
Macrogenomics Reveals Effects on Marine Microbial Communities during Oplegnathus punctatus Enclosure Farming

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area and Sample Collection
2.2. DNA Extraction and Metagenomic Sequencing
2.3. Read-Based Phylogenetic Annotation
2.4. Metagenomic De Novo Assembly, Gene Prediction, and Gene Abundance
2.5. Gene Function Annotation Based on Unique Genes
2.6. Data Statistics and Visualization
3. Results
3.1. Environmental Factors
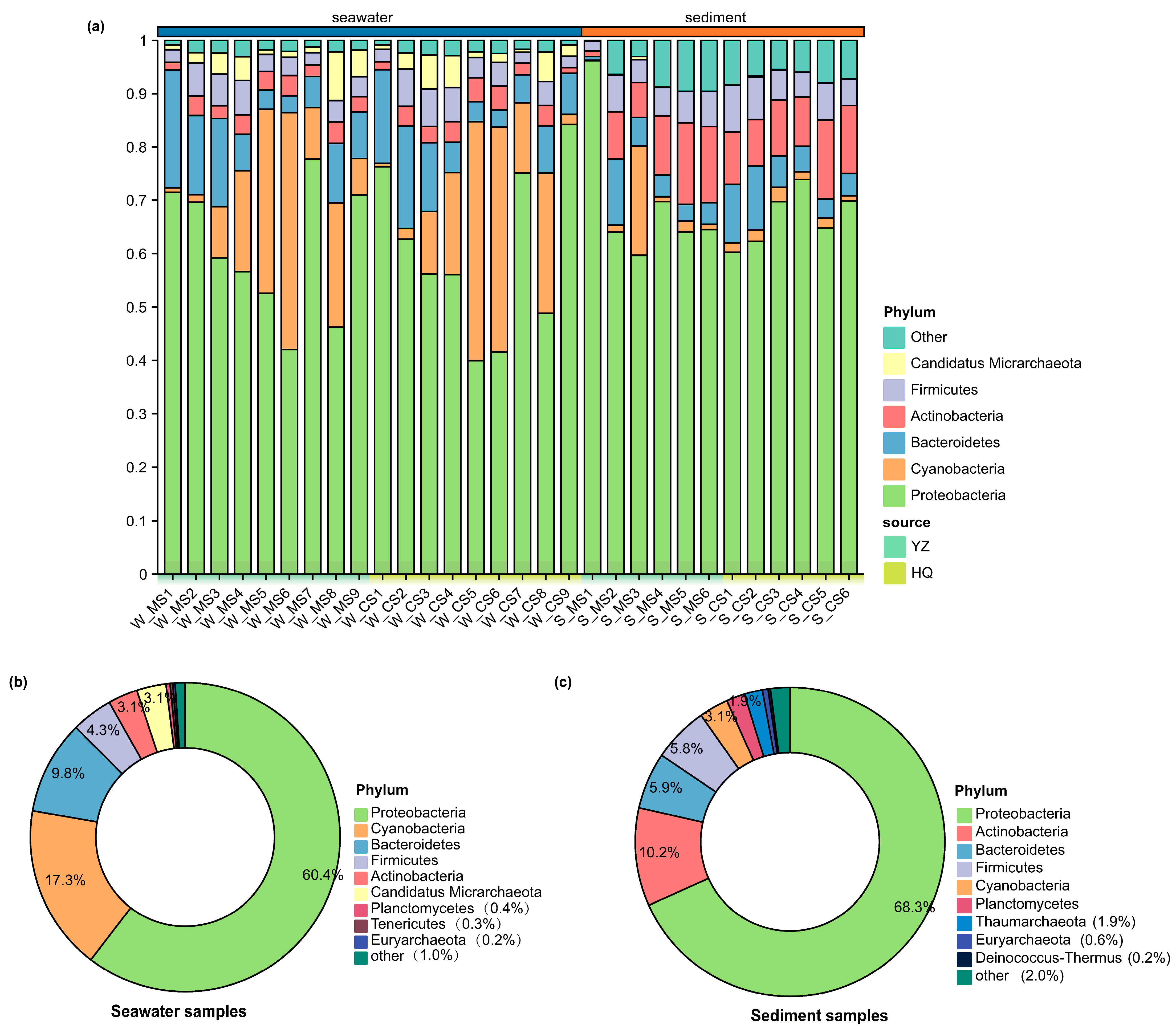
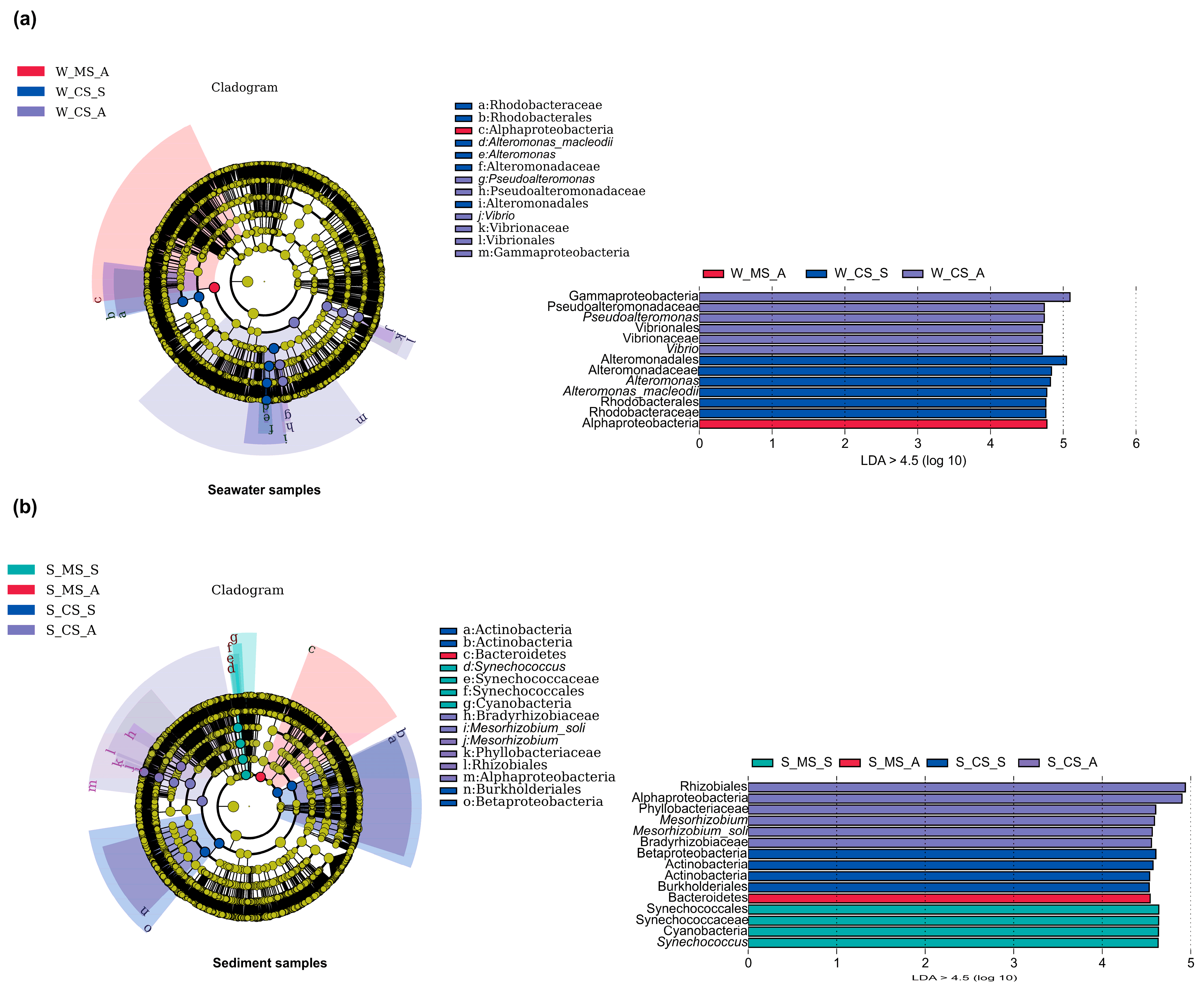
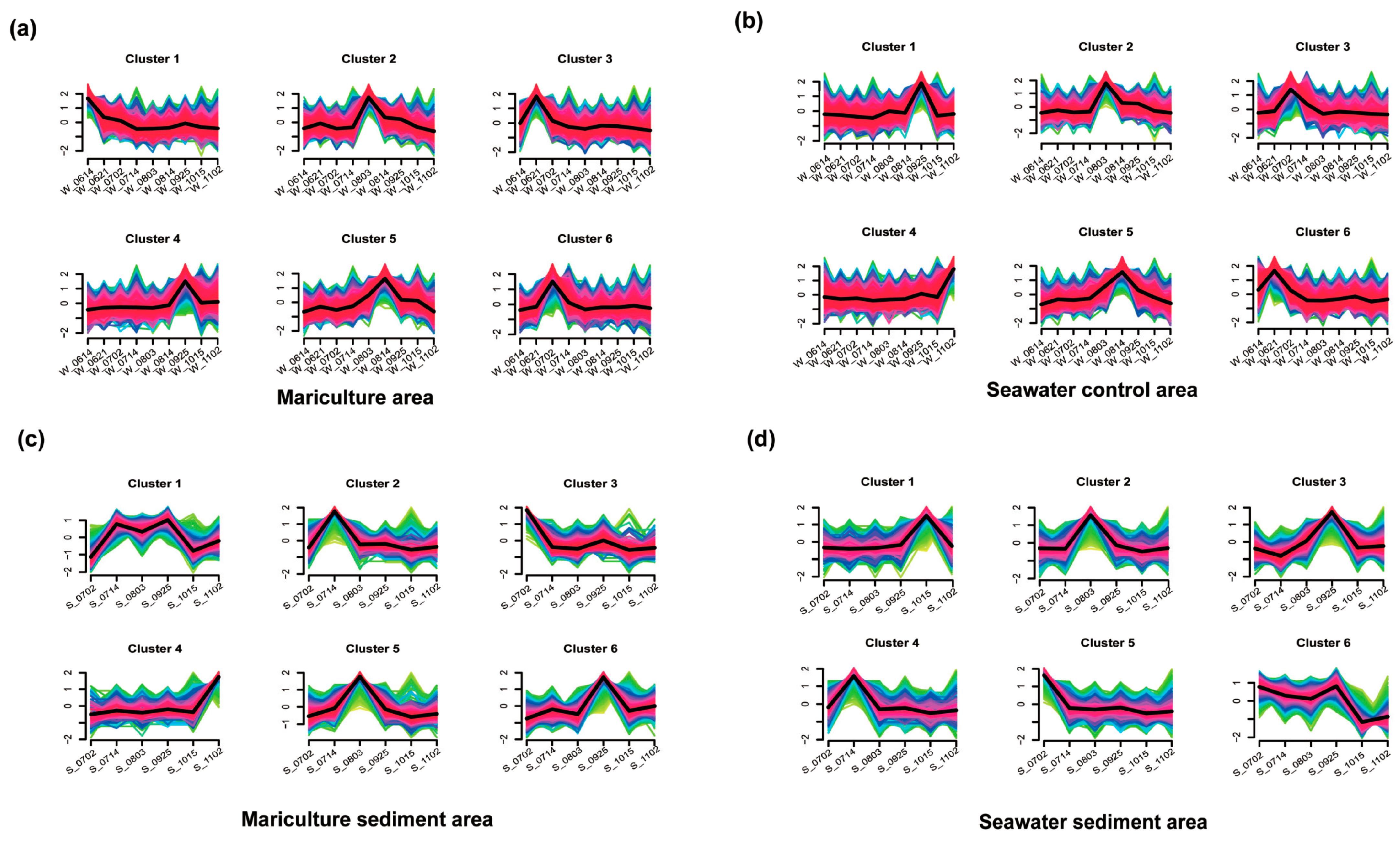
3.2. Microbial Community Composition
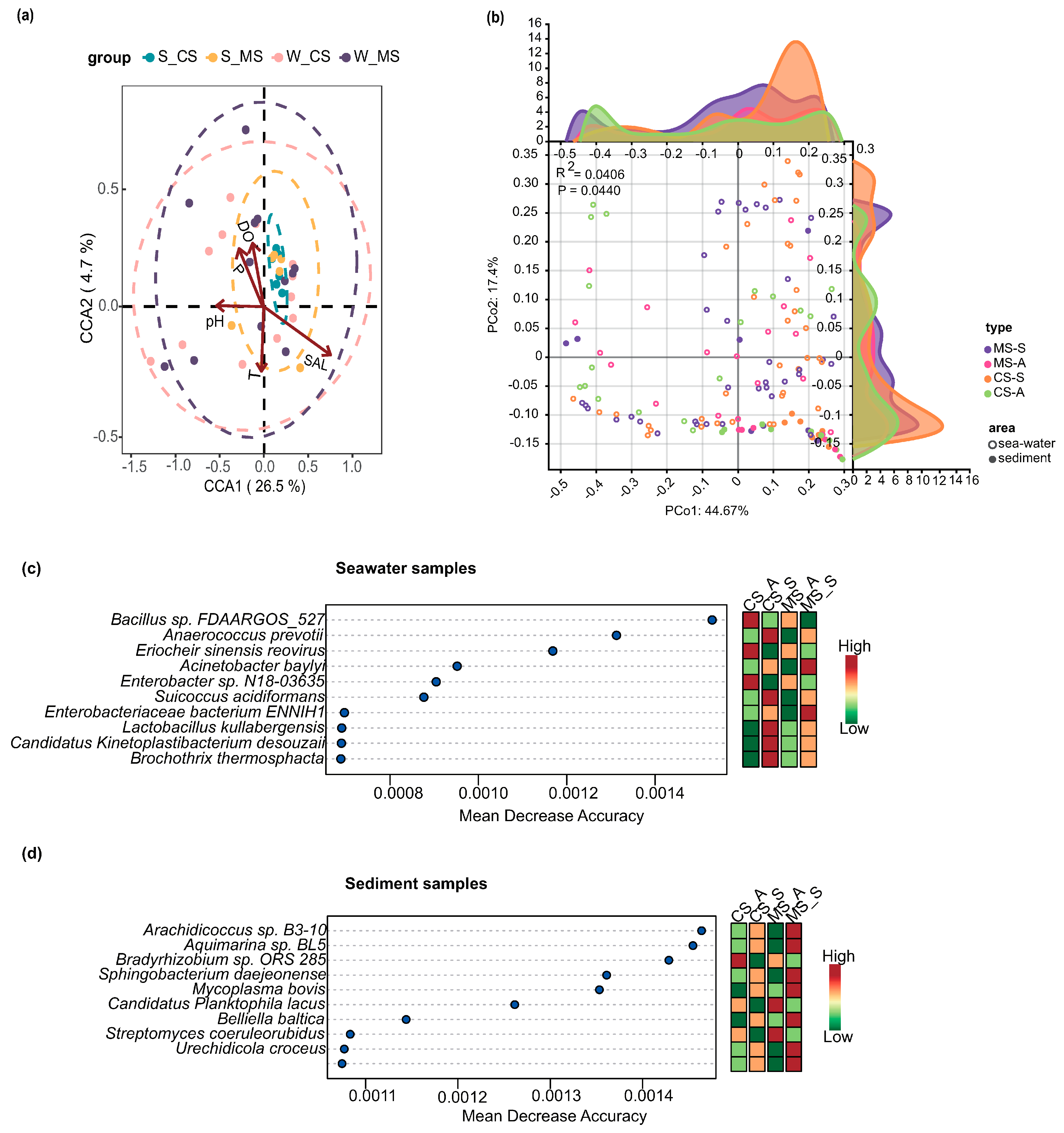
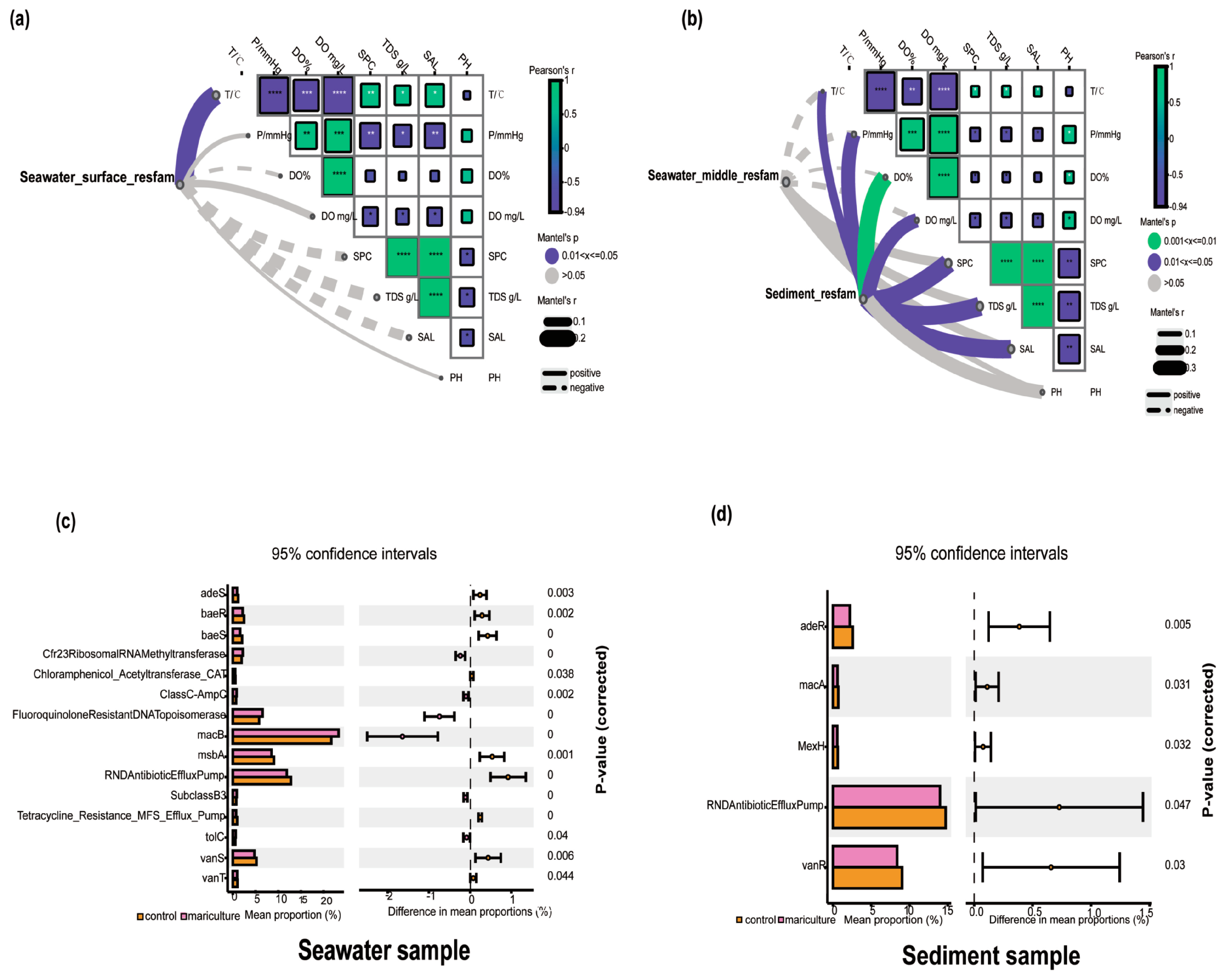
3.3. Relationships between Bacteria and Environmental Factors
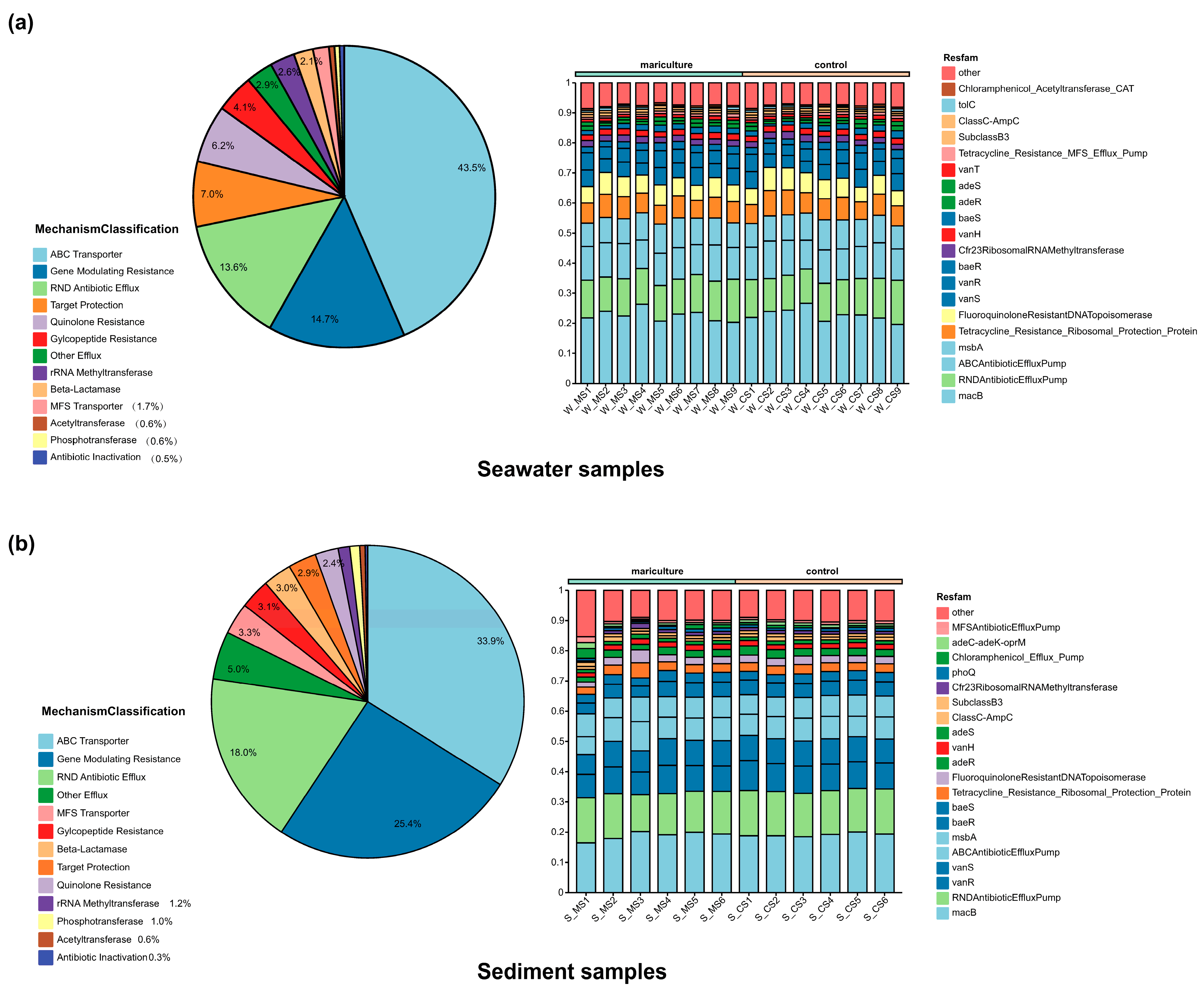
3.4. Occurrence of Antibiotics in Mariculture
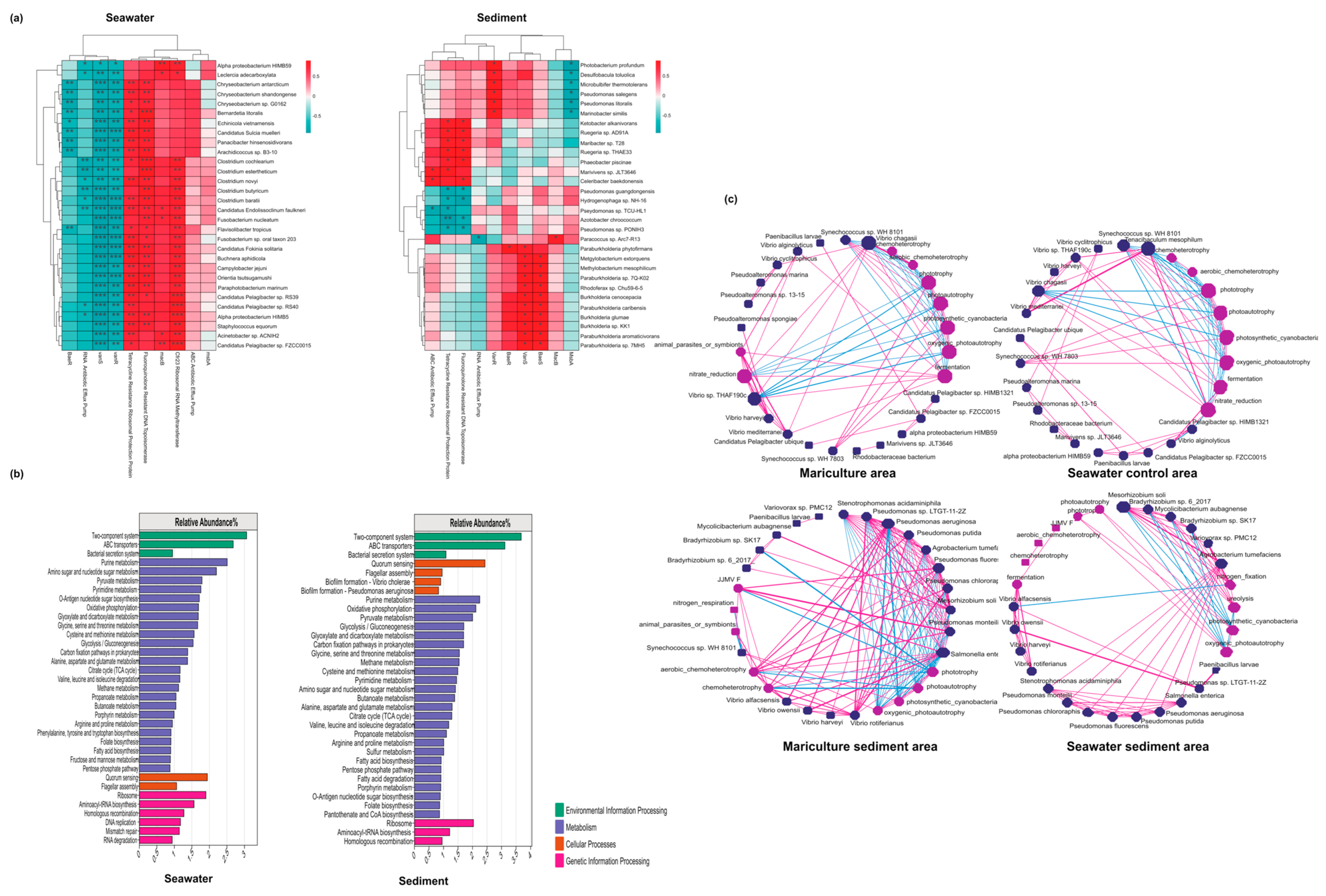
3.5. Associations between Antibiotic Resistance Genes and Microbiomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FAO. The State of World Fisheries and Aquaculture 2022. Towards Blue Transformation; FAO: Rome, Italy, 2022. [Google Scholar] [CrossRef]
- Liu, H.; Su, J. Vulnerability of China’s nearshore ecosystems under intensive mariculture development. Environ. Sci. Pollut. Res. Int. 2017, 24, 8957–8966. [Google Scholar] [CrossRef] [PubMed]
- Naylor, R.L.; Goldburg, R.J.; Primavera, J.H.; Kautsky, N.; Beveridge, M.C.M.; Clay, J.; Folke, C.; Lubchenco, J.; Mooney, H.; Troell, M. Effect of aquaculture on world fish supplies. Nature 2000, 405, 1017–1024. [Google Scholar] [CrossRef]
- Liu, X.; Steele, J.C.; Meng, X.Z. Usage, residue, and human health risk of antibiotics in Chinese aquaculture: A review. Environ. Pollut. 2017, 223, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Guardone, L.; Tinacci, L.; Armani, A.; Trevisani, M. Residues of veterinary drugs in fish and fish products: An analysis of RASFF data over the last 20 years. Food Control 2022, 135, 108780. [Google Scholar] [CrossRef]
