
Natural Selection Shaped Codon Usage Patterns in Wheat Dwarf Virus in Triticale
 ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Acquisition of Genetic Information of WDV in Triticale
2.2. Codon Base Composition Analysis
2.3. Calculation of Codon Usage Indexes
2.4. Neutrality Plot Analysis
2.5. ENC-Plot Analysis
2.6. PR2-Plot Analysis
2.7. Identification of the Optimal Codons
2.8. RSCU Clustering and Phylogenetic Analysis
3. Results
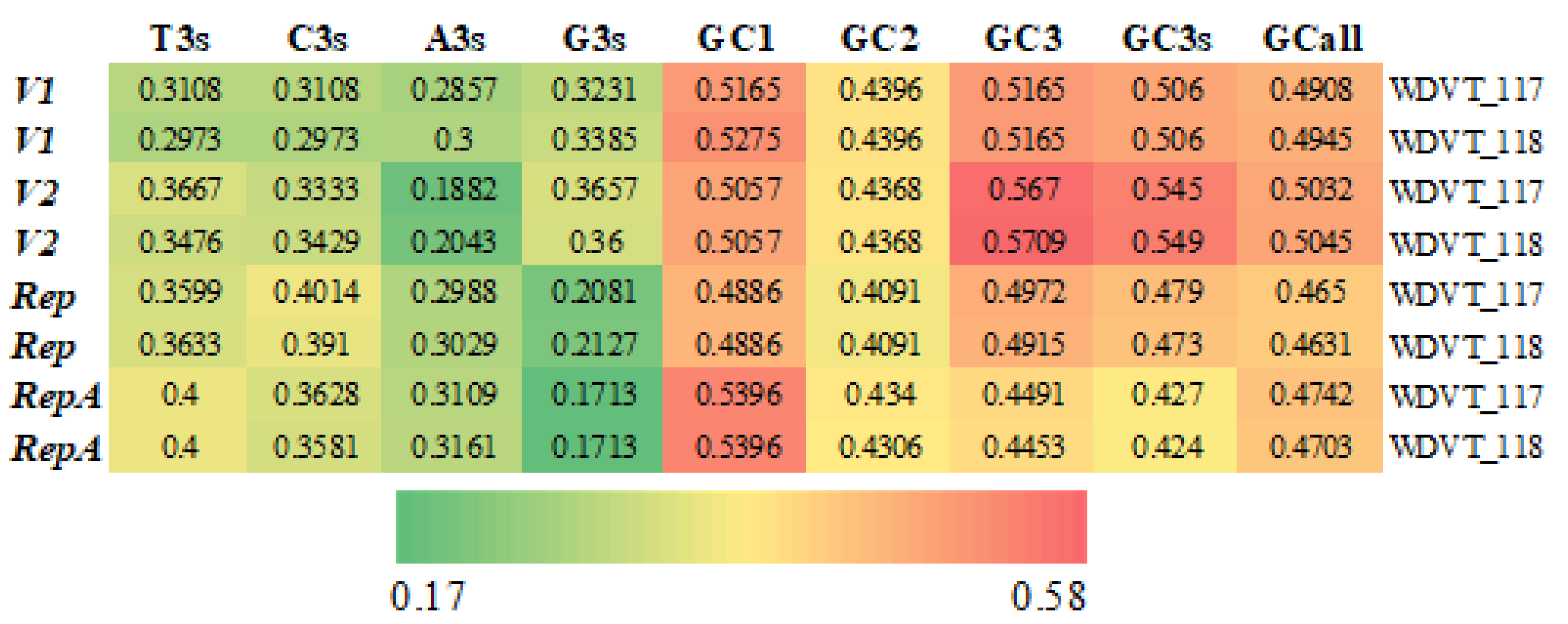
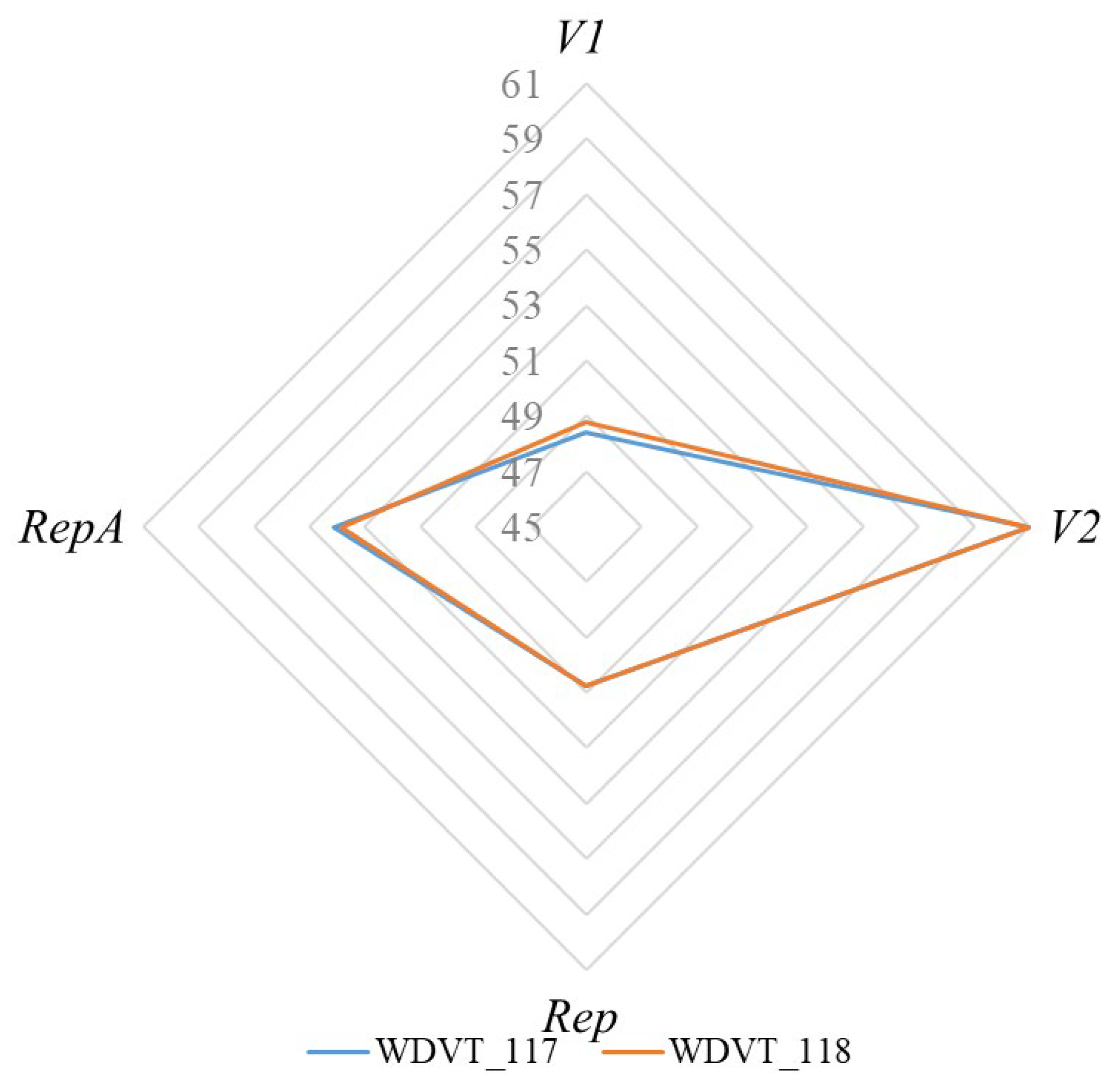
3.1. Codon Composition
3.2. Synonymous Codon Usage Features
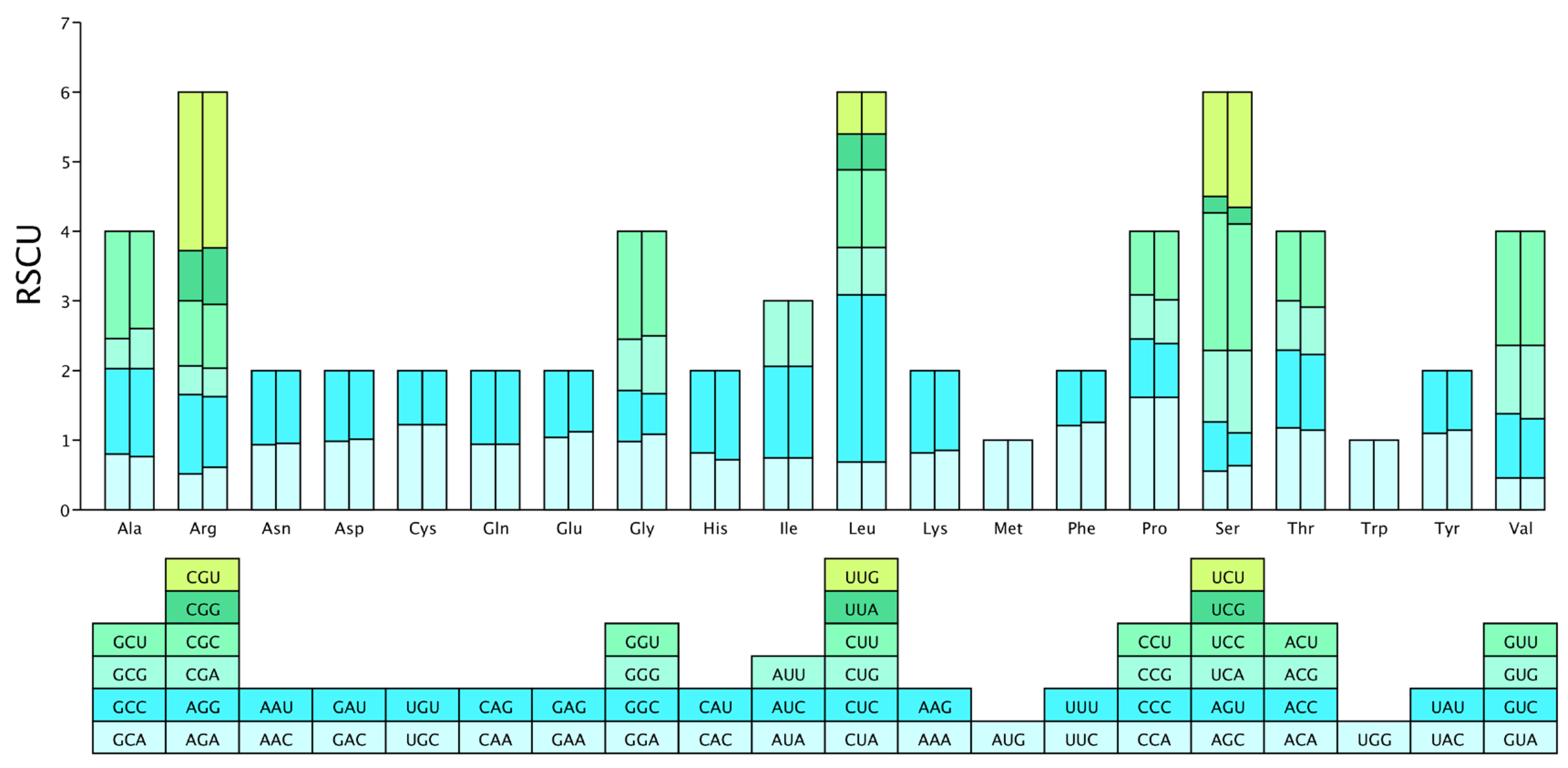
