

Identification of Genomic Variants and Candidate Genes for Reproductive Traits and Growth Traits in Pishan Red Sheep Using Whole-Genome Resequencing


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Sample Collection and Whole-Genome Resequencing
2.3. Quality Control and Read Mapping
2.4. Population Genetic Diversity Analysis
2.5. Analysis of Genome-Wide Selective Sweep Regions
2.6. GWAS
3. Results
3.1. Sequencing, Mapping, and SNP/InDel Variation Annotation
3.2. Population Genetic Structure and Linkage Disequilibrium
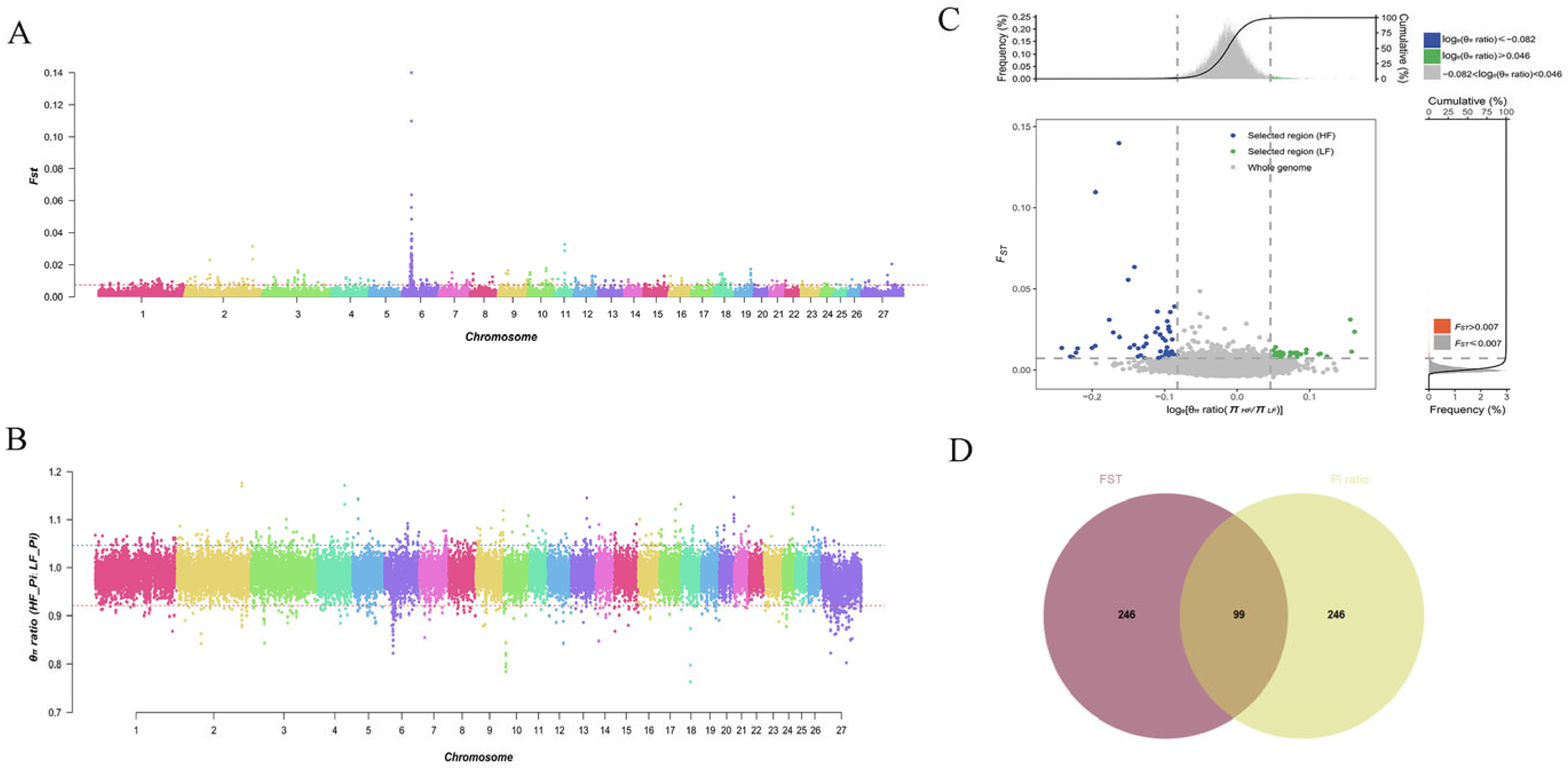
3.3. Selective Imprints of Reproductive Traits in Pishan Red Sheep
3.4. GWAS of Growth and Reproductive Traits in Pishan Red Sheep
3.5. Candidate Gene Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kong, L.; Yue, Y.; Li, J.; Yang, B.; Chen, B.; Liu, J.; Lu, Z. Transcriptomics and metabolomics reveal improved performance of Hu sheep on hybridization with Southdown sheep. Food Res. Int. 2023, 173, 113240. [Google Scholar] [CrossRef] [PubMed]
- Nel, C.; Gurman, P.; Swan, A.; Werf, J.; Snyman, M.; Dzama, K.; Olivier, W.; Scholtz, A.; Cloete, S. Including genomic information in the genetic evaluation of production and reproduction traits in South African Merino sheep. J. Anim. Breed. Genet. 2024, 141, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tang, J.; He, X.; Di, R.; Zhang, X.; Zhang, J.; Guo, X.; Hu, W.; Chu, M. Key mRNAs and lncRNAs of pituitary that affect the reproduction of FecB + + small tail han sheep. BMC Genom. 2024, 25, 392. [Google Scholar] [CrossRef]
- Zhang, C.L.; Zhang, J.; Tuersuntuoheti, M.; Zhou, W.; Han, Z.; Li, X.; Yang, R.; Zhang, L.; Zheng, L.; Liu, S. Landscape genomics reveals adaptive divergence of indigenous sheep in different ecological environments of Xinjiang, China. Sci. Total Environ. 2023, 15, 166698. [Google Scholar] [CrossRef] [PubMed]
- Lan, S.; Ma, M.; Muhetapa, M.; Yang, H.; Yiming, S. Study on the Genetic Structure and Litter Size Candidate Genes of Pishan Red Sheep Population Based on Whole Genome Resequencing. China Anim. Husb. Vet. Med. 2024, 51, 624–638. [Google Scholar]
- Xu, S.S.; Ren, X.; Yang, G.L.; Xie, X.L.; Zhao, Y.X.; Zhang, M.; Shen, Z.Q.; Ren, Y.L.; Gao, L.; Shen, M.; et al. Genome-wide association analysis identifies the genetic basis of fat deposition in the tails of sheep (Ovis aries). Anim. Genet. 2017, 48, 560–569. [Google Scholar] [CrossRef]
- Lv, F.H.; Cao, Y.H.; Liu, G.J.; Luo, L.Y.; Lu, R.; Liu, M.J.; Li, W.R.; Zhou, P.; Wang, X.H.; Shen, M.; et al. Whole-Genome Resequencing of Worldwide Wild and Domestic Sheep Elucidates Genetic Diversity, Introgression, and Agronomically Important Loci. Mol. Biol. Evol. 2022, 39, msab353. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.; Shen, M.; Xie, X.L.; Liu, G.J.; Xu, Y.X.; Lv, F.H.; Yang, H.; Yang, Y.L.; Liu, C.B.; et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef]