- Han, Q.F.; Zhang, X.R.; Xu, X.Y.; Wang, X.L.; Yuan, X.Z.; Ding, Z.J.; Zhao, S.; Wang, S.G. Antibiotics in marine aquaculture farms surrounding Laizhou Bay, Bohai Sea: Distribution characteristics considering various culture modes and organism species. Sci. Total Environ. 2021, 760, 143863. [Google Scholar] [CrossRef] [PubMed]
- Binh, V.N.; Dang, N.; Anh, N.T.K.; Ky, L.X.; Thai, P.K. Antibiotics in the aquatic environment of Vietnam: Sources, concentrations, risk and control strategy. Chemosphere 2018, 197, 438–450. [Google Scholar] [CrossRef]
- Lennon, J.T.; Jones, S.E. Microbial seed banks: The ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 2011, 9, 119–130. [Google Scholar] [CrossRef]
- Zheng, X.; Xu, K.; Naoum, J.; Lian, Y.; Wu, B.; He, Z.; Yan, Q. Deciphering microeukaryotic–bacterial co-occurrence networks in coastal aquaculture ponds. Mar. Life Sci. Technol. 2023, 5, 44–55. [Google Scholar] [CrossRef]
- Pierella Karlusich, J.J.; Ibarbalz, F.M.; Bowler, C. Phytoplankton in the Tara Ocean. Ann. Rev. Mar. Sci. 2020, 12, 233–265. [Google Scholar] [CrossRef]
- Wu, J.; Su, Y.; Deng, Y.; Guo, Z.; Cheng, C.; Ma, H.; Liu, G.; Xu, L.; Feng, J. Spatial and temporal variation of antibiotic resistance in marine fish cage-culture area of Guangdong, China. Environ. Pollut. 2019, 246, 463–471. [Google Scholar] [CrossRef]
- Choi, S.; Sim, W.; Jang, D.; Yoon, Y.; Ryu, J.; Oh, J.; Woo, J.-S.; Kim, Y.M.; Lee, Y. Antibiotics in coastal aquaculture waters: Occurrence and elimination efficiency in oxidative water treatment processes. J. Hazard. Mater. 2020, 396, 122585. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.A.; Corbett, D.R.; Fitzgerald, A.M.; Lemley, D.A.; Quigg, A.; Steppe, C.N. Impacts of urbanization and development on estuarine ecosystems and water quality. Estuar. Coast. 2019, 42, 1821–1838. [Google Scholar] [CrossRef]
- Grigorakis, K.; Rigos, G. Aquaculture effects on environmental and public welfare—The case of Mediterranean mariculture. Chemosphere 2011, 85, 899–919. [Google Scholar] [CrossRef]