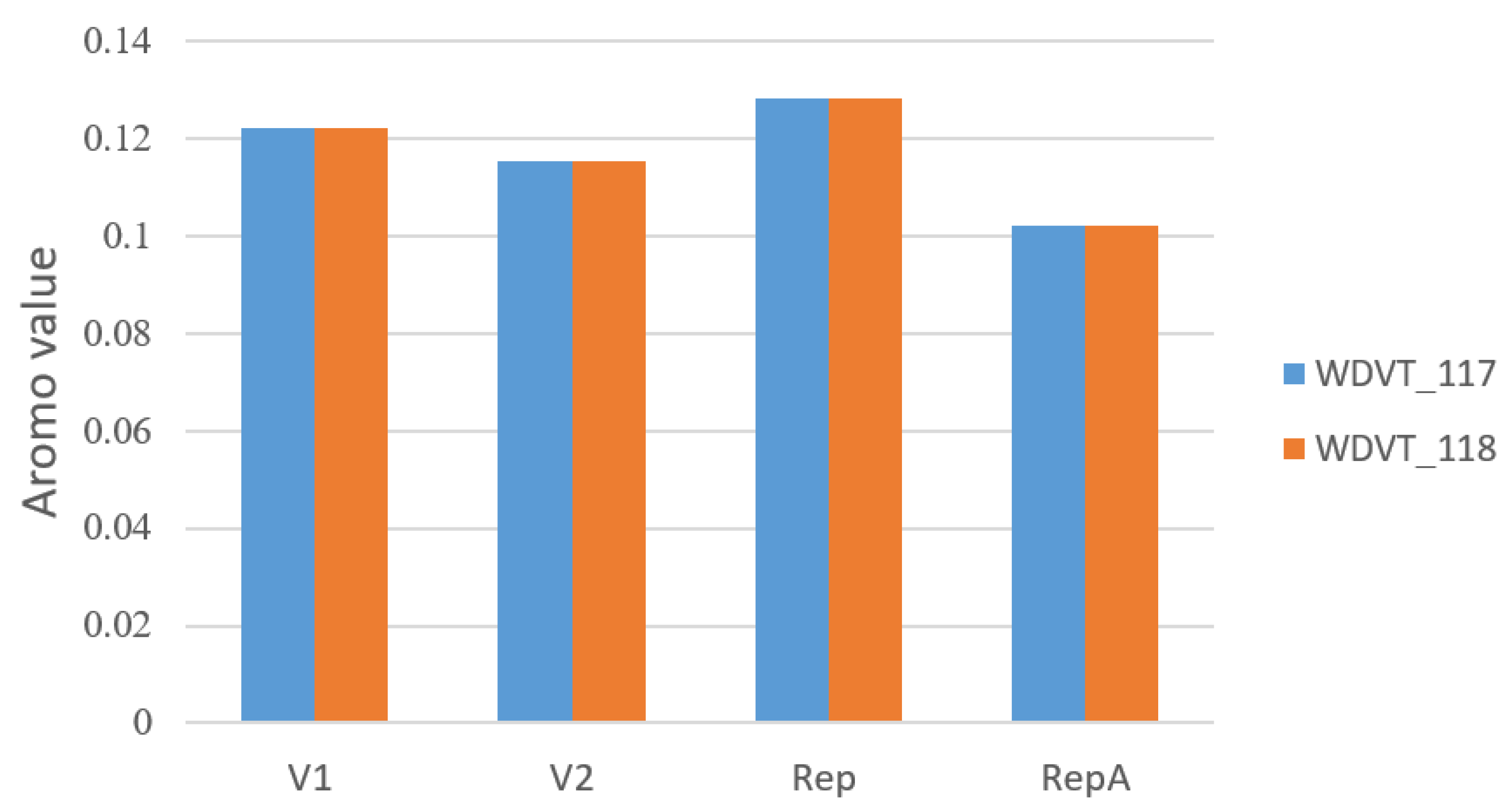
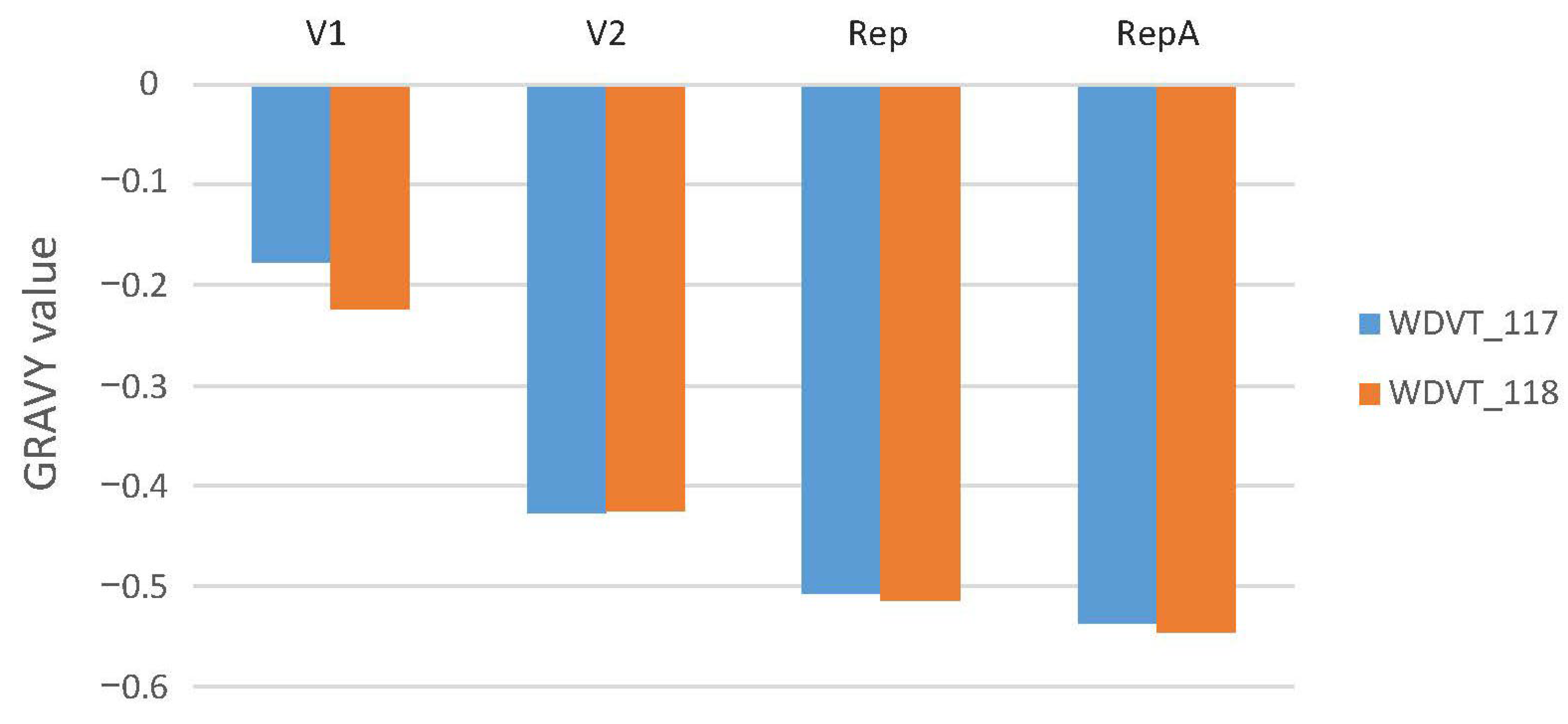
3.3. Amino Acid Usage
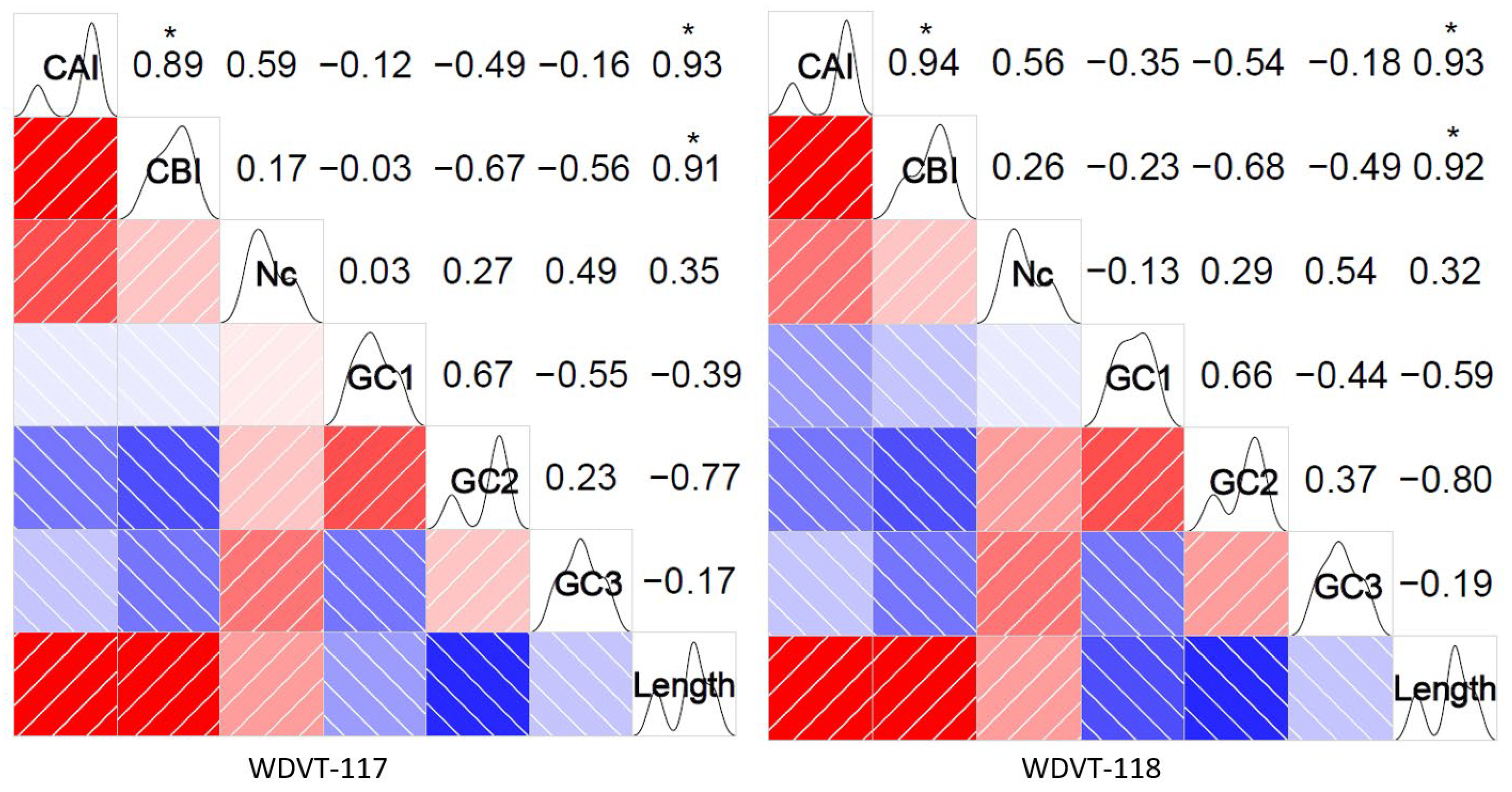
3.4. Correlation Among Indexes
- (a)
- CAI is very strongly positively correlated with CBI and length (significant at p < 0.05, t-tests), moderately positively correlated with ENC (significant at p < 0.05), and moderately negatively correlated with GC2. It has very weak negative correlations with GC1 and GC2.
- (b)
- CBI is very weakly positively correlated with ENC, very weakly negatively correlated with GC1, strongly negatively correlated with GC2, moderately negatively correlated with GC3, and very strongly positively correlated with length (significant at p < 0.05).
- (c)
- ENC has very weak positive and negative correlations with GC1, weak positive correlations with GC2 and length, and a moderate positive correlation with GC3.
- (d)
- GC1 is strongly positively correlated with GC2, moderately negatively correlated with GC3, and weakly negatively correlated with length.
- (e)
- GC2 has weak positive correlations with GC3 and length and strong negative correlations with length.
- (f)
- GC3 has very weak negative correlations with length.
