
Predicting Treatment Outcomes in Glioblastoma: A Risk Score Model for TMZ Resistance and Immune Checkpoint Inhibition

 , , , , , ,
, , , , , ,
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Data Collection and Processing
2.2. Differential Expression Analysis
2.3. Identification of Prognostic Genes
2.4. Establishment of the Risk Score Model for Prognosis and Treatment Response Prediction
2.5. Kaplan–Meier Survival Analysis
2.6. Predictive Effect for Alternative Drugs According to the Risk Score
2.7. Clinical Response to Anti-PD1 Therapies: Model Development
2.8. Drugs
2.9. Cell Culture Reagents
2.10. Cell Culture
2.11. Generation of TMZ-Resistant Cell Lines
2.12. MTT Cell Viability Assay
2.13. Flow Cytometry
2.14. Statistical Analyses
3. Results
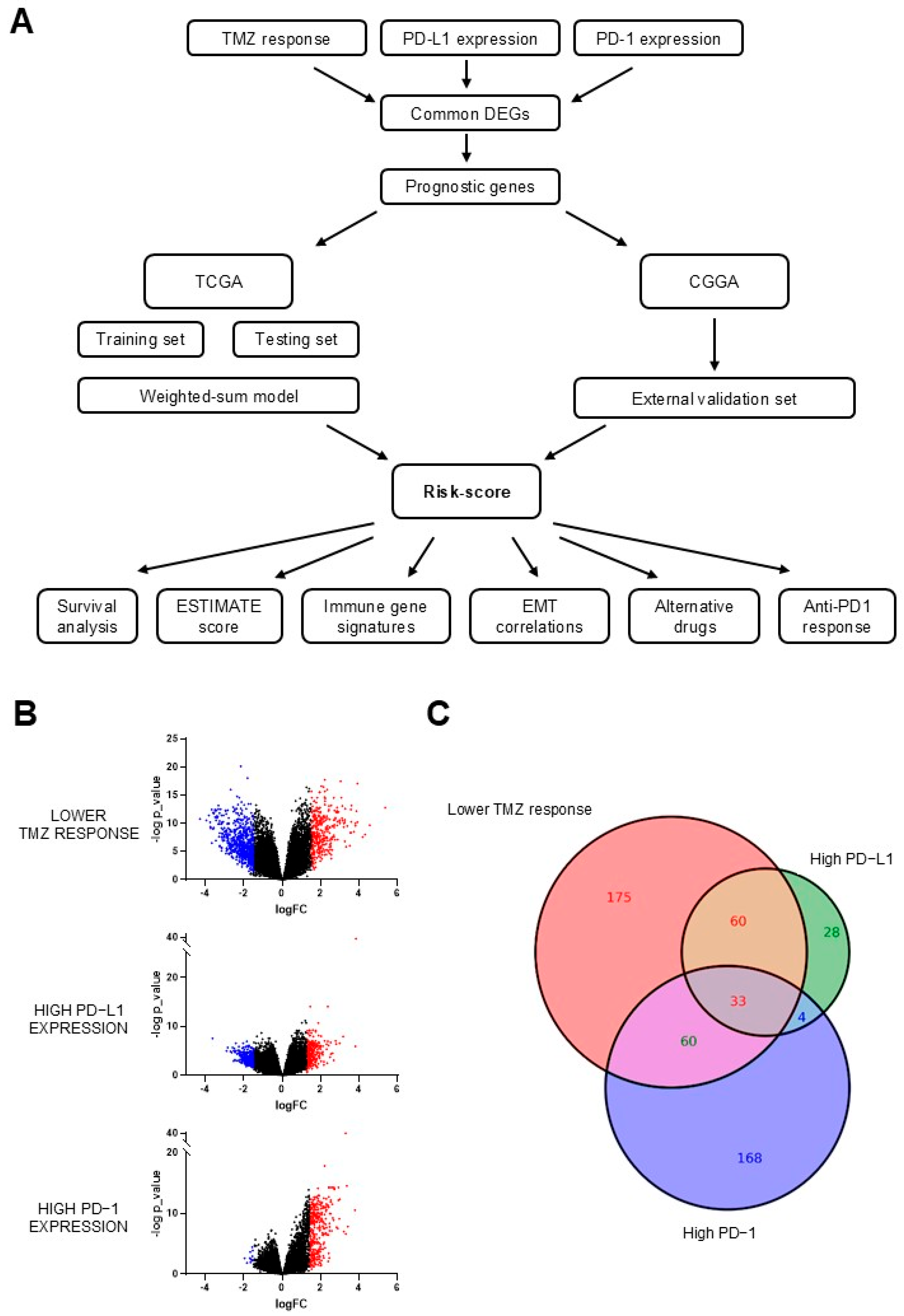
3.1. Identification of Differentially Expressed Genes (DEGs)
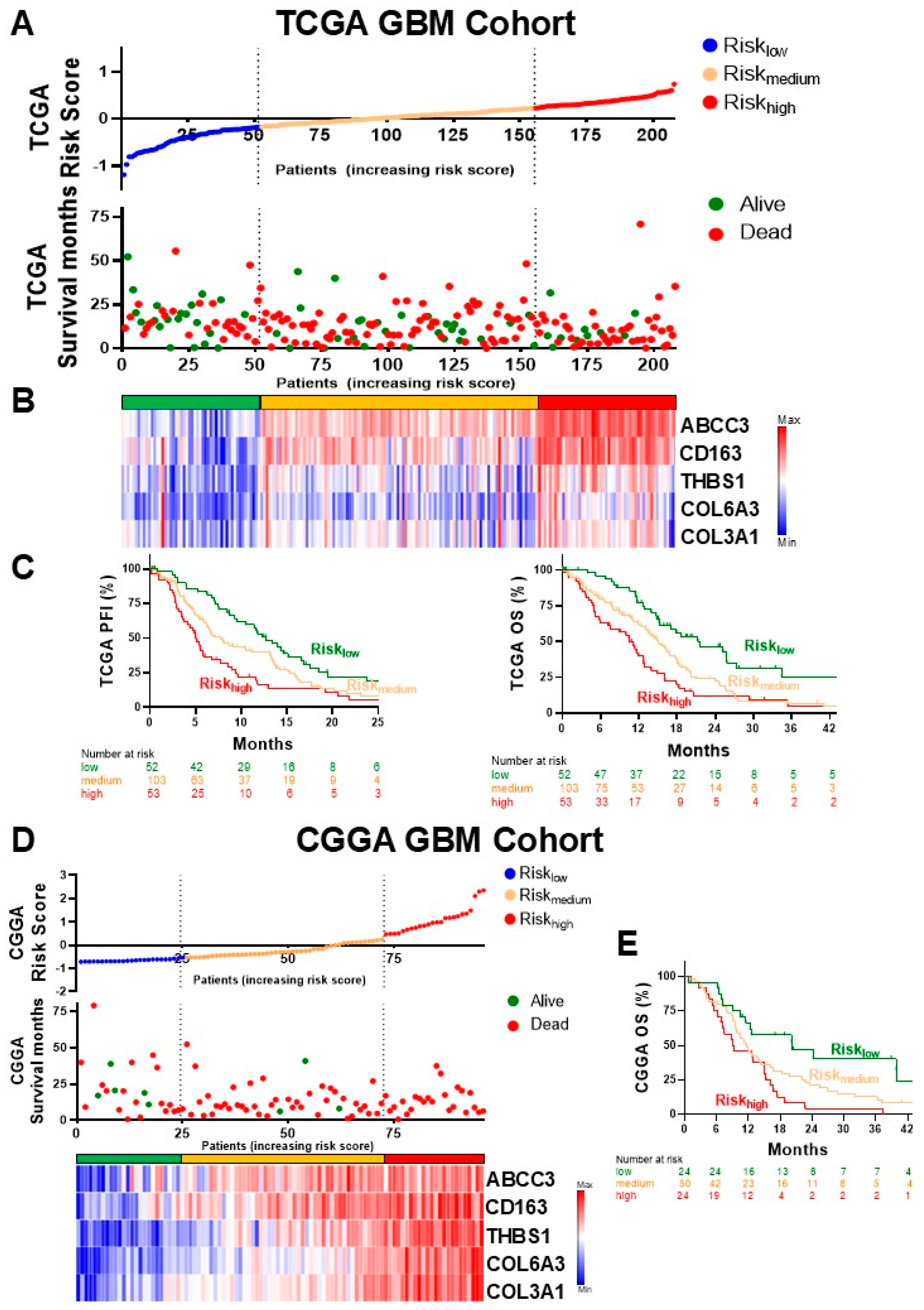
3.2. Construction of the Risk Score Model
3.3. Correlation with Oncogenic Features
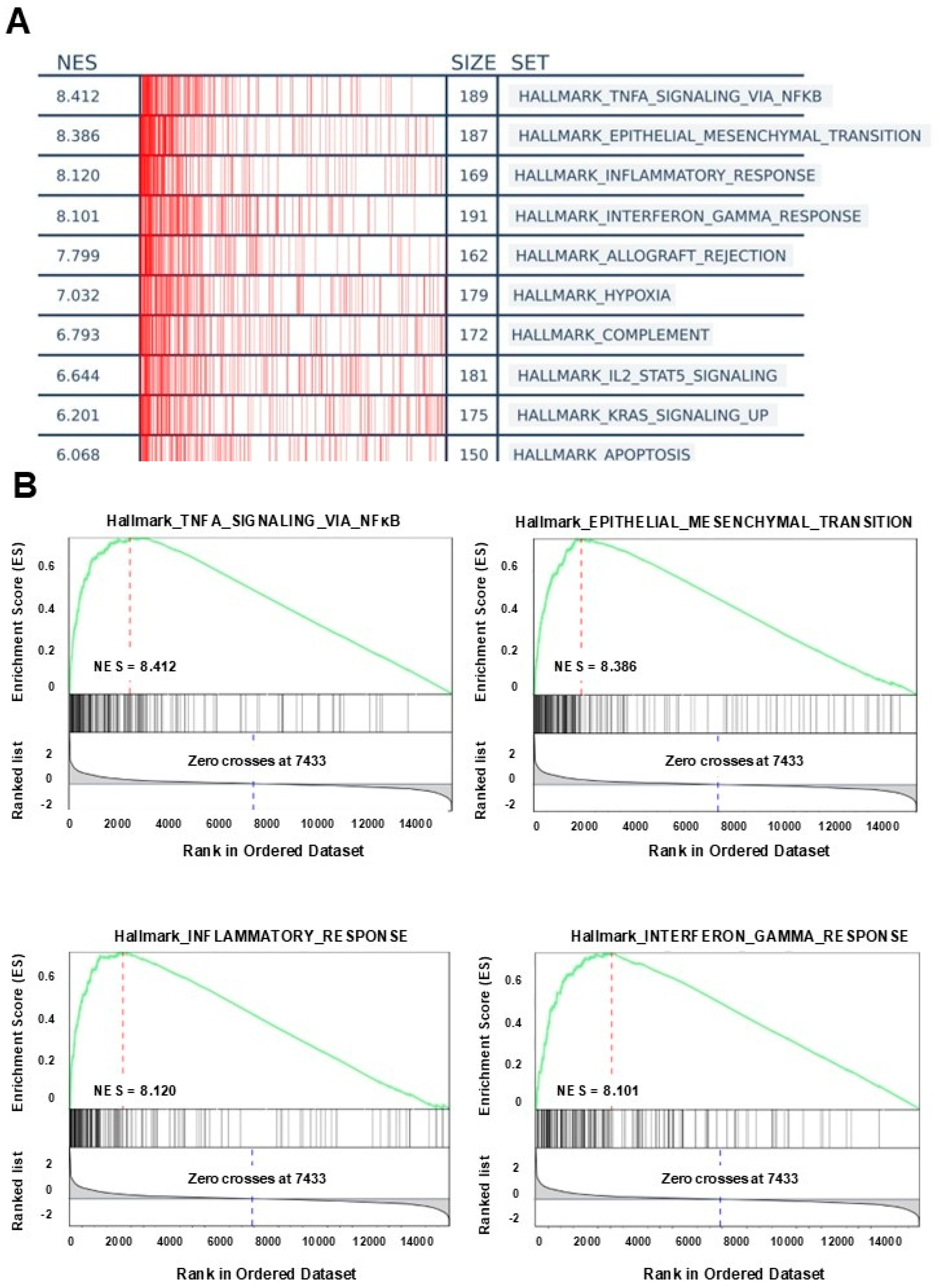
3.4. GSEA Analysis
3.5. Prediction of Drug Sensitivity According to the Risk Score
3.6. Prediction of Clinical Response to Anti-PD−1 Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016–2020. Neuro Oncol. 2023, 25, iv1–iv99. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Chen, W.; Zhu, Y.; Zhang, P. Clinical features associated with the efficacy of chemotherapy in patients with glioblastoma (GBM): A surveillance, epidemiology, and end results (SEER) analysis. BMC Cancer 2021, 21, 81. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.A.; Sosnovtseva, A.O.; Valikhov, M.P.; Chernysheva, A.A.; Abramova, O.V.; Pavlov, K.A.; Chekhonin, V.P. Systemic and local immunosuppression in glioblastoma and its prognostic significance. Front. Immunol. 2024, 15, 1326753. [Google Scholar] [CrossRef]
- Lin, H.; Liu, C.; Hu, A.; Zhang, D.; Yang, H.; Mao, Y. Understanding the immunosuppressive microenvironment of glioma: Mechanistic insights and clinical perspectives. J. Hematol. Oncol. 2024, 17, 31. [Google Scholar] [CrossRef]
- Hao, C.; Chen, G.; Zhao, H.; Li, Y.; Chen, J.; Zhang, H.; Li, S.; Zhao, Y.; Chen, F.; Li, W.; et al. PD−L1 Expression in Glioblastoma, the Clinical and Prognostic Significance: A Systematic Literature Review and Meta-Analysis. Front. Oncol. 2020, 10, 1015. [Google Scholar] [CrossRef]
- Park, J.H.; Kang, I.; Lee, H.K. The immune landscape of high-grade brain tumor after treatment with immune checkpoint blockade. Front. Immunol. 2022, 13, 1044544. [Google Scholar] [CrossRef]
- Sun, Q.; Hong, Z.; Zhang, C.; Wang, L.; Han, Z.; Ma, D. Immune checkpoint therapy for solid tumours: Clinical dilemmas and future trends. Signal Transduct. Target. Ther. 2023, 8, 320. [Google Scholar] [CrossRef]