- Dou, M.; Li, M.; Zheng, Z.; Chen, Q.; Wu, Y.; Wang, J.; Shan, H.; Wang, F.; Dai, X.; Li, Y.; et al. A missense mutation in RRM1 contributes to animal tameness. Sci. Adv. 2023, 9, eadf4068. [Google Scholar] [CrossRef]
- Gu, J.; Chen, S.; Zhou, Y.; Chen, Y. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, H.; Liu, G.; Zhao, F.; Kijas, J.W.; Ma, Y.; Lu, J.; Zhang, L.; Cao, J.; Wu, M.; et al. Genome-wide analysis reveals adaptation to high altitudes in Tibetan sheep. Sci. Rep. 2016, 6, 26770. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar]
- Jiao, X.; Sherman, B.T.; Huangda, W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [PubMed]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z. GAPIT Version 3: Boosting Power and Accuracy for Genomic Association and Prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Goeman, J.J.; Solari, A. Multiple hypothesis testing in genomics. Stat. Med. 2014, 33, 1946–1978. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, G.S.; Rognoni, E.; Lichtenberger, B.M.; Driskell, R.R.; Kretzschmar, K.; Hoste, E.; Watt, F.M. Fate of Prominin-1 Expressing Dermal Papilla Cells during Homeostasis, Wound Healing and Wnt Activation. J. Investig. Dermatol. 2015, 135, 2926–2934. [Google Scholar] [CrossRef]
- Symoens, S.; Barnes, A.M.; Gistelinck, C.; Malfait, F.; Guillemyn, B.; Steyaert, W.; Syx, D.; D’hondt, S.; Biervliet, M.; De Backer, J.; et al. Genetic Defects in TAPT1 Disrupt Ciliogenesis and Cause a Complex Lethal Osteochondrodysplasia. Am. J. Hum. Genet. 2015, 97, 521–534. [Google Scholar] [CrossRef]
- Liu, T.M.; Lee, E.H. Transcriptional regulatory cascades in Runx2-dependent bone development. Tissue Eng. Part B Rev. 2013, 19, 254–263. [Google Scholar] [CrossRef]
- Hoepfner, S.; Severin, F.; Cabezas, A.; Habermann, B.; Runge, A.; Gillooly, D.; Stenmark, H.; Zerial, M. Modulation of Receptor Recycling and Degradation by the Endosomal Kinesin KIF16B. Cell 2005, 121, 437–450. [Google Scholar] [CrossRef]
- Leon, L.; Giannoukas, A.; Dodd, D.; Chan, P.; Labropoulos, N. Clinical Significance of Superficial Vein Thrombosis. Eur. J. Vasc. Endovasc. 2005, 29, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Relationship Between the Evolution of Mammalian Hippo Signaling Pathway-Related Genes and Body Size. Master’s Thesis, Nanjing Normal University, Nanjing, China, 2019. [Google Scholar]
- Si, X.; Xu, F.; Xu, F.; Wei, M.; Ge, Y.; Chenge, S. CADM1 inhibits ovarian cancer cell proliferation and migration by potentially regulating the PI3K/Akt/mTOR pathway. Biomed. Pharmacother. 2020, 123, 109717. [Google Scholar] [CrossRef]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 Senses and Restricts Influenza A Virus by Ubiquitination of PB1 Polymerase. PLoS Pathog. 2015, 11, e1004960. [Google Scholar] [CrossRef]