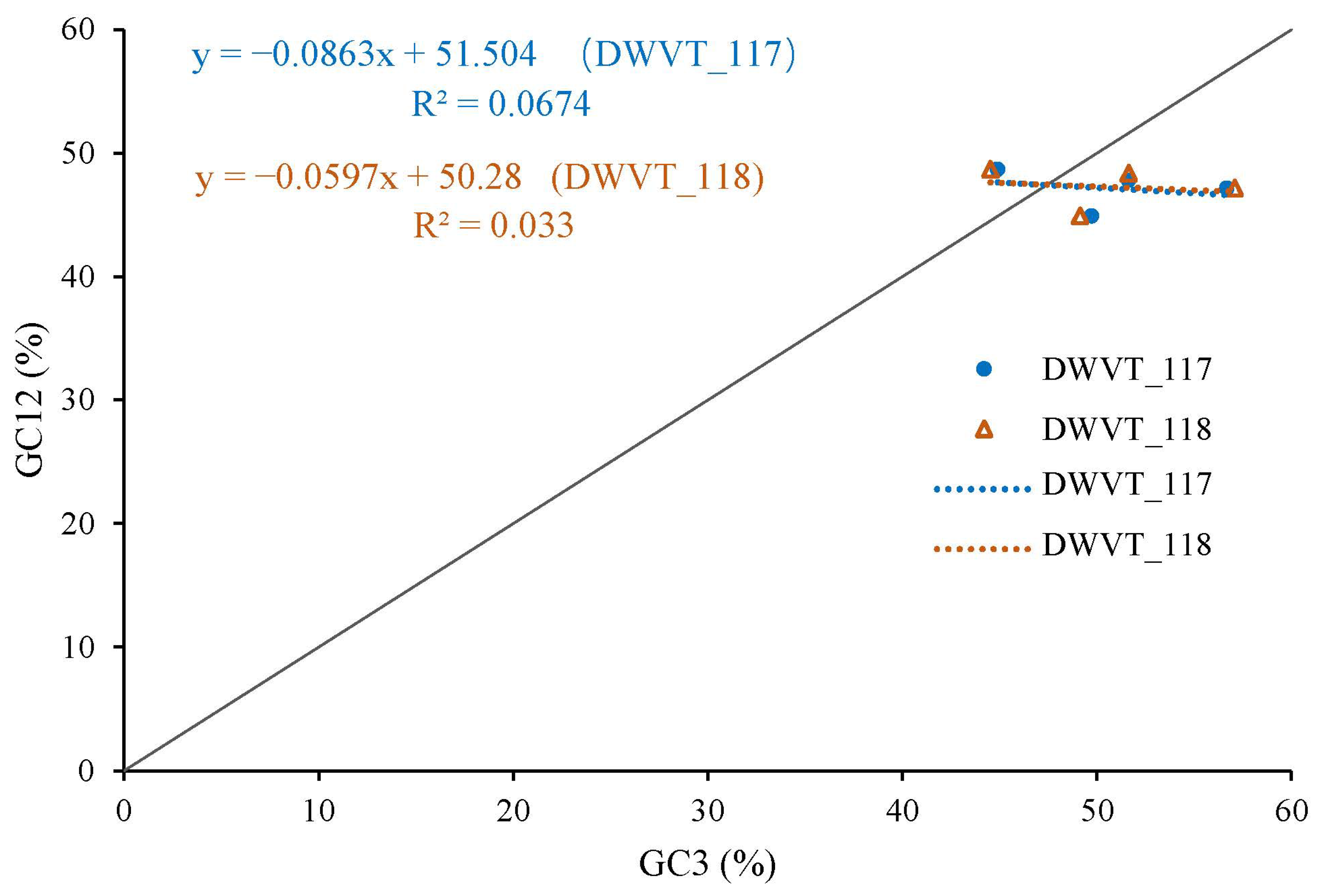
3.5. Neutrality Plot Analysis Reveals Natural Selection Dominance
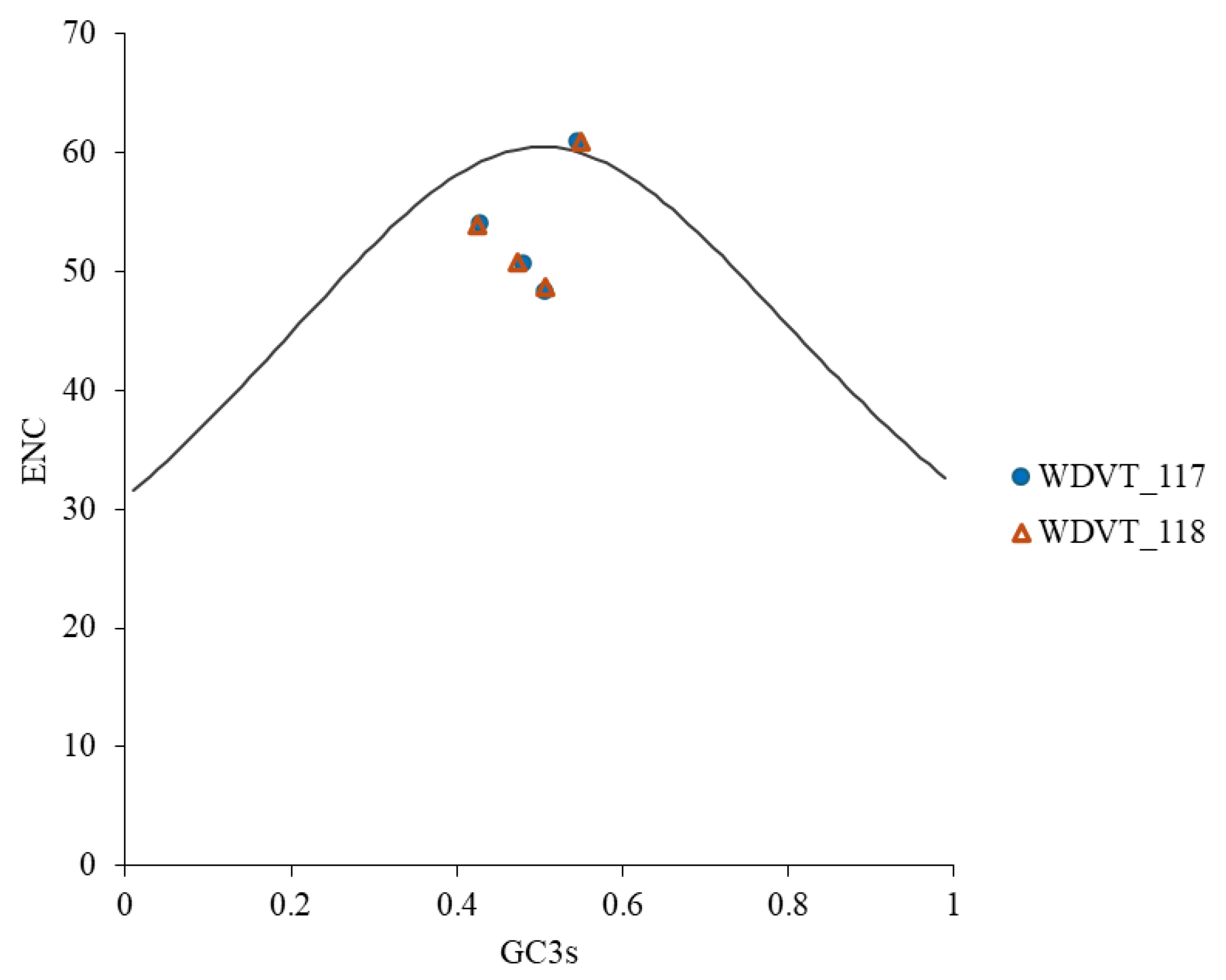
3.6. ENC-Plot Analysis Reveals Selection Pressure in WDV Codon Usage
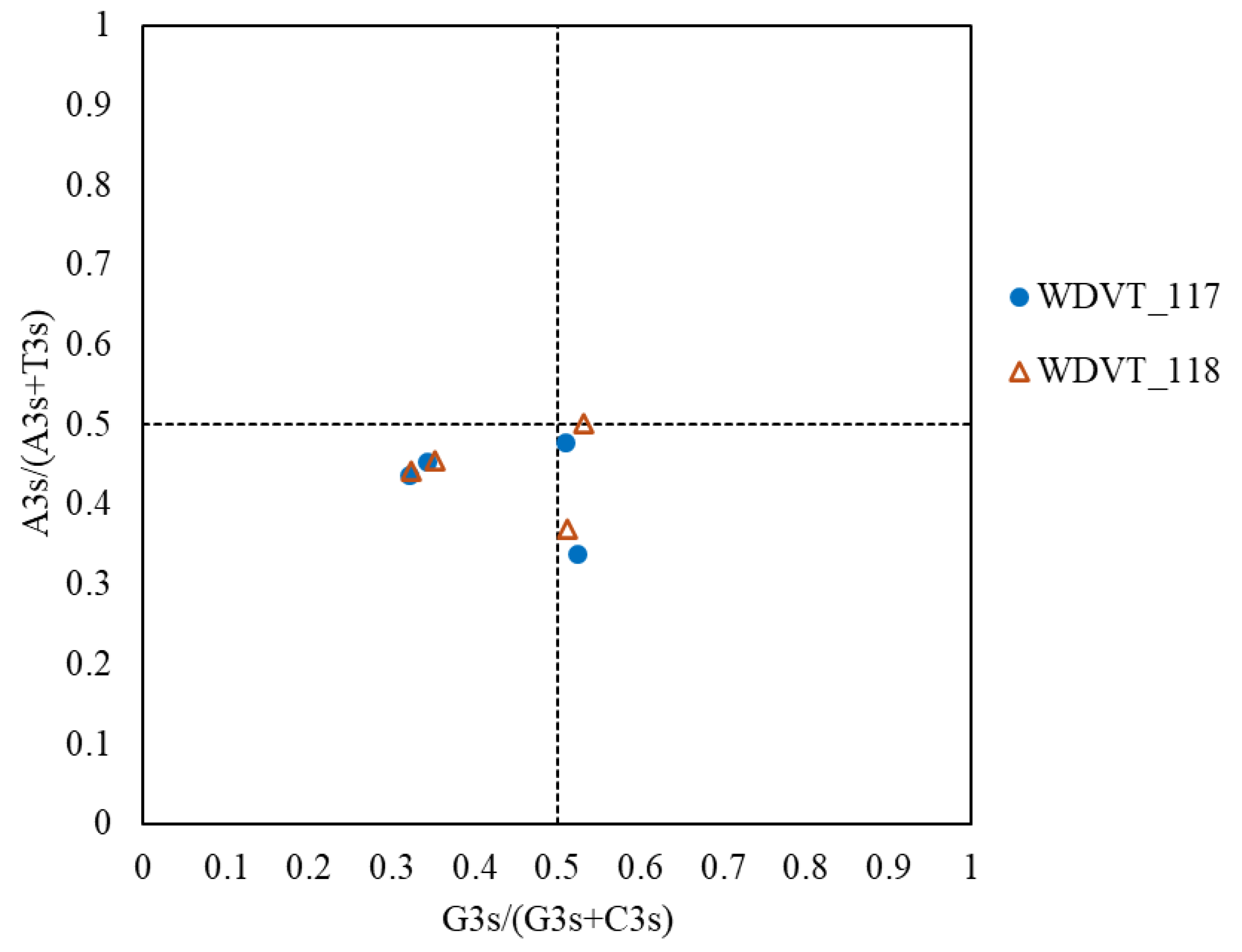
3.7. PR2-Plot Analysis Reveals Codon Usage Bias
3.8. Optimal Codons
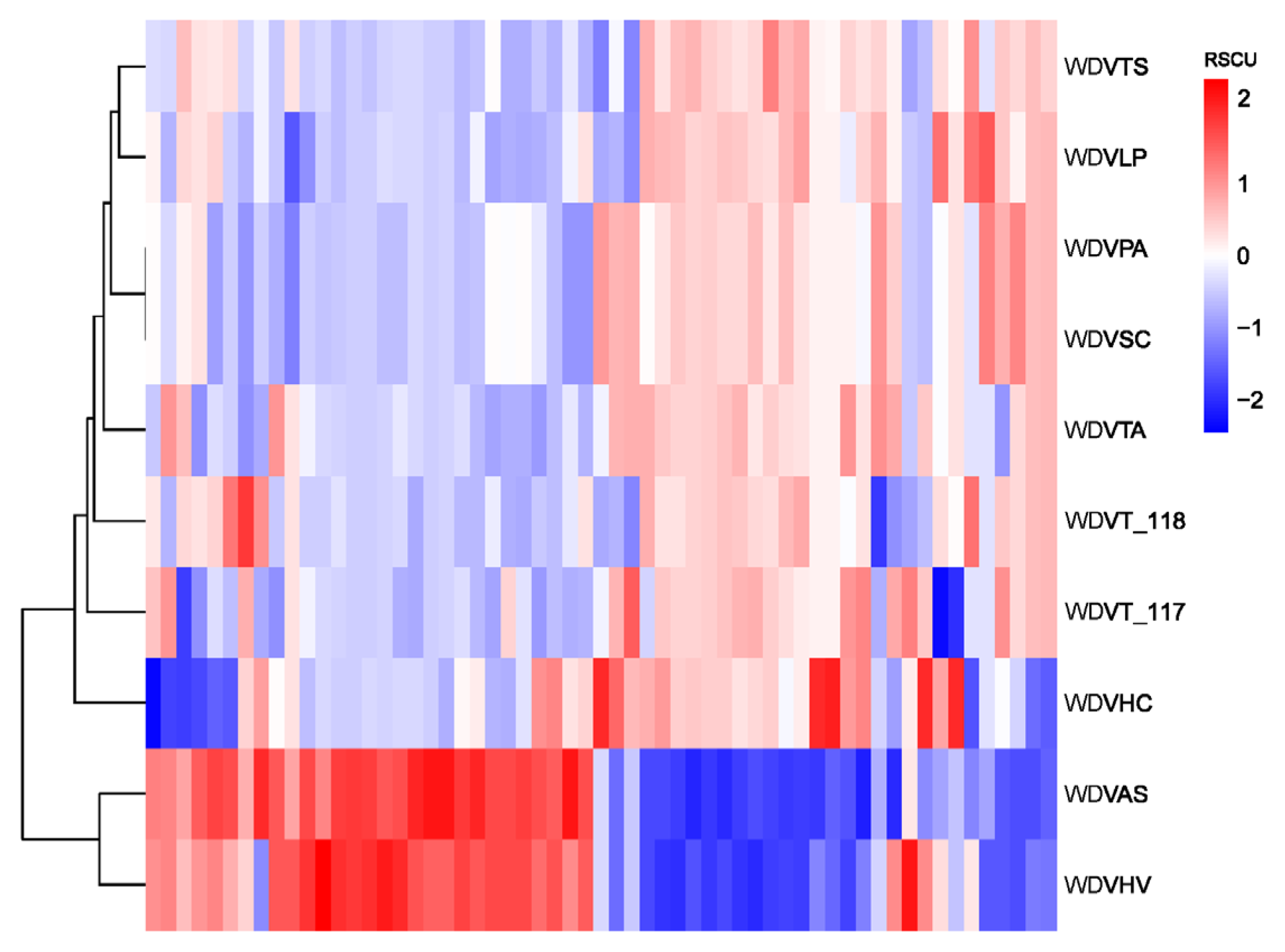
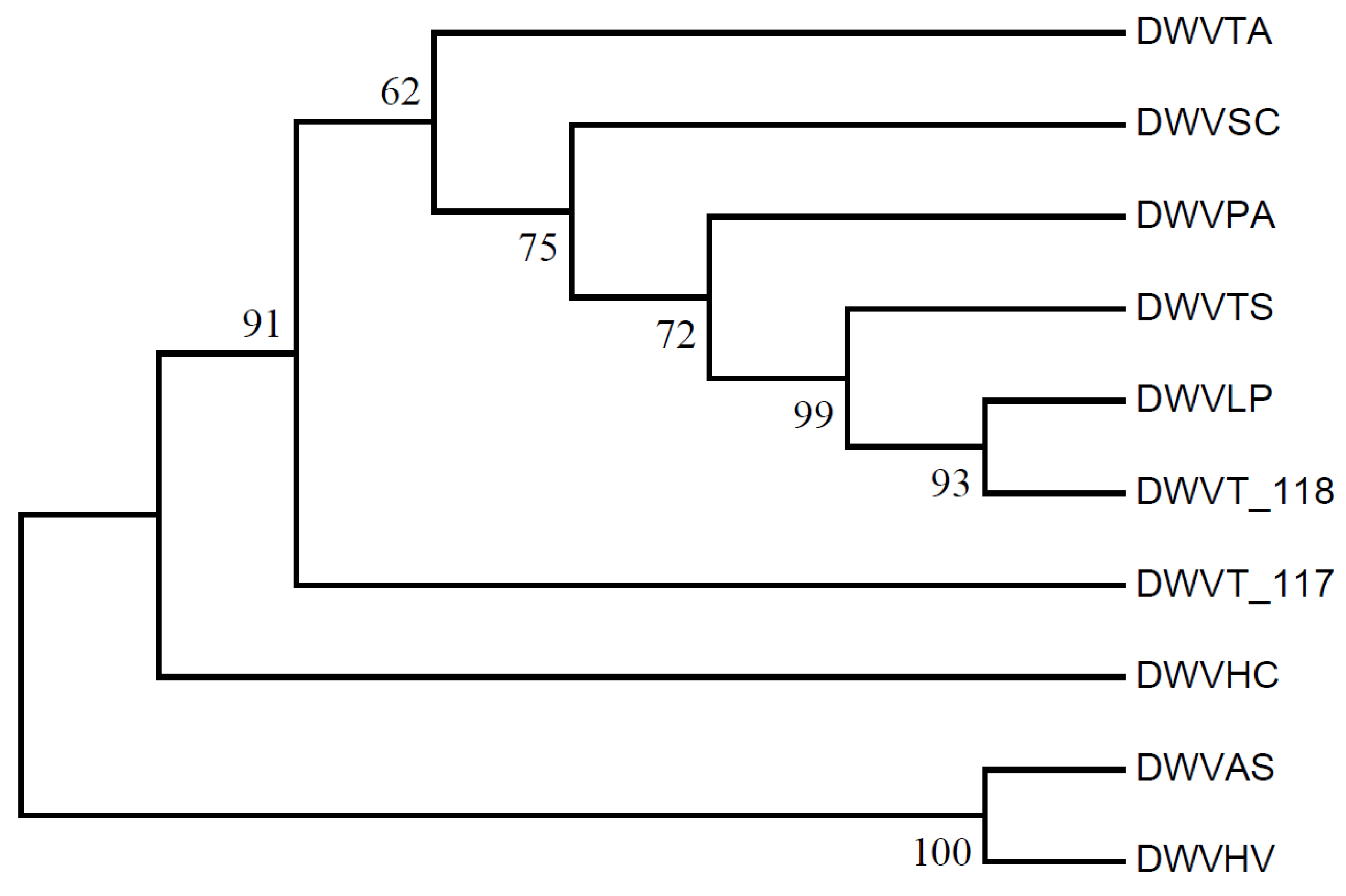
3.9. Phylogenetic Analysis and RSCU Clustering
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| RSCU | Relative synonymous codon usage |
| WDV | Wheat dwarf virus |
| WDVT | Wheat dwarf virus isolates from triticale |
| CAI | Codon adaptation index |
| SCUB | Synonymous codon usage bias |
| CBI | Codon bias index |
| ENC | Effective number of codons |
| CDS | Coding sequences |
| Aromo | Aromatic amino acid proportion |
| GRAVY | Grand average of hydropathicity |
| MP | Maximum parsimony |
References
- Mergoum, M.; Singh, P.K.; Pena, R.J.; Pea, R.J.; Lozano-del Río, A.J.; Gómez, H. Triticale: A “new” crop with old challenges. In Cereals; Springer: New York, NY, USA, 2009; pp. 267–287. [Google Scholar]
- Zhu, F. Triticale: Nutritional composition and food uses. Food Chem. 2018, 241, 468–479. [Google Scholar] [CrossRef] [PubMed]
- McGoverin, C.M.; Snyders, F.; Muller, N.; Botes, W.; Fox, G.; Manley, M. A review of triticale uses and the effect of growth environment on grain quality. J. Sci. Food Agric. 2011, 91, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Q.; Liu, J.; Tian, X.H.; Du, W.H. Evaluations on the adaptability of Triticosecale wittmack “Gannong NO.4” in different regions of Qinghai Province. Acta Agrestia Sin. 2020, 28, 1626–1634. [Google Scholar]
- Hammouda, D.; Baaziz, N.; Khalfallah, N. Genetic characterization of octoploid (AABBDDRR) and hexaploid (AABBRR) triticales. Eur. Sci. J. 2015, 11, 284–296. [Google Scholar]
- Wang, B.L.; Chen, Q.F.; Men, W.Q.; Tian, X.B.; Miao, J.N.; He, J.Q.; Zhao, Y.; Li, H.H.; Liu, W.X. Analysis of chromosome compasition of different plant heights. J. Henan Agric. Univ. 2021, 55, 631–638. [Google Scholar]
- Yang, T.; Chen, X.Y.; Sui, J.S.; Wang, W.; Yang, C.M.; Peng, Z.; Geng, G.D.; Zhang, Q.Q.; Zhang, S.Q. Chromosomic analysis of hexaploid triticale by fluorescence in situ hybridization. J. Southwest Univ. (Nat. Sci. Ed.) 2019, 41, 42–48. [Google Scholar]
- Parizipour, M.H.G.; Ramazani, L.; Sardrood, B.P. Temperature affected Transmission, Symptom Development and Accumulation of Wheat Dwarf Virus. Plant Prot. Sci. 2018, 54, 222–233. [Google Scholar] [CrossRef]
- Mishchenko, L.T.; Dunich, A.A.; Mishchenko, I.A.; Dashchenko, A.V.; Kozub, N.O.; Kyslykh, T.M.; Molodchenkova, O.O. Wheat dwarf virus in Ukraine: Occurrence, molecular characterization and impact on the yield. J. Plant Dis. Prot. 2022, 129, 107–116. [Google Scholar] [CrossRef]
- Vacke, J. Wheat dwarf virus disease. Biol. Plant. 1961, 3, 228–233. [Google Scholar] [CrossRef]
- Pfrieme, A.K.; Will, T.; Pillen, K.; Stahl, A. The Past, Present, and Future of Wheat Dwarf Virus Management—A Review. Plants 2023, 12, 3633. [Google Scholar] [CrossRef]
- Wang, X.F.; Liu, Y.; Han, C.G.; Wu, Y.F.; Zhao, Z.H. Present situation and development strategies for the research and control of wheat viral diseases. Plant Prot. 2010, 36, 13–19. [Google Scholar]
- Murray, E.E.; Lotzer, J.; Eberle, M. Codon usage in plant genes. Nucleic Acids Res. 1989, 17, 477–498. [Google Scholar] [CrossRef] [PubMed]
- Quax, T.E.F.; Claassens, N.J.; Söll, D.; Oost, J. Codon bias as a means to fine-tune gene expression. Mol. Cell 2015, 59, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Kurland, C.G. Codon bias and gene expression. FEBS Lett. 1991, 285, 165–169. [Google Scholar] [CrossRef]