- Yu, C.; Hsieh, K.; Cherry, D.R.; Nehlsen, A.D.; Resende Salgado, L.; Lazarev, S.; Sindhu, K.K. Immune Escape in Glioblastoma: Mechanisms of Action and Implications for Immune Checkpoint Inhibitors and CAR T-Cell Therapy. Biology 2023, 12, 1528. [Google Scholar] [CrossRef]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef]
- Zhao, X.; Yan, H.; Yan, X.; Chen, Z.; Zhuo, R. A Novel Prognostic Four-Gene Signature of Breast Cancer Identified by Integrated Bioinformatics Analysis. Dis. Markers 2022, 2022, 5925982. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Tang, J.; Liao, X.; Yang, Q.; Wu, Y.; Wu, G. An immune-related prognostic signature for predicting breast cancer recurrence. Cancer Med. 2020, 9, 7672–7685. [Google Scholar] [CrossRef] [PubMed]
- Atay, S. A 15-Gene-Based Risk Signature for Predicting Overall Survival in SCLC Patients Who Have Undergone Surgical Resection. Cancers 2023, 15, 5219. [Google Scholar] [CrossRef]
- Zhou, N.; Zhou, M.; Ding, N.; Li, Q.; Ren, G. An 11-Gene Signature Risk-Prediction Model Based on Prognosis-Related miRNAs and Their Target Genes in Lung Adenocarcinoma. Front. Oncol. 2021, 11, 726742. [Google Scholar] [CrossRef]
- Wang, J.; Kong, P.F.; Wang, H.Y.; Song, D.; Wu, W.Q.; Zhou, H.C.; Weng, H.Y.; Li, M.; Kong, X.; Meng, B.; et al. Identification of a Gene-Related Risk Signature in Melanoma Patients Using Bioinformatic Profiling. J. Oncol. 2020, 2020, 7526204. [Google Scholar] [CrossRef]
- Meyer, S.; Buser, L.; Haferkamp, S.; Berneburg, M.; Maisch, T.; Klinkhammer-Schalke, M.; Pauer, A.; Vogt, T.; Garbe, C. Identification of high-risk patients with a seven-biomarker prognostic signature for adjuvant treatment trial recruitment in American Joint Committee on Cancer v8 stage I–IIA cutaneous melanoma. Eur. J. Cancer 2023, 182, 77–86. [Google Scholar] [CrossRef]
- Chen, M.; Bie, L.; Ying, J. Cancer cell-intrinsic PD−1: Its role in malignant progression and immunotherapy. Biomed. Pharmacother. 2023, 167, 115514. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Jiao, H.; Miao, X. The prognostic significance of PD−L1 expression in patients with glioblastoma: A meta-analysis. Front. Oncol. 2022, 12, 925560. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Reifenberger, G.; Weber, R.G.; Riehmer, V.; Kaulich, K.; Willscher, E.; Wirth, H.; Gietzelt, J.; Hentschel, B.; Westphal, M.; Simon, M.; et al. Molecular characterization of long-term survivors of glioblastoma using genome- and transcriptome-wide profiling. Int. J. Cancer 2014, 135, 1822–1831. [Google Scholar] [CrossRef]
- Kawaguchi, A.; Yajima, N.; Tsuchiya, N.; Homma, J.; Sano, M.; Natsumeda, M.; Takahashi, H.; Fujii, Y.; Kakuma, T.; Yamanaka, R. Gene expression signature-based prognostic risk score in patients with glioblastoma. Cancer Sci. 2013, 104, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Umbach, D.M.; Krahn, J.M.; Shats, I.; Li, X.; Li, L. Predicting tumor response to drugs based on gene-expression biomarkers of sensitivity learned from cancer cell lines. BMC Genom. 2021, 22, 272. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD−1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD−1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Li, B.; Zhou, C.; Yi, L.; Xu, L.; Xu, M. Effect and molecular mechanism of mTOR inhibitor rapamycin on temozolomide-induced autophagic death of U251 glioma cells. Oncol. Lett. 2018, 15, 2477–2484. [Google Scholar] [CrossRef]
