromatic groups to fully utilize the aromatic interactions with the residues [
76]. These observations are supported by the results shown in
Figure 7.
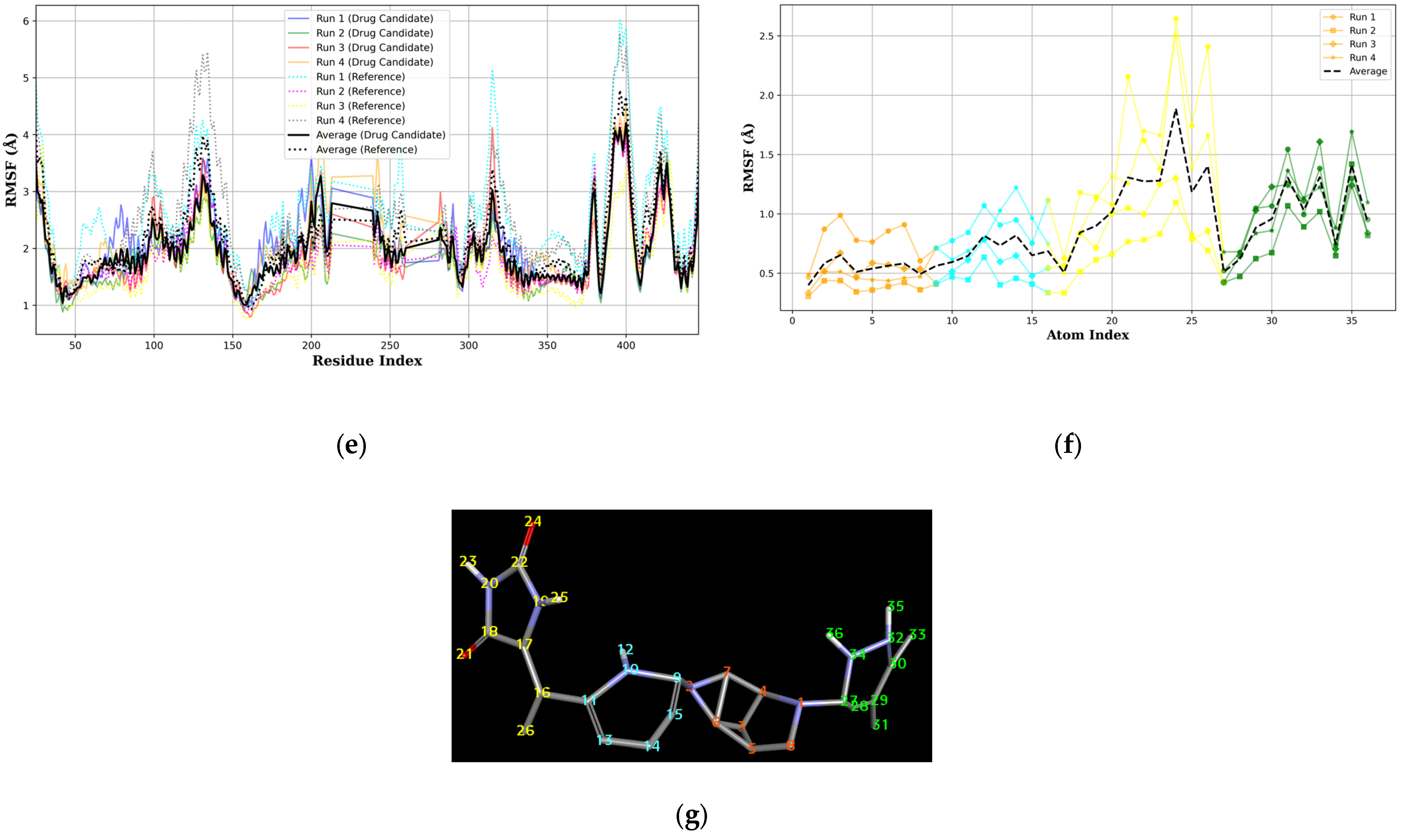
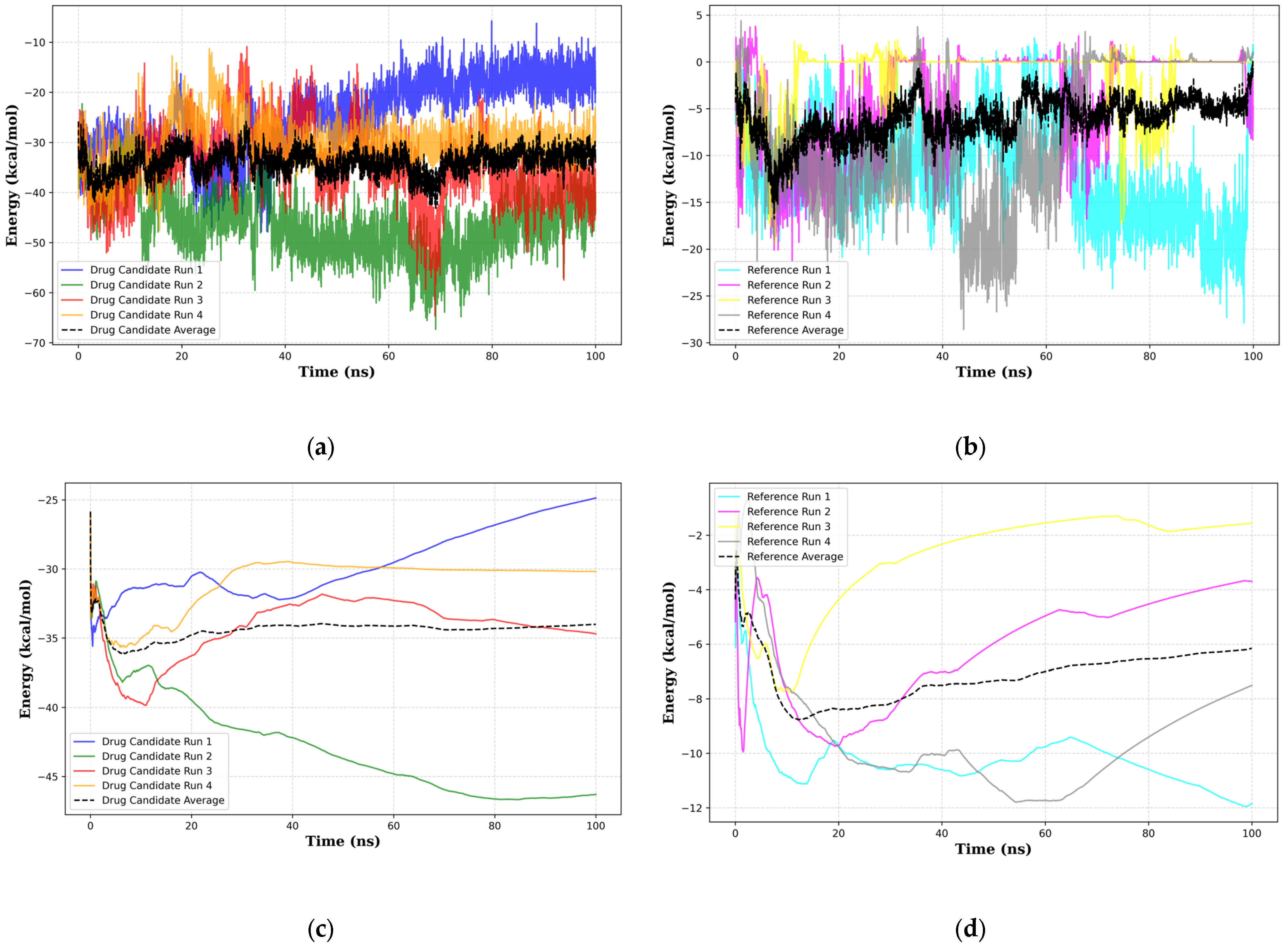
When considering the properties of the top 1 final candidate drug precursor, it is important to note that the molecular dynamics simulation showed the ligand to be highly flexible within the binding pocket. The fact that CLU is a chaperone protein designed to accommodate all sorts of ligands means that it is unlikely to have a very firm binding site for a very specific type of ligand [
46]. Although this runs counter to the principle of ligand-based design, flexible ligands do have potential uses that may prove useful to various treatment opportunities. This can be achieved through the functionalization of the peripheral sections, as shown in
Figure 10f,g, of the top 1 final candidate drug precursor with a chemical group with proven effects. One such application is serving as a drug delivery system [
77] for certain chemotherapy drugs such as gemcitabine, whilst simultaneously providing some degree of CLU suppression. Another potential route of application for flexible ligands is to serve as facilitators for certain protein-protein interactions, such as reported by Liu, B. et al. (2024), in which they suggested using small molecule ligands as potential facilitators for interactions with certain ubiquitin ligases, which directly degrade the protein [