- Li, X.Z.; Song, H.; Zhang, Z.H.; Xu, H.F.; Liu, X.; Li, Y.L.; Li, C.J. Analysis of codon usage bias in the genome of Epichloë gansuensis. Acta Prataculturase Sin. 2020, 29, 67–77. [Google Scholar]
- Gao, S.Y.; Li, Y.Y.; Yang, Z.Q.; Dong, K.H.; Xia, F.S. Codon usage bias analysis of the chloroplast genome of Bothriochloa ischaemum. Acta Prataculturase Sin. 2023, 32, 85–95. [Google Scholar]
- Yang, G.F.; Su, K.L.; Zhao, Y.R.; Song, Z.B.; Sun, J. Analysis of codon usage in the chloroplast genome of Medicago truncatula. Acta Prataculturase Sin. 2015, 24, 171–179. [Google Scholar]
- Komar, A.A. The Yin and Yang of codon usage. Hum. Mol. Genet. 2016, 25, R77–R85. [Google Scholar] [CrossRef]
- Schaefer, S. Hepatitis B virus genotypes in Europe. Hepatol. Res. 2007, 37, S20–S26. [Google Scholar] [CrossRef]
- Wu, W.Q.; Hu, Y.; Hu, J.L.; Huang, A.L.; Tu, Z. Codon usage bias of the hepatitis B virus and factors influencing it. J. Pathog. Biol. 2020, 15, 1404–1410. [Google Scholar]
- Meade, J.C.; Shah, P.H.; Lushbaugh, W.B. Trichomonas vaginalis: Analysis of codon usage. Exp. Parasitol. 1997, 87, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Hall, B.D. Codon selection in yeast. J. Biol. Chem. 1982, 257, 3026–3031. [Google Scholar] [CrossRef]
- Biswas, K.K.; Palchoudhury, S.; Chakraborty, P.; Bhattacharyya, U.K.; Ghosh, D.K.; Debnath, P.; Ramadugu, C.; Keremane, M.K.; Khetarpal, R.K.; Lee, R.F. Codon usage bias analysis of Citrus tristeza virus: Higher codon adaptation to Citrus reticulata host. Viruses 2019, 11, 331. [Google Scholar] [CrossRef]
- Sueoka, N. Translation-coupled violation of Parity Rule 2 in human genes is not the cause of heterogeneity of the DNA G+C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef]
- Sueaoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef]
- Lin, Z.C.; Yang, S.H.; Mallavia, L.P. Codon usage and nucleotide composition in Coxiella burnetii. Gene 1997, 198, 171–180. [Google Scholar] [CrossRef]
- Ling, L.Z.; Zhang, S.D.; Yang, T. Analysis of Codon Usage Bias in Chloroplast Genomes of Dryas octopetala var. asiatica (Rosaceae). Genes 2024, 15, 899. [Google Scholar] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Chamary, J.V.; Parmley, J.L.; Hurst, L.D. Hearing silence: Non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 2006, 7, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.K.; Xu, M.Z.; Yang, J.R.; Lu, J. Genome-wide impact of codon usage bias on translation optimization in Drosophila melanogaster. Nat. Commun. 2024, 15, 8329. [Google Scholar] [CrossRef]
- Chen, S.L.; Lee, W.; Hottes, A.K.; Shapiro, L.; Mcadams, H.H. Codon usage between genomes is constrained by genome-wide mutational processes. Proc. Natl. Acad. Sci. USA 2004, 101, 3480–3485. [Google Scholar] [CrossRef]
- Galtier, N.; Piganeau, G.; Mouchiroud, D.; Duret, L. GC-content evolution in mammalian genomes: The biased gene conversion hypothesis. Genetics 2001, 159, 907–911. [Google Scholar] [CrossRef]
- Lin, Y.S.; Byrnes, J.K.; Hwang, J.K.; Li, W.H. Codon-usage bias versus gene conversion in the evolution of yeast duplicate genes. Proc. Natl. Acad. Sci. USA 2006, 103, 14412–14416. [Google Scholar] [CrossRef]
- Sharp, P.M.; Averof, M.; Lloyd, A.T.; Matassi, G.; Peden, J.F. DNA sequence evolution: The sounds of silence. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1995, 349, 241–247. [Google Scholar]
- Duret, L. Evolution of synonymous codon usage in metazoans. Curr. Opin. Genet. Dev. 2002, 12, 640–649. [Google Scholar] [CrossRef]