- Wang, K. Identification and Functional Verification of Genes Related to Pig Growth Traits Based on Multiple Association Analysis. Ph.D. Thesis, China Agricultural University, Beijing, China, 2017. [Google Scholar]
- Bliskunova, T.; Genis-Mendoza, A.D.; Martínez-Magaña, J.J.; Vega-Sevey, J.G.; Jiménez-Genchi, J.; Roche, A.; Guzmán, R.; Zapata, L.; Castro-Chavira, S.; Fernández, T.; et al. Association of MGAT4C with major neurocognitive disorder in the Mexican population. Gene 2021, 778, 145484. [Google Scholar] [CrossRef]
- Quénet, D.; Mark, M.; Govin, J.; van Dorsselear, A.; Schreiber, V.; Khochbin, S.; Dantzer, F. Parp2 is required for the differentiation of post-meiotic germ cells: Identification of a spermatid-specific complex containing Parp1, Parp2, TP2 and HSPA2. Exp. Cell Res. 2009, 315, 2824–2834. [Google Scholar] [CrossRef]
- Hirasawa, R.; Feil, R. A KRAB domain zinc finger protein in imprinting and disease. Dev. Cell 2008, 15, 487–488. [Google Scholar] [CrossRef]
- Hidaru, G.G. Genomic Analysis of Important Economic Traits of Wool Color and Fat Tail in Tan Sheep. Ph.D. Thesis, Chinese Academy of Agricultural Sciences, Beijing, China, 2021. [Google Scholar]
- Dong, J. Analysis of Egg Production Characteristics and Development of Related Molecular Markers in Guangxi Yao Chicken. Master’s Thesis, Henan Agricultural University, Zhengzhou, China, 2023. [Google Scholar]
- Surendran, S.; Szucs, S.; Tyring, S.K.; Matalon, R. Aspartoacylase gene knockout in the mouse: Impact on reproduction. Reprod. Toxicol. 2005, 20, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z. Study on the Function and Evolution Characteristics of Vertebrate H-P Axis Based on the Hypothalamus/Pituitary Cell Map of Large Yellow Croaker. Master’s Thesis, Zhejiang Ocean University, Zhoushan, China, 2024. [Google Scholar]
- Elsayed, A.; von Hardenberg, S.; Atschekzei, F.; Siek, P.; Witte, T.; Sogkas, G.; Ringshausen, F.C. A novel hemizygous nonsense variant in DOCK11 causes systemic inflammation and immunodeficiency. Clin. Immunol. 2025, 276, 110504. [Google Scholar] [CrossRef]
- Yousuf, S.; Malik, W.A.; Feng, H.; Liu, T.; Xie, L.; Miao, X. Genome wide identification and characterization of fertility associated novel CircRNAs as ceRNA reveal their regulatory roles in sheep fecundity. J. Ovarian Res. 2023, 16, 115. [Google Scholar] [CrossRef] [PubMed]
- Lan, S.; Du, J.; Nuerabudula, W.; Han, Y.; Yiming, S.; Yang, B.; Ailijiang, A. Study on the growth and development law of Pishan red sheep twins and its correlation with body size. Chin. J. Anim. Sci. 2023, 59, 144–149. [Google Scholar]
- Naval-Sanchez, M.; Nguyen, Q.; Mcwilliam, S.; Porto-Neto, L.R.; Tellam, R.; Vuocolo, T.; Reverter, A.; Perez-Enciso, M.; Brauning, R.; Clarke, S. Sheep genome functional annotation reveals proximal regulatory elements contributed to the evolution of modern breeds. Nat. Commun. 2018, 9, 859. [Google Scholar] [CrossRef]