- He, L.-X.; He, L.-Y.; Gao, F.-Z.; Wu, D.-L.; Ye, P.; Cheng, Y.-X.; Chen, Z.-Y.; Hu, L.-X.; Liu, Y.-S.; Chen, J.; et al. Antibiotics, antibiotic resistance genes and microbial community in grouper mariculture. Sci. Total Environ. 2022, 808, 152042. [Google Scholar] [CrossRef] [PubMed]
- Elgendy, M.Y.; Ali, S.E.; Abbas, W.T.; Algammal, A.M.; Abdelsalam, M. The role of marine pollution on the emergence of fish bacterial diseases. Chemosphere 2023, 344, 140366. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.Z. The application status and strategy of fishery medicine in China. J. Shanghai Fish. Univ. 2007, 374–380. Available online: http://shhydxxb.ijournals.cn/shhyen/article/abstract/20070484 (accessed on 13 August 2024).
- Han, Q.F.; Song, C.; Sun, X.; Zhao, S.; Wang, S.G. Spatiotemporal distribution, source apportionment and combined pollution of antibiotics in natural waters adjacent to mariculture areas in the Laizhou Bay, Bohai Sea. Chemosphere 2021, 279, 130381. [Google Scholar] [CrossRef] [PubMed]
- Kemper, N. Veterinary antibiotics in the aquatic and terrestrial environment. Ecol. Indic. 2008, 8, 1–13. [Google Scholar] [CrossRef]
- Lu, S.; Lin, C.; Lei, K.; Xin, M.; Gu, X.; Lian, M.; Wang, B.; Liu, X.; Ouyang, W.; He, M. Profiling of the spatiotemporal distribution, risks, and prioritization of antibiotics in the waters of Laizhou Bay, northern China. J. Hazard. Mater. 2022, 424 Pt B, 127487. [Google Scholar] [CrossRef]
- Lulijwa, R.; Rupia, E.J.; Alfaro, A.C. Antibiotic use in aquaculture, policies and regulation, health and environmental risks: A review of the top 15 major producers. Rev. Aquac. 2020, 12, 640–663. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, Y.; Wu, J. Continental-scale spatio-temporal distribution of antibiotic resistance genes in coastal waters along coastline of China. Chemosphere 2020, 247, 125908. [Google Scholar] [CrossRef] [PubMed]