- Melaiu, O.; Chierici, M.; Lucarini, V.; Jurman, G.; Conti, L.A.; De Vito, R.; Boldrini, R.; Cifaldi, L.; Castellano, A.; Furlanello, C.; et al. Cellular and gene signatures of tumor-infiltrating dendritic cells and natural-killer cells predict prognosis of neuroblastoma. Nat. Commun. 2020, 11, 5992. [Google Scholar] [CrossRef]
- Gonzalez, N.; Asad, A.S.; Gómez Escalante, J.; Peña Agudelo, J.A.; Nicola Candia, A.J.; García Fallit, M.; Seilicovich, A.; Candolfi, M. Potential of IDH mutations as immunotherapeutic targets in gliomas: A review and meta-analysis. Expert. Opin. Ther. Targets 2021, 25, 1045–1060. [Google Scholar] [CrossRef]
- Moon, C.I.; Elizarraras, J.M.; Lei, J.T.; Jia, B.; Zhang, B. ClinicalOmicsDB: Exploring molecular associations of oncology drug responses in clinical trials. Nucleic Acids Res. 2023, 52, D1201–D1209. [Google Scholar] [CrossRef]
- Yan, F.; Pang, J.; Peng, Y.; Molina, J.R.; Yang, P.; Liu, S. Elevated Cellular PD1/PD−L1 Expression Confers Acquired Resistance to Cisplatin in Small Cell Lung Cancer Cells. PLoS ONE 2016, 11, e0162925. [Google Scholar] [CrossRef] [PubMed]
- Rom-Jurek, E.M.; Kirchhammer, N.; Ugocsai, P.; Ortmann, O.; Wege, A.K.; Brockhoff, G. Regulation of Programmed Death Ligand 1 (PD−L1) Expression in Breast Cancer Cell Lines In Vitro and in Immunodeficient and Humanized Tumor Mice. Int. J. Mol. Sci. 2018, 19, 563. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, X.; Zhang, C.; Wang, Y.; Cheng, T.; Duan, L.; Tong, Z.; Tan, S.; Zhang, H.; Saw, P.E.; et al. Tumor cell-intrinsic PD−1 receptor is a tumor suppressor and mediates resistance to PD−1 blockade therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 6640–6650. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Zhu, H.; Tan, J.; Xin, Z.; Zhou, Q.; Cao, Y.; Wu, Z.; Wang, L.; Zhao, M.; Jiang, X.; et al. Identification of collagen genes related to immune infiltration and epithelial-mesenchymal transition in glioma. Cancer Cell Int. 2021, 21, 276. [Google Scholar] [CrossRef]
- Caglar, H.O.; Duzgun, Z. Identification of upregulated genes in glioblastoma and glioblastoma cancer stem cells using bioinformatics analysis. Gene 2023, 848, 146895. [Google Scholar] [CrossRef]
- Cha, J.; Ding, E.A.; Carvalho, E.M.; Fowler, A.; Aghi, M.K.; Kumar, S. Glioma Cells Secrete Collagen VI to Facilitate Invasion. bioRxiv 2023. [Google Scholar] [CrossRef]