- Zhu, M.; Yang, Y.; Yang, H.; Zhao, Z.; Zhang, H.; Blair, H.T.; Zheng, W.; Wang, M.; Fang, C.; Yu, Q. Whole-genome resequencing of the native sheep provides insights into the microevolution and identifies genes associated with reproduction traits. BMC Genom. 2023, 24, 392. [Google Scholar] [CrossRef]
- Guo, X.; Wang, X.; Di, R.; Liu, Q.; Hu, W.; He, X.; Yu, J.; Zhang, X.; Zhang, J.; Broniowska, K.; et al. Metabolic Effects of FecB Gene on Follicular Fluid and Ovarian Vein Serum in Sheep (Ovis aries). Int. J. Mol. Sci. 2018, 19, 539. [Google Scholar] [CrossRef]
- Sechman, A.; Grzegorzewska, A.K.; Grzesiak, M.; Kozubek, A.; Katarzynska-Banasik, D.; Kowalik, K.; Hrabia, A. Nitrophenols suppress steroidogenesis in prehierarchical chicken ovarian follicles by targeting STAR, HSD3B1, and CYP19A1 and downregulating LH and estrogen receptor expression. Domest. Anim. Endocrinol. 2020, 70, 106378. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, H.; Yang, H.; Zhao, Z.; Blair, H.T.; Liang, H.; Wu, P.; Yu, Q. Targeting GNAQ in hypothalamic nerve cells to regulate seasonal estrus in sheep. Theriogenology 2022, 181, 79–88. [Google Scholar] [CrossRef]
- Saravanan, K.A.; Manjit, P.; Kumar, H.; Bhushan, B.; Dutt, T.; Mishra, B.P. Genome-wide analysis of genetic diversity and selection signatures in three Indian sheep breeds. Livest. Sci. 2021, 243, 104367. [Google Scholar] [CrossRef]
- Dou, D.; Shen, L.; Zhou, J.; Cao, Z.; Luan, P.; Li, Y.; Xiao, F.; Guo, H.; Li, H.; Zhang, H. Genome-wide association studies for growth traits in broilers. BMC Genom. Data 2022, 23, 1. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Huang, X.; Tanaka, Y.; Hirokawa, N. KIF16B/Rab14 Molecular Motor Complex Is Critical for Early Embryonic Development by Transporting FGF Receptor. Dev. Cell 2011, 20, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Pogoda, H.M.; Irlenbusch, S.; Smyth, N.; Brüning, J.C. PLRG1 Is an Essential Regulator of Cell Proliferation and Apoptosis during Vertebrate Development and Tissue Homeostasis. Mol. Cell. Biol. 2009, 29, 3173. [Google Scholar] [CrossRef]
- Klimczak, A. Biological Characteristics and Osteogenic Differentiation of Ovine Bone Marrow Derived Mesenchymal Stem Cells Stimulated with FGF-2 and BMP-2. Int. J. Mol. Sci. 2020, 21, 9726. [Google Scholar]
- Singh, A. Effects of BMP15 and GDF9 on Ovine Oocytes in an In Vitro Maturation System. Master’s Thesis, Victoria University of Wellington, Wellington, New Zealand, 2020. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Chr | POS (Mb) | p Value | Genes | Function |
|---|---|---|---|---|---|
| BH | 6 | 110,844,020–110,844,500 | 2.92 × 10−7 | PROM1, TAPT1 | Nipple size Hair follicle development [23]; skeletal development [24] |