- Larsson, D.G.J.; Flach, C.F. Antibiotic resistance in the environment. Nat. Rev. Microbiol. 2022, 20, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Hu, J.; Magnuson, J.T.; Greer, J.; Yang, M.; Chen, Q.; Fang, M.; Zheng, C.; Schlenk, D. Evidence linking exposure of fish primary macrophages to antibiotics activates the NF-kB pathway. Environ. Int. 2020, 138, 105624. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhou, Y.; Zhu, Q.; Xia, B.; Ma, W.; Xiao, X.; Shi, H.; Zhang, Y. Determination of antibiotic concentration in meconium and its association with fetal growth and development. Environ. Int. 2019, 123, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Gaw, S.; Thomas, K.V.; Hutchinson, T.H. Sources, impacts and trends of pharmaceuticals in the marine and coastal environment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130572. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Chen, Q.; Li, Y.; Liu, Y.; Zhang, Y.; Huang, Y.; Zhu, L. Status of antibiotic residues and detection techniques used in Chinese milk: A systematic review based on cross-sectional surveillance data. Food Res. Int. 2021, 147, 110450. [Google Scholar] [CrossRef]
- Naylor, R.L.; Hardy, R.W.; Buschmann, A.H.; Bush, S.R.; Cao, L.; Klinger, D.H.; Little, D.C.; Lubchenco, J.; Shumway, S.E.; Troell, M. A 20-year retrospective review of global aquaculture. Nature 2021, 591, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.C.; Pan, L.Q.; Liu, T.; Li, Z.Y. Source risk, ecological risk, and bioeffect assessment for polycyclic aromatic hydrocarbons (PAHs) in Laizhou Bay and Jiaozhou Bay of Shandong Peninsula, China. Environ. Sci. Pollut. Res. Int. 2022, 29, 56705–56726. [Google Scholar] [CrossRef]
- Zhang, M.; Lu, Q.Y.; Wang, D.W.; Ding, D.; Cui, Z.; Shi, H. Spatiotemporal evolution of nutrients and the influencing factors in Laizhou Bay over the past 40 years. Mar. Pollut. Bull. 2022, 184, 114186. [Google Scholar] [CrossRef]