- Lau, S.K.; Chu, P.G.; Weiss, L.M. CD163: A specific marker of macrophages in paraffin-embedded tissue samples. Am. J. Clin. Pathol. 2004, 122, 794–801. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, C.; Maimela, N.R.; Yang, L.; Zhang, Z.; Ping, Y.; Huang, L.; Zhang, Y. Molecular and clinical characterization of CD163 expression via large-scale analysis in glioma. Oncoimmunology 2019, 8, 1601478. [Google Scholar] [CrossRef]
- Chen, T.; Chen, J.; Zhu, Y.; Li, Y.; Wang, Y.; Chen, H.; Wang, J.; Li, X.; Liu, Y.; Li, B.; et al. CD163, a novel therapeutic target, regulates the proliferation and stemness of glioma cells via casein kinase 2. Oncogene 2019, 38, 1183–1199. [Google Scholar] [CrossRef]
- Zeng, C.; Fan, D.; Xu, Y.; Li, X.; Yuan, J.; Yang, Q.; Zhou, X.; Lu, J.; Zhang, C.; Han, J.; et al. Curcumol enhances the sensitivity of doxorubicin in triple-negative breast cancer via regulating the miR-181b-2-3p-ABCC3 axis. Biochem. Pharmacol. 2020, 174, 113795. [Google Scholar] [CrossRef]
- Liu, X.; Yao, D.; Liu, C.; Cao, Y.; Yang, Q.; Sun, Z.; Liu, D. Overexpression of ABCC3 promotes cell proliferation, drug resistance, and aerobic glycolysis and is associated with poor prognosis in urinary bladder cancer patients. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 8367–8374. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Funayama, R.; Ohnuma, S.; Unno, M.; Nakayama, K. Wnt-beta-catenin signaling regulates ABCC3 (MRP3) transporter expression in colorectal cancer. Cancer Sci. 2016, 107, 1776–1784. [Google Scholar] [CrossRef]
- Ramírez-Cosmes, A.; Reyes-Jiménez, E.; Zertuche-Martínez, C.; Hernández-Hernández, C.A.; García-Román, R.; Romero-Díaz, R.I.; Manuel-Martínez, A.E.; Elizarrarás-Rivas, J.; Vásquez-Garzón, V.R. The implications of ABCC3 in cancer drug resistance: Can we use it as a therapeutic target? Am. J. Cancer Res. 2021, 11, 4127–4140. [Google Scholar] [PubMed]
- Wang, F.; Zheng, Z.; Guan, J.; Qi, D.; Zhou, S.; Shen, X.; Wang, F.; Wenkert, D.; Kirmani, B.; Solouki, T.; et al. Identification of a panel of genes as a prognostic biomarker for glioblastoma. EBioMedicine 2018, 37, 68–77. [Google Scholar] [CrossRef]
- Fang, D.D.; Huang, W.; Cheng, G.; Liu, X.N.; Liu, S.M.; Hou, B.S.; Mao, J.; Zhou, H. Clinicopathological and Prognostic Significance of ABCC3 in Human Glioma. J. Oncol. 2021, 2021, 1827992. [Google Scholar] [CrossRef]
- Ruiz-Lopez, E.; Jovcevska, I.; Gonzalez-Gomez, R.; Tejero, H.; Al-Shahrour, F.; Muyldermans, S.; Schuhmacher, A.J. Nanobodies targeting ABCC3 for immunotargeted applications in glioblastoma. Sci. Rep. 2022, 12, 22581. [Google Scholar] [CrossRef]
- Huang, T.; Sun, L.; Yuan, X.; Qiu, H. Thrombospondin-1 is a multifaceted player in tumor progression. Oncotarget 2017, 8, 84546–84558. [Google Scholar] [CrossRef]
- Daubon, T.; Leon, C.; Clarke, K.; Andrique, L.; Salabert, L.; Darbo, E.; Pineau, R.; Guerit, S.; Maitre, M.; Dedieu, S.; et al. Deciphering the complex role of thrombospondin-1 in glioblastoma development. Nat. Commun. 2019, 10, 1146. [Google Scholar] [CrossRef]