- Lu, J.; Wu, C.I. Weak selection revealed by the whole-genome comparison of the X chromosome and autosomes of human and chimpanzee. Proc. Natl. Acad. Sci. USA 2005, 102, 4063–4067. [Google Scholar] [CrossRef]
- Shen, X.K.; Song, S.L.; Li, C.; Zhang, J.Z. Synonymous mutations in representative yeast genes are mostly strongly non-neutral. Nature 2022, 606, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Akashi, H.; Eyre-Walker, A. Translational selection and molecular evolution. Curr. Opin. Genet. Dev. 1998, 8, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 1977, 267, 275–276. [Google Scholar] [CrossRef]
- Shah, P.; Gilchrist, M.A. Explaining complex codon usage patterns with selection for translational efficiency, mutation bias, and genetic drift. Proc. Natl. Acad. Sci. USA 2011, 108, 10231–10236. [Google Scholar] [CrossRef]
- Wong, E.H.; Smith, D.K.; Rabadan, R.; Poon, P.L.L. Codon usage bias and the evolution of influenza A viruses. Codon Usage Biases of Influenza Virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef]
- Xu, X.; Ding, T.Y.; Yu, W.; Jiang, Y.H.; Quan, Y.P. Analysis of codon usage bias in porcine circovirus gene. Chin. J. Prev. Vet. Med. 2018, 40, 471–475. [Google Scholar]
- Zhan, J.F.; Luo, H.; Hu, S.S.; Wu, Q.; Yao, H.P. Genome codon bias analysis of Zika virus. Genom. Appl. Biol. 2017, 36, 426–432. [Google Scholar]
- Wang, G.C.; Qiao, J.; Meng, Q.L.; Liu, Y.C.; Yang, H.B.; He, Z.H.; Peng, Y.L.; Xie, K.; Chen, C.F. Analysis of synonymous codon preferences of poly protease genes in three viruses of Flaviviridae Pestivirus genus. J. Northwest A&F Univ. (Nat. Sci. Ed.) 2015, 43, 7–13. [Google Scholar]
- Shi, S.L.; Li, M.M.; Jiang, Y.R.; Yang, R.S.; Qin, L. Analysis on codon and codon-pair usage pattern in five Silkworm viruses. Sci. Seric. 2012, 38, 0273–0280. [Google Scholar]
- Rahman, S.U.; Nawaz, S.; Ullah, S.; Rahman, I.U.; Haq, M.I.U.; Khan, M.A.; Al-Ghamdi, A.A.; Al-Hemaid, F.M.; Elshikh, M.S.; Aljowaie, R.M.; et al. Study of codon usage patterns and influencing factors in rice yellow mottle virus based on coding sequence data. Agronomy 2022, 12, 1990. [Google Scholar] [CrossRef]
- Yuan, Y.Y.; Huang, S.H.; Wang, C.K.; Zhi, H.J. Analysis on codon usage and evolution of Soybean Mosaic virus. Soybean Sci. 2014, 33, 801–807. [Google Scholar]
- Simón, D.; Cristina, J.; Musto, H. Nucleotide composition and codon usage across viruses and their respective hosts. Front. Microbiol. 2021, 12, 646300. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Q.; Zhao, J.H.; Chen, Q.; Zhang, Z.H.; Li, J.; Guo, Z.X.; Xie, Q.; Ding, S.W.; Guo, H.S. DNA geminivirus infection induces an imprinted E3 ligase gene to epigenetically activate viral gene transcription. Plant Cell 2020, 32, 3256–3272. [Google Scholar] [CrossRef]
- Hu, T.; Li, C.; Liu, H.; Su, C.; Wang, Y.; Li, F.; Zhou, X. Geminivirus βV1 protein activates bZIP17/28-mediated UPR signaling to facilitate viral pathogenicity but its activity is attenuated by autophagic degradation in plants. Plant Commun. 2024, 26, 101198. [Google Scholar] [CrossRef]
- Boulton, M.I.; Pallaghy, C.K.; Chatani, M.; MacFarlane, S.; Davies, J.W. Replication of maize streak virus mutants in maize protoplasts: Evidence for a movement protein. Virology 1993, 192, 85–93. [Google Scholar] [CrossRef]
- Kotlizky, G.; Boulton, M.I.; Pitaksutheepong, C.; Davies, J.W.; Epel, B.L. Intracellular and intercellular movement of maize streak geminivirus V1 and V2 proteins transiently expressed as green fluorescent protein fusions. Virology 2000, 274, 32–38. [Google Scholar] [CrossRef]