- Wei, Y.; Cui, H.; Hu, Q.; Bai, Y.; Qu, K.; Sun, J.; Cui, Z. Eutrophication status assessment in the Laizhou Bay, Bohai Sea: Further evidence for the ecosystem degradation. Mar. Pollut. Bull. 2022, 181, 113867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fu, H.; Lou, H.; Sun, X.; Zhang, D.; Sun, P.; Wang, X.; Li, Y.; Lu, J.; Bao, M. Assessment of eutrophication from Xiaoqing River estuary to Laizhou Bay: Further warning of ecosystem degradation in typically polluted estuary. Mar. Pollut. Bull. 2023, 193, 115209. [Google Scholar] [CrossRef]
- Xian, S.J.; Xiu, J.S.; Xian, S.L.; Jun, W.; Yi, C.; Tao, Z. Long-term changes in the fishery ecosystem structure of Laizhou Bay, China. Sci. China Earth 2013, 56, 366–374. [Google Scholar] [CrossRef]
- Wang, F.L.; Gao, Y.T.; Gao, Y.H.; Li, M.Y.; Zhang, B.Z.; Guan, C.T.; Jia, Y.D. Preliminary study on land-sea alternate aquaculture of spotted knifejaw Oplegnathus punctatus. Fish. Mod. 2022, 7, 8–14. [Google Scholar] [CrossRef]
- Fu, L.M.; Niu, B.F.; Zhu, Z.W.; Wu, S.T.; Li, W.Z. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Sichert, A.; Corzett, C.H.; Schechter, M.S.; Unfried, F.; Markert, S.; Becher, D.; Fernandez-Guerra, A.; Liebeke, M.; Schweder, T.; Polz, M.F.; et al. Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan. Nat. Microbiol. 2020, 5, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Han, T.; Xu, S.; Huang, H.; Qi, Z.; Zhu, Q. Bacterial community responses to the redox profile changes of mariculture sediment. Mar. Pollut. Bull. 2021, 166, 112250. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Xue, J.Q.; Jia, S.; Li, Y.Z.; Ke, Y. Microbial structure and function of activated sludge in landfill leachate treatment plant. Acta Nat. Sci. 2021, 57, 927–937. [Google Scholar] [CrossRef]
- Barczynska, R.; Kapusniak, J.; Litwin, M.; Slizewska, K.; Szalecki, M. Dextrins from maize starch as substances activating the growth of bacteroidetes and actinobacteria simultaneously inhibiting the growth of Firmicutes, responsible for the occurrence of obesity. Plant Foods Hum. Nutr. 2016, 71, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Bo, Z.; Xiang, Y.X.; Liang, Z. Structure and function of the microbial consortia of activated sludge in typical municipal wastewater treatment plants in winter. Sci. Rep. 2017, 7, 17930. [Google Scholar] [CrossRef]