- Gao, Y.F.; Mao, X.Y.; Zhu, T.; Mao, C.X.; Liu, Z.X.; Wang, Z.B.; Li, L.; Li, X.; Yin, J.Y.; Zhang, W.; et al. COL3A1 and SNAP91: Novel glioblastoma markers with diagnostic and prognostic value. Oncotarget 2016, 7, 70494–70503. [Google Scholar] [CrossRef]
- Michiels, S.; Ternes, N.; Rotolo, F. Statistical controversies in clinical research: Prognostic gene signatures are not (yet) useful in clinical practice. Ann. Oncol. 2016, 27, 2160–2167. [Google Scholar] [CrossRef]
- Liu, W.; He, H.; Chicco, D. Gene signatures for cancer research: A 25-year retrospective and future avenues. PLoS Comput. Biol. 2024, 20, e1012512. [Google Scholar] [CrossRef] [PubMed]
- Chimal-Ramírez, G.K.; Espinoza-Sánchez, N.A.; Fuentes-Pananá, E.M. Protumor Activities of the Immune Response: Insights in the Mechanisms of Immunological Shift, Oncotraining, and Oncopromotion. J. Oncol. 2013, 2013, 835956. [Google Scholar] [CrossRef]
- Lin, W.; Qiu, X.; Sun, P.; Ye, Y.; Huang, Q.; Kong, L.; Lu, J.J. Association of IDH mutation and 1p19q co-deletion with tumor immune microenvironment in lower-grade glioma. Mol. Ther.-Oncolytics 2021, 21, 288–302. [Google Scholar] [CrossRef]
- Zhang, L.; Sorensen, M.D.; Kristensen, B.W.; Reifenberger, G.; McIntyre, T.M.; Lin, F. D-2-Hydroxyglutarate Is an Intercellular Mediator in IDH-Mutant Gliomas Inhibiting Complement and T Cells. Clin. Cancer Res. 2018, 24, 5381–5391. [Google Scholar] [CrossRef]
- Marinari, E.; Allard, M.; Gustave, R.; Widmer, V.; Philippin, G.; Merkler, D.; Tsantoulis, P.; Dutoit, V.; Dietrich, P.Y. Inflammation and lymphocyte infiltration are associated with shorter survival in patients with high-grade glioma. Oncoimmunology 2020, 9, 1779990. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef]
- Zamora-Salas, S.X.; Macias-Silva, M.; Tecalco-Cruz, A.C. Upregulation of the canonical signaling pathway of interferon-gamma is associated with glioblastoma progression. Mol. Biol. Rep. 2024, 51, 64. [Google Scholar] [CrossRef]
- Liu, T.; Hu, J.; Han, B.; Tan, S.; Jia, W.; Xin, Y. A positive feedback loop of lncRNA-RMRP/ZNRF3 axis and Wnt/beta-catenin signaling regulates the progression and temozolomide resistance in glioma. Cell Death Dis. 2021, 12, 952. [Google Scholar] [CrossRef]
- Samant, C.; Kale, R.; Pai, K.S.R.; Nandakumar, K.; Bhonde, M. Role of Wnt/beta-catenin pathway in cancer drug resistance: Insights into molecular aspects of major solid tumors. Biochem. Biophys. Res. Commun. 2024, 729, 150348. [Google Scholar] [CrossRef]
- Yildirim, Z.; Dogan, E.; Guler Kara, H.; Kosova, B.; Bozok, V. STING activation increases the efficiency of temozolomide in PTEN harbouring glioblastoma cells. Turk. J. Med. Sci. 2024, 54, 607–614. [Google Scholar] [CrossRef]
- Cao, M.; Cai, J.; Yuan, Y.; Shi, Y.; Wu, H.; Liu, Q.; Yao, Y.; Chen, L.; Dang, W.; Zhang, X.; et al. A four-gene signature-derived risk score for glioblastoma: Prospects for prognostic and response predictive analyses. Cancer Biol. Med. 2019, 16, 595–605. [Google Scholar] [CrossRef]