78]. This strategy also applies to any protein-protein interactions, especially in the case of chaperone proteins such as CLU as previously highlighted in this section [
79]. Flexible ligands also have a role in multi-directed ligands whereby the non-specific structure may serve to benefit the drug due to being applicable to related proteins or even other proteins that have similar binding site characteristics [
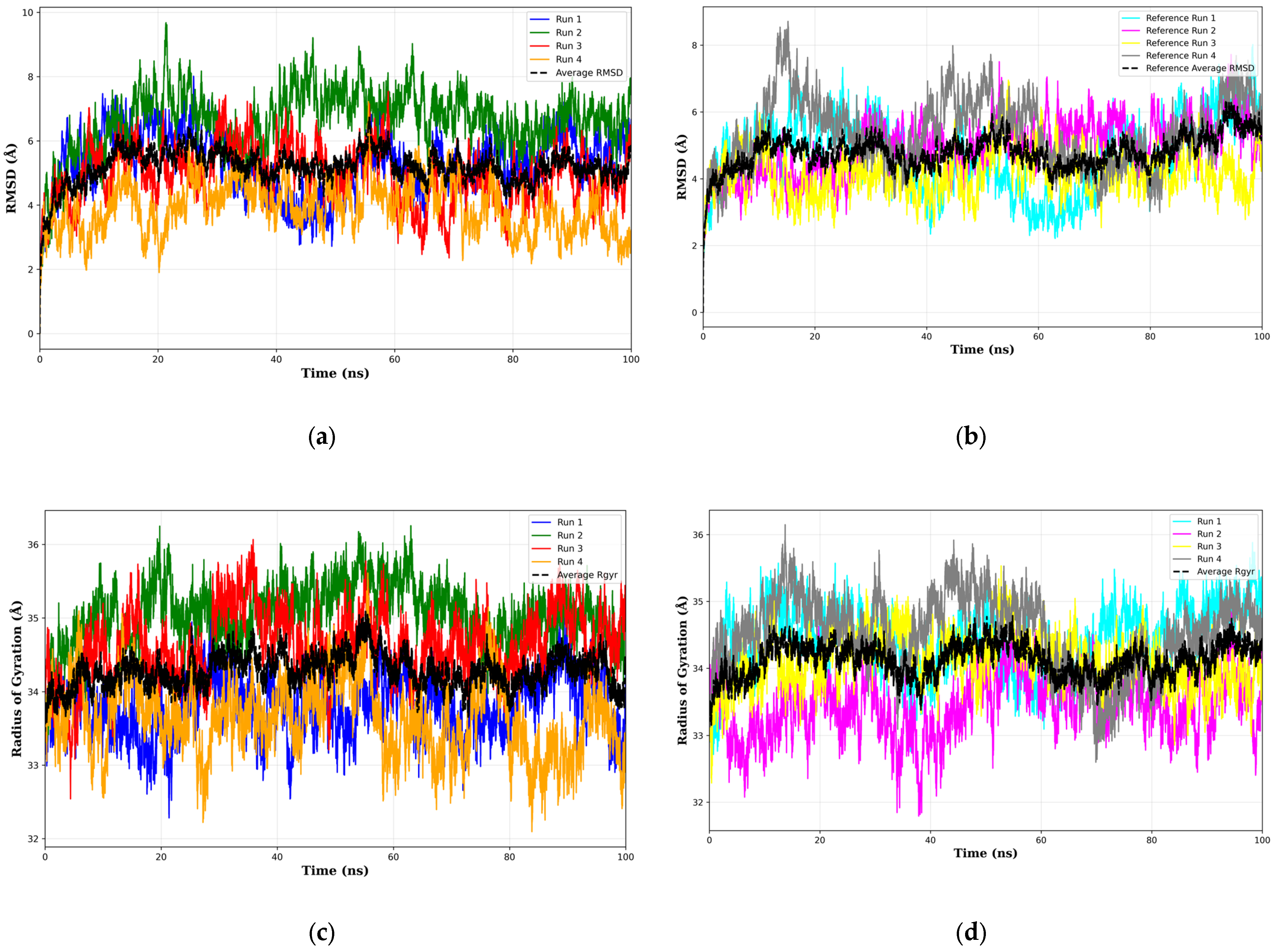
80]. Although the results of the molecular dynamics simulations show many potential scenarios of the bound top 1 final candidate drug precursor and the sCLU complex, it is apparent that there are still many conformational states that have not been covered. This can be seen in how Kruskal–Wallis [
81] (
Supplementary Materials Table S19) and Dunn’s Pairwise tests [
82] (
Supplementary Materials Table S20) show significant differences across all runs in RMSD, radius of gyration, and all energy terms from MMGBSA. We recommend more simulations in future work to properly ascertain the full range of behavior across many possible initial conditions. Another notable limitation is that MMGBSA does not account for explicit entropy and ligand dissolution. In this study, MMGBSA was primarily employed to compare the relative stability of the complexes, rather than for comprehensive free energy prediction. For future research, we recommend integrating wet-lab experiments to ascertain therapeutic effects, as these cannot be fully captured through in-silico predictions alone. The complete MMGBSA results are detailed in
Supplementary Materials Table S21.
Although FBDD also has its shortcomings in terms of chemical space coverage, being biased and limited in some cases, initial fragment binding causing relatively low binding scores, which potentially filter out better drugs once grown, the presence of potential false negatives or false positives among the candidate drugs, synthesizability, and specificity [
47], these results serve to illustrate the capability of the FBDD pipeline to produce biologically relevant results, with success stories in the form of vemurafenib [
83], erdafitinib [
84], pexidartinib [
85], and venetoclax [
86], as well as at least 40 different fragment-based candidate drugs for all types of conditions [
87]. It is clear that this pipeline is capable of developing potential drugs at a much lower cost. This study provides insight into the capabilities of FBDD by identifying and characterizing the main ligand binding sites as well as potential allosteric sites. In addition, the candidate drugs in this study may be used as the basis for potential drugs, probes for biomarkers, and even delivery systems for other types of treatment. This highlights the flexibility of the method in producing compounds with a wide array of potential functions depending on the circumstance.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}