- Fang, D.; Zhao, G.; Xu, X.; Zhang, Q.; Shen, Q.; Fang, Z.; Huang, L.; Ji, F. Microbial community structures and functions of wastewater treatment systems in plateau and cold regions. Bioresour. Technol. 2018, 249, 684–693. [Google Scholar] [CrossRef]
- Gupta, A.; Sainis, J.K.; Bhagwat, S.G.; Chittela, R.K. Modulation of photosynthesis in Synechocystis and Synechococcus grown with chromium (VI). J. Biosci. 2021, 46, 1. [Google Scholar] [CrossRef]
- Marston, M.F.; Polson, S.W. Whole-Genome Sequence of the Cyanobacterium Synechococcus sp. Strain WH 8101. Microbiol. Resour. Announc. 2020, 9, 10–1128. [Google Scholar] [CrossRef]
- Kurmayer, R.; Deng, L.; Entfellner, E. Role of toxic and bioactive secondary metabolites in colonization and bloom formation by filamentous cyanobacteria Planktothrix. Harmful Algae 2016, 54, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Pan, L.; He, Z.; Zhang, M. Identification, interactions, nitrogen removal pathways and performances of culturable heterotrophic nitrification-aerobic denitrification bacteria from mariculture water by using cell culture and metagenomics. Sci. Total Environ. 2020, 732, 139268. [Google Scholar] [CrossRef]
- Calcio Gaudino, E.; Canova, E.; Liu, P.; Wu, Z.; Cravotto, G. Degradation of antibiotics in wastewater: New advances in cavitational treatments. Molecules 2021, 26, 617. [Google Scholar] [CrossRef]
- Watanabe, N.; Bergamaschi, B.A.; Loftin, K.A.; Meyer, M.T.; Harter, T. Use and environmental occurrence of antibiotics in freestall dairy farms with manured forage fields. Environ. Sci. Technol. 2010, 44, 6591–6600. [Google Scholar] [CrossRef]