- Chen, F.; Wu, P.; Deng, S.Y.; Zhang, H.; Hou, Y.T.; Hu, Z.; Zhang, J.Z.; Chen, X.S.; Yang, J.R. Dissimilation of synonymous codon usage bias in virus–host coevolution due to translational selection. Nat. Ecol. Evol. 2020, 4, 589–600. [Google Scholar] [CrossRef]
- Hao, J.; Liang, Y.Y.; Wang, T.; Su, Y.J. Correlations of gene expression, codon usage bias, and evolutionary rates of the mitochondrial genome show tissue differentiation in Ophioglossum vulgatum. BMC Plant Biol. 2025, 25, 134. [Google Scholar] [CrossRef]
- Stanković, I.; Zečević, K.; Ristić, D.; Vučurović, I.; Krstić, B. Molecular characterization of wheat dwarf virus isolates from Serbia based on complete genome sequences. Front. Microbiol. 2024, 15, 1469453. [Google Scholar] [CrossRef]
- Pintó, R.M.; Burns, C.C.; Moratorio, G. Codon usage and dinucleotide composition of virus genomes: From the virus-host interaction to the development of vaccines. Front. Microbiol. 2021, 12, 791750. [Google Scholar] [CrossRef] [PubMed]
- Mordstein, C.; Cano, L.; Morales, A.C.; Young, B.; Ho, A.T.; Rice, A.M.; Liss, M.; Hurst, L.D.; Kudla, G. Transcription, mRNA export, and immune evasion shape the codon usage of viruses. Genome Biol. Evol. 2021, 13, evab106. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Ni, Z.; Lan, T.; Ping, P.; Tang, J.; Yu, Z.; Hutvagner, G.; Li, J. Predicting viral host codon fitness and path shifting through tree-based learning on codon usage biases and genomic characteristics. Sci. Rep. 2025, 15, 12251. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host | Accession Number | Genome Length | Designation |
|---|---|---|---|
| Triticum aestivum | HF968638.1 | 2750 nt | WDVTA |
| Hordeum vulgare | HG422315.1 | 2735 nt | WDVHV |
| Lolium perenne | HG422316.1 | 2750 nt | WDVLP |
| Secale cereale | HG422318.1 | 2749 nt | WDVSC |
| Psammotettix alienus | AM040733.1 | 2750 nt | WDVPA |
| Avena sativa | AM296018.1 | 2734 nt | WDVAS |
| Triticum spelta | KJ473696.1 | 2748 nt | WDVTS |
| Hordeum vulgare var. coeleste | KJ536149.1 | 2750 nt | WDVHC |
| × Triticosecale | KJ473702.1 | 2750 nt | WDVT_117 |
| × Triticosecale | KJ473703.1 | 2748 nt | WDVT_118 |
| Gene | Production | Length (nt) |
|---|---|---|
| V1 | movement protein | 273 |
| V2 | coat protein | 783 |
| Rep | replication-associated protein | 1056 |
| RepA | replication-associated protein A | 795 |
| Gene | WDVT_117 | WDVT_118 | ||
|---|---|---|---|---|
| CAI | CBI | CAI | CBI | |
| V1 | 0.155 | −0.026 | 0.145 | −0.042 |
| V2 | 0.261 | 0.035 | 0.262 | 0.05 |
| Rep | 0.267 | 0.1 | 0.269 | 0.106 |
| RepA | 0.266 | 0.098 | 0.269 | 0.111 |
| Average | 0.237 | 0.052 | 0.236 | 0.056 |
| Amino Acid | Codon | WDVT_117 | WDVT_118 | ||||
|---|---|---|---|---|---|---|---|
| hRSCU | lRSCU | ΔRSCU | hRSCU | lRSCU | ΔRSCU | ||
| Ala | GCA | 0.5 | 0.8889 | −0.3889 | 0.5 | 0.7059 | −0.2059 |
| Ala | GCC | 2 | 0.8889 | 1.1111 | 2 | 0.9412 | 1.0588 |
| Ala | GCG | 0.5 | 0.6667 | −0.1667 | 0.5 | 0.9412 | −0.4412 |
| Ala | GCU | 1 | 1.5556 | −0.5556 | 1 | 1.4118 | −0.4118 |
| Cys | UGC | 0 | 1.2 | −1.2 | 0 | 1.2 | −1.2 |
| Cys | UGU | 2 | 0.8 | 1.2 | 2 | 0.8 | 1.2 |
| Asp | GAC | 1.5 | 0.875 | 0.625 | 1.5 | 0.875 | 0.625 |
| Asp | GAU | 0.5 | 1.125 | −0.625 | 0.5 | 1.125 | −0.625 |
| Glu | GAA | 0.6667 | 1.1 | −0.4333 | 0.6667 | 1.2 | −0.5333 |
| Glu | GAG | 1.3333 | 0.9 | 0.4333 | 1.3333 | 0.8 | 0.5333 |
| Phe | UUC | 1 | 1.0909 | −0.0909 | 1 | 1.0909 | −0.0909 |
| Phe | UUU | 1 | 0.9091 | 0.0909 | 1 | 0.9091 | 0.0909 |
| Gly | GGA | 0.5714 | 1.5 | −0.9286 | 1.3333 | 1.5 | −0.1667 |
| Gly | GGC | 0.5714 | 1 | −0.4286 | 0 | 1 | −1 |