- Zuo, S.; Zhang, X.; Wang, L. A RNA sequencing-based six-gene signature for survival prediction in patients with glioblastoma. Sci. Rep. 2019, 9, 2615. [Google Scholar] [CrossRef]
- Wang, G.; Hu, J.Q.; Liu, J.Y.; Zhang, X.M. Angiogenesis-Related Gene Signature-Derived Risk Score for Glioblastoma: Prospects for Predicting Prognosis and Immune Heterogeneity in Glioblastoma. Front. Cell Dev. Biol. 2022, 10, 778286. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Yan, B.; Liu, Y.; Jiang, S.; He, H.; Huang, J.; Liu, W.; Xie, L. FOXP3 Contributes to TMZ Resistance, Prognosis, and Immune Infiltration in GBM from a Novel Pyroptosis-Associated Risk Signature. Dis. Markers 2022, 2022, 4534080. [Google Scholar] [CrossRef]
- Yu, Z.; Du, M.; Lu, L. A Novel 16-Genes Signature Scoring System as Prognostic Model to Evaluate Survival Risk in Patients with Glioblastoma. Biomedicines 2022, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Tang, G.; Zhou, Q.; Cao, Y.; Li, H.; Fu, X.; Wu, Z.; Jiang, X. Expression Profile Analysis Identifies a Novel Five-Gene Signature to Improve Prognosis Prediction of Glioblastoma. Front. Genet. 2019, 10, 419. [Google Scholar] [CrossRef]
- Huang, J.; Wang, G.; Zhang, J.; Liu, Y.; Shen, Y.; Chen, G.; Ji, W.; Shao, J. A novel ARHGAP family gene signature for survival prediction in glioma patients. J. Cell Mol. Med. 2024, 28, e18555. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, L.; Wu, W.; Chen, Z.; Xie, Z.; Shi, D.; Cai, N.; Zhuo, S. Prognosis and therapeutic significance of IGF-1R-related signaling pathway gene signature in glioma. Front. Cell Dev. Biol. 2024, 12, 1375030. [Google Scholar] [CrossRef]
- Weller, M.; Wen, P.Y.; Chang, S.M.; Dirven, L.; Lim, M.; Monje, M.; Reifenberger, G. Glioma. Nat. Rev. Dis. Primers 2024, 10, 33. [Google Scholar] [CrossRef]
- Franson, A.; McClellan, B.L.; Varela, M.L.; Comba, A.; Syed, M.F.; Banerjee, K.; Zhu, Z.; Gonzalez, N.; Candolfi, M.; Lowenstein, P.; et al. Development of immunotherapy for high-grade gliomas: Overcoming the immunosuppressive tumor microenvironment. Front. Med. 2022, 9, 966458. [Google Scholar] [CrossRef]
- Lan, Z.; Li, X.; Zhang, X. Glioblastoma: An Update in Pathology, Molecular Mechanisms and Biomarkers. Int. J. Mol. Sci. 2024, 25, 3040. [Google Scholar] [CrossRef] [PubMed]
- Chyuan, I.T.; Chu, C.L.; Hsu, P.N. Targeting the Tumor Microenvironment for Improving Therapeutic Effectiveness in Cancer Immunotherapy: Focusing on Immune Checkpoint Inhibitors and Combination Therapies. Cancers 2021, 13, 1188. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD−1 and PD−L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Xu, Y.; Wan, B.; Chen, X.; Zhan, P.; Zhao, Y.; Zhang, T.; Liu, H.; Afzal, M.Z.; Dermime, S.; Hochwald, S.N.; et al. The association of PD−L1 expression with the efficacy of anti-PD−1/PD−L1 immunotherapy and survival of non-small cell lung cancer patients: A meta-analysis of randomized controlled trials. Transl. Lung Cancer Res. 2019, 8, 413–428. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, Y.; Zhou, S.; Jiang, H.; Zhu, K.; Wang, R. Predictive effect of PD−L1 expression for immune checkpoint inhibitor (PD−1/PD−L1 inhibitors) treatment for non-small cell lung cancer: A meta-analysis. Int. Immunopharmacol. 2020, 80, 106214. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Bibeau, F.; Greillier, L.; Fumet, J.D.; Ilie, A.; Monville, F.; Lauge, C.; Catteau, A.; Boquet, I.; Majdi, A.; et al. Immunoscore immune checkpoint using spatial quantitative analysis of CD8 and PD−L1 markers is predictive of the efficacy of anti- PD1/PD−L1 immunotherapy in non-small cell lung cancer. EBioMedicine 2023, 92, 104633. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overall Survival | ||||
|---|---|---|---|---|
| Univariate Analysis | Multivariate Analysis | |||
| HR (95% CI) | p-Value | HR (95% CI) | p-Value | |
| COL6A3 | 1.189 (1.061–1.332) | 0.003 | 4.794 (1.796–12.797) | 0.002 |
| CD163 | 1.124 (0.979–1.292) | 0.097 | 0.515 (0.295–0.901) | 0.020 |
| ABCC3 | 1.108 (0.995–1.234) | 0.061 | 1.381 (1.050–1.815) | 0.021 |
| COL3A1 | 1.190 (1.053–1.344) | 0.005 | 0.331 (0.115–0.950) | 0.040 |
| THBS1 | 1.341 (1.168–1.540) | <0.001 | 1.620 (1.014–2.590) | 0.044 |
| GBP5 | 1.024 (0.0884–1.187) | 0.751 | 0.745 (0.548–1.011) | 0.059 |
| ITK | 1.162 (1.024–1.317) | 0.020 | 1.225 (0.986–1.522) | 0.066 |
| MARCO | 1.166 (1.006–1.352) | 0.042 | 1.677 (0.936–3.003) | 0.082 |
| UBD | 0.980 (0.861–1.115) | 0.756 | 0.748 (0.526–1.064) | 0.106 |
| MMP7 | 1.100 (0.941–1.286) | 0.232 | 1.224 (0.948–1.580) | 0.121 |
| IDO1 | 1.070 (0.899–1.275) | 0.446 | 1.171 (0.948–1.446) | 0.142 |
| FCGR2C | 1.289 (1.109–1.499) | <0.001 | 1.479 (0.872–2.509) | 0.146 |
| PTPN22 | 1.176 (1.023–1.353) | 0.022 | 0.717 (0.456–1.127) | 0.149 |
| PLA2G2A | 0.989 (0.855–1.144) | 0.883 | 0.818 (0.603–1.110) | 0.196 |
| COL1A1 | 1.252 (1.095–1.431) | <0.001 | 0.516 (0.187–1.422) | 0.200 |
| LYZ | 1.231 (1.058–1.434) | 0.007 | 1.249 (0.884–1.764) | 0.207 |
| MYO1G | 1.361 (1.170–1.582) | <0.001 | 1.317 (0.786–2.206) | 0.295 |
| IL21R | 1.233 (1.066–1.426) | 0.004 | 1.277 (0.768–2.121) | 0.345 |
| TREM1 | 1.058 (0.948–1.180) | 0.314 | 1.231 (0.794–1.908) | 0.353 |
| FCGR2B | 1.316 (1.129–1.533) | <0.001 | 1.276 (0.761–2.138) | 0.355 |
| GALNT5 | 1.126 (0.992–1.279 | 0.066 | 0.893 (0.691–1.155) | 0.389 |
| SAA2 | 1.027 (0.899–1.173) | 0.699 | 1.113 (0.821–1.508) | 0.491 |
| IL2RA | 1.082 (0.929–1.259) | 0.309 | 0.834 (0.494–1.406) | 0.494 |
| IBSP | 1.131 (0.972–1.317) | 0.111 | 1.125 (0.795–1.593) | 0.505 |
| F13A1 | 1.251 (1.068–1.465) | 0.005 | 0.817 (0.449–1.487) | 0.507 |
| RNASE2 | 1.092 (0.957–1.246) | 0.188 | 0.885 (0.537–1.458) | 0.632 |