- Seo, C.; Shin, J.; Lee, M.; Lee, W.; Yoom, H.; Son, H.; Jang, S.; Lee, Y. Elimination efficiency of organic UV filters during ozonation and UV/H2O2 treatment of drinking water and wastewater effluent. Chemosphere 2019, 230, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Yi, Q.D.; Li, W.X.; Ya, Q.Z.; Zhi, X.G.; Juan, F. Analysis of virulence genes and antibiotic resistance of Photobacterium damselae isolated from marine fishes in coastal South China. Acta Microbiol. Sin. 2020, 60, 2606–2619. [Google Scholar] [CrossRef]
- Gomez-Gil, B.; Roque, A.; Chimetto, L.; Moreira, A.P.B.; Lang, E.; Thompson, F.L. Vibrio alfacsensis sp. nov., isolated from marine organisms. Int. J. Syst. Evol. Microbiol. 2012, 62 Pt 12, 2955–2961. [Google Scholar] [CrossRef]
- Zhang, X.H.; He, X.; Austin, B. Vibrio harveyi: A serious pathogen of fish and invertebrates in mariculture. Mar. Life Sci. Technol. 2020, 2, 231–245. [Google Scholar] [CrossRef]
- Chen, J.; Lu, Y.; Ye, X.; Emam, M.; Zhang, H.; Wang, H. Current advances in Vibrio harveyi quorum sensing as drug discovery targets. Eur. J. Med. Chem. 2020, 207, 112741. [Google Scholar] [CrossRef]
- Liang, X.; Zhou, L.; Yan, S.; Wang, Y. Correction to: Complete genome sequence analysis of the Vibrio owensii strain SH-14 isolated from shrimp with acute hepatopancreatic necrosis disease. Arch. Microbiol. 2020, 202, 2019. [Google Scholar] [CrossRef] [PubMed]
- Montánchez, I.; Kaberdin, V.R. Vibrio harveyi: A brief survey of general characteristics and recent epidemiological traits associated with climate change. Mar. Environ. Res. 2020, 154, 104850. [Google Scholar] [CrossRef]
- Zhang, Y.; Deng, Y.; Feng, J.; Hu, J.; Chen, H.; Guo, Z.; Gao, R.; Su, Y. ToxR modulates biofilm formation in fish pathogen Vibrio harveyi. Lett. Appl. Microbiol. 2022, 74, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Lukjancenko, O.; Ussery, D.W. Vibrio chromosome-specific families. Front. Microbiol. 2014, 5, 73. [Google Scholar] [CrossRef]
- Xiao, T.W.; Yu, F.L.; Yang, Z.; Fan, P.M. Antibiotics in mariculture systems: A review of occurrence, environmental behavior, and ecological effects. Environ. Pollut. 2022, 293, 118541. [Google Scholar] [CrossRef]