| Gly | GGG | 0.5714 | 0.5 | 0.0714 | 0.6667 | 0.5 | 0.1667 |
| Gly | GGU | 2.2857 | 1 | 1.2857 | 2 | 1 | 1 |
| His | CAC | 0 | 0.75 | −0.75 | 0 | 0.6667 | −0.6667 |
| His | CAU | 0 | 1.25 | −1.25 | 0 | 1.3333 | −1.3333 |
| Ile | AUA | 1.8 | 0.75 | 1.05 | 1.8 | 0.75 | 1.05 |
| Ile | AUC | 0 | 1.5 | −1.5 | 0 | 1.5 | −1.5 |
| Ile | AUU | 1.2 | 0.75 | 0.45 | 1.2 | 0.75 | 0.45 |
| Lys | AAA | 0.5 | 1.3333 | −0.8333 | 0.5 | 1.3333 | −0.8333 |
| Lys | AAG | 1.5 | 0.6667 | 0.8333 | 1.5 | 0.6667 | 0.8333 |
| Leu | CUA | 0.6 | 0.5 | 0.1 | 0.6 | 0.5 | 0.1 |
| Leu | CUC | 0.6 | 3 | −2.4 | 0.6 | 3 | −2.4 |
| Leu | CUG | 0.6 | 0.25 | 0.35 | 1.2 | 0.25 | 0.95 |
| Leu | CUU | 1.2 | 1.5 | −0.3 | 1.2 | 1.5 | −0.3 |
| Leu | UUA | 1.8 | 0.25 | 1.55 | 1.8 | 0.25 | 1.55 |
| Leu | UUG | 1.2 | 0.5 | 0.7 | 0.6 | 0.5 | 0.1 |
| Met | AUG | 1 | 1 | 0 | 1 | 1 | 0 |
| Asn | AAC | 0 | 1 | −1 | 0 | 1.1111 | −1.1111 |
| Asn | AAU | 2 | 1 | 1 | 2 | 0.8889 | 1.1111 |
| Pro | CCA | 0 | 2 | −2 | 0 | 2 | −2 |
| Pro | CCC | 1.1429 | 0.6667 | 0.4762 | 1.1429 | 0.6667 | 0.4762 |
| Pro | CCG | 1.7143 | 0.2222 | 1.4921 | 1.7143 | 0.2222 | 1.4921 |
| Pro | CCU | 1.1429 | 1.1111 | 0.0318 | 1.1429 | 1.1111 | 0.0318 |
| Gln | CAA | 1.3333 | 1.2727 | 0.0606 | 1.3333 | 1.2727 | 0.0606 |
| Gln | CAG | 0.6667 | 0.7273 | −0.0606 | 0.6667 | 0.7273 | −0.0606 |
| Arg | AGA | 1 | 0.75 | 0.25 | 0.8571 | 0.75 | 0.1071 |
| Arg | AGG | 3 | 0.75 | 2.25 | 2.5714 | 0.75 | 1.8214 |
| Arg | CGA | 1 | 0 | 1 | 0.8571 | 0 | 0.8571 |
| Arg | CGC | 0 | 1.125 | −1.125 | 0 | 1.125 | −1.125 |
| Arg | CGG | 1 | 0.375 | 0.625 | 1.7143 | 0.375 | 1.3393 |
| Arg | CGU | 0 | 3 | −3 | 0 | 3 | −3 |
| Ser | AGC | 0 | 0.6207 | −0.6207 | 0 | 0.6207 | −0.6207 |
| Ser | AGU | 0 | 0.6207 | −0.6207 | 0 | 0.4138 | −0.4138 |
| Ser | UCA | 0 | 1.0345 | −1.0345 | 0 | 1.2414 | −1.2414 |
| Ser | UCC | 6 | 1.8621 | 4.1379 | 6 | 1.6552 | 4.3448 |
| Ser | UCG | 0 | 0 | 0 | 0 | 0 | 0 |
| Ser | UCU | 0 | 1.8621 | −1.8621 | 0 | 2.069 | −2.069 |
| Thr | ACA | 1.3333 | 1.4118 | −0.0785 | 1.3333 | 1.3333 | 0 |
| Thr | ACC | 2 | 0.9412 | 1.0588 | 2 | 0.8889 | 1.1111 |
| Thr | ACG | 0 | 0.4706 | −0.4706 | 0 | 0.4444 | −0.4444 |
| Thr | ACU | 0.6667 | 1.1765 | −0.5098 | 0.6667 | 1.3333 | −0.6666 |
| Val | GUA | 0.8 | 0.3636 | 0.4364 | 0.8 | 0.3636 | 0.4364 |
| Val | GUC | 0 | 0.3636 | −0.3636 | 0 | 0.3636 | −0.3636 |
| Val | GUG | 1.6 | 0.7273 | 0.8727 | 1.6 | 0.7273 | 0.8727 |
| Val | GUU | 1.6 | 2.5455 | −0.9455 | 1.6 | 2.5455 | −0.9455 |
| Trp | UGG | 1 | 1 | 0 | 1 | 1 | 0 |
| Tyr | UAC | 1.6 | 1.1667 | 0.4333 | 1.6 | 1.1667 | 0.4333 |
| Tyr | UAU | 0.4 | 0.8333 | −0.4333 | 0.4 | 0.8333 | −0.4333 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Lu, X.; Dong, J.; Liu, J.; Guo, B.; Zhang, C.; Liu, J.; Wang, H. Natural Selection Shaped Codon Usage Patterns in Wheat Dwarf Virus in Triticale. Biology 2025, 14, 524. https://doi.org/10.3390/biology14050524
Wang J, Lu X, Dong J, Liu J, Guo B, Zhang C, Liu J, Wang H. Natural Selection Shaped Codon Usage Patterns in Wheat Dwarf Virus in Triticale. Biology. 2025; 14(5):524. https://doi.org/10.3390/biology14050524
Chicago/Turabian StyleWang, Jiuli, Xinhang Lu, Jiaying Dong, Jiaqian Liu, Borui Guo, Chen Zhang, Jing Liu, and Hongxia Wang. 2025. "Natural Selection Shaped Codon Usage Patterns in Wheat Dwarf Virus in Triticale" Biology 14, no. 5: 524. https://doi.org/10.3390/biology14050524
APA StyleWang, J., Lu, X., Dong, J., Liu, J., Guo, B., Zhang, C., Liu, J., & Wang, H. (2025). Natural Selection Shaped Codon Usage Patterns in Wheat Dwarf Virus in Triticale. Biology, 14(5), 524. https://doi.org/10.3390/biology14050524