| 110,844,501–110,845,000 | 1.19 × 10−7 | TAPT1, LDB2 | Skeletal development [24] | ||
| 110,845,001–110,847,000 | 3.46 × 10−7 | TAPT1, LDB2 | Skeletal development [24] | ||
| 20 | 18,934,759–18,934,991 | 2.39 × 10−7 | RUNX2 | Bone development, osteogenic differentiation [25] | |
| CC | 13 | 10,002,600–10,002,800 | 6.79 × 10−7 | KIF16B, SNRPB2, OTOR | Fat deposition [26] |
| 17 | 2,870,800–2,871,000 | 3.36 × 10−7 | FGA, FGB, PLRG1, DCHS2 | Early embryonic development [27]; organ development [28] | |
| TL | 15 | 25,526,000–25,540,000 | 1.18 × 10−8 | CADM1 | Bone development [29] |
| LW | 2 | 6,990,000–9,780,000 | 4.68 × 10−8 | TRIM32, COL27A1, AMBP, ZNF618 | Early growth and development, osteogenic development [30]; lamb birth weight [31] |
| 3 | 122,161,200–122,161,300 | 5.14 × 10−7 | MGAT4C | Neurodevelopment [32] | |
| 7 | 24,044,700–24,044,800 | 5.43 × 10−7 | PARP2, TTC5 | Germ cell development [33] | |
| 14 | 57,979,500–57,979,600 | 1.66 × 10−8 | DPRX, ZNF331 | Immunoregulation [34] | |
| 19 | 48,471,500–48,471,600 | 5.43 × 10−7 | DNAH1, NISCH, NT5DC2, WDR82, PHF7, SEMA3G, GLYCTK, STAB1, TLR9, BAP1, TNNC1, PPM1M, TWF2 | Embryonic development [35] | |
| 27 | 49,928,000–49,929,000 | 4.01 × 10−7 | GPR173, TSPYL2, KDM5C | Follicular development [36] | |
| LS | 8 | 5,677,600–5,677,700 | 7.14 × 10−7 | PHIP, IRAK1BP1 | Follicular development [36] |
| 11 | 23,580,200–23,584,400 | 6.73 × 10−7 | ASPA, RAP1GAP2 | Litter size [37] | |
| 13 | 36,904,100–36,904,200 | 3.24 × 10−7 | BFSP1, PCSK2, RRBP1, DSTN | Neuronal development [38] | |
| 27 | 81,156,000–110,377,700 | 2.88 × 10−8 | IDS, DOCK11 | Immunodeficiency [39] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhetapa, M.; Zhu, M.; Qi, A.; Yiming, S. Identification of Genomic Variants and Candidate Genes for Reproductive Traits and Growth Traits in Pishan Red Sheep Using Whole-Genome Resequencing. Biology 2025, 14, 636. https://doi.org/10.3390/biology14060636
Muhetapa M, Zhu M, Qi A, Yiming S. Identification of Genomic Variants and Candidate Genes for Reproductive Traits and Growth Traits in Pishan Red Sheep Using Whole-Genome Resequencing. Biology. 2025; 14(6):636. https://doi.org/10.3390/biology14060636
Chicago/Turabian StyleMuhetapa, Maimaitijiang, Mengting Zhu, Aladaer Qi, and Sulaiman Yiming. 2025. "Identification of Genomic Variants and Candidate Genes for Reproductive Traits and Growth Traits in Pishan Red Sheep Using Whole-Genome Resequencing" Biology 14, no. 6: 636. https://doi.org/10.3390/biology14060636
APA StyleMuhetapa, M., Zhu, M., Qi, A., & Yiming, S. (2025). Identification of Genomic Variants and Candidate Genes for Reproductive Traits and Growth Traits in Pishan Red Sheep Using Whole-Genome Resequencing. Biology, 14(6), 636. https://doi.org/10.3390/biology14060636