| FPR2 | 1.107 (0.954–1.284) | 0.181 | 0.840 (0.405–1.745) | 0.640 |
| CCR2 | 1.181 (1.028–1.356) | 0.018 | 1.091 (0.750–1.589) | 0.648 |
| LTF | 1.003 (0.874–1.153) | 0.961 | 0.965 (0.784–1.187) | 0.733 |
| CCL2 | 1.160 (1.018–1.322) | 0.026 | 0.974 (0.655–1.449) | 0.896 |
| CCL8 | 1.083 (0.941–1.248) | 0.267 | 1.016 (0.754–1.367) | 0.919 |
| AIM1 | 1.144 (1.009–1.296) | 0.035 | 0.979 (0.755–1.269) | 0.872 |
| EMR1 | 1.189 (1.022–1.383) | 0.025 | 0.982 (0.646–1.492) | 0.931 |
| Progression-Free Interval | Overall Survival | |||||||
|---|---|---|---|---|---|---|---|---|
| Univariate Analysis | Multivariate Analysis | Univariate Analysis | Multivariate Analysis | |||||
| HR (95% CI) | p-Value | HR (95% CI) | p-Value | HR (95% CI) | p-Value | HR (95% CI) | p-Value | |
| Risk Score | 2.071 | 0.004 | 1.877 | 0.017 | 2.531 | <0.001 | 2.141 | 0.003 |
| (1.256–3.416) | (1.121–3.142) | (1.569–4.082) | (1.298–3.530) | |||||
| Gender | 1.313 | 0.0977314 | 1.216 | 0.2467 | 1.128 | 0.48344 | 1.017 | 0.9238035 |
| (0.951–1.813) | (0.873–1.694) | (0.805–1.580) | (0.716–1.445) | |||||
| Age | 1.016 | 0.0146262 | 10.123 | 0.0805 | 1.030 | <0.001 | 1.025 | 0.002 |
| (1.003–1.029) | (0.999–1.026) | (1.015–1.046) | (1.009–1.041) | |||||
| Karnofsky performance score | 0.995 | 0.4448754 | 0.998 | 0.7798 | 0.982 | 0.009 | 0.990 | 0.1893078 |
| (0.982–1.008) | (0.984–1.012) | (0.969–0.996) | (0.976–1.005) | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez, N.; Perez Küper, M.; Garcia Fallit, M.; Nicola Candia, A.J.; Peña Agudelo, J.A.; Suarez Velandia, M.; Romero, A.C.; Videla-Richardson, G.A.; Candolfi, M. Predicting Treatment Outcomes in Glioblastoma: A Risk Score Model for TMZ Resistance and Immune Checkpoint Inhibition. Biology 2025, 14, 572. https://doi.org/10.3390/biology14050572
Gonzalez N, Perez Küper M, Garcia Fallit M, Nicola Candia AJ, Peña Agudelo JA, Suarez Velandia M, Romero AC, Videla-Richardson GA, Candolfi M. Predicting Treatment Outcomes in Glioblastoma: A Risk Score Model for TMZ Resistance and Immune Checkpoint Inhibition. Biology. 2025; 14(5):572. https://doi.org/10.3390/biology14050572
Chicago/Turabian StyleGonzalez, Nazareno, Melanie Perez Küper, Matias Garcia Fallit, Alejandro J. Nicola Candia, Jorge A. Peña Agudelo, Maicol Suarez Velandia, Ana Clara Romero, Guillermo Agustin Videla-Richardson, and Marianela Candolfi. 2025. "Predicting Treatment Outcomes in Glioblastoma: A Risk Score Model for TMZ Resistance and Immune Checkpoint Inhibition" Biology 14, no. 5: 572. https://doi.org/10.3390/biology14050572
APA StyleGonzalez, N., Perez Küper, M., Garcia Fallit, M., Nicola Candia, A. J., Peña Agudelo, J. A., Suarez Velandia, M., Romero, A. C., Videla-Richardson, G. A., & Candolfi, M. (2025). Predicting Treatment Outcomes in Glioblastoma: A Risk Score Model for TMZ Resistance and Immune Checkpoint Inhibition. Biology, 14(5), 572. https://doi.org/10.3390/biology14050572