- Xing, L.; Liu, H.; Zhou, J.G. Numerical study of the antibiotic transport and distribution in the Laizhou Bay, China. Environ. Sci. Pollut. Res. Int. 2020, 27, 37760–37772. [Google Scholar] [CrossRef]
- Almeida, Â.; Solé, M.; Soares, A.; Freitas, R. Anti-inflammatory drugs in the marine environment: Bioconcentration, metabolism and sub-lethal effects in marine bivalves. Environ. Pollut. 2020, 263 Pt A, 114442. [Google Scholar] [CrossRef]
- Suzuki, S.; Nakanishi, S.; Tamminen, M.; Yokokawa, T.; Sato-Takabe, Y.; Ohta, K.; Chou, H.-Y.; Muziasari, W.I.; Virta, M. Occurrence of sul and tet(M) genes in bacterial community in Japanese marine aquaculture environment throughout the year: Profile comparison with Taiwanese and Finnish aquaculture waters. Sci. Total Environ. 2019, 669, 649–656. [Google Scholar] [CrossRef]
- Gao, J.; Pedersen, J.A. Adsorption of sulfonamide antimicrobial agents to clay minerals. Environ. Sci. Technol. 2005, 39, 9509–9516. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.; Qiao, X.; Wang, Y.; Cai, X.; Zhou, C.; Zhang, Y.; Ding, G. DOM from mariculture ponds exhibits higher reactivity on photodegradation of sulfonamide antibiotics than from offshore seawaters. Water Res. 2018, 144, 365–372. [Google Scholar] [CrossRef]
- Dopson, M.; Johnson, D.B. Biodiversity, metabolism and applications of acidophilic sulfur-metabolizing microorganisms. Environ. Microbiol. 2012, 14, 2620–2631. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, P.; Wei, H.; Li, H.; Li, J.; Qiu, X.; Ding, R.; Guo, X. Alteration in microbial community and antibiotic resistance genes mediated by microplastics during wastewater ultraviolet disinfection. Sci. Total Environ. 2022, 825, 153918. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.N.; Anderson, M.T.; Bachman, M.A.; Mobley, H.L.T. The arcAB two-component system: Function in metabolism, redox control, and infection. Microbiol. Mol. Biol. Rev. 2022, 86, e0011021. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Zhou, X.; Tang, Y.; Jiang, Z.; Chen, J.; Mohsin, M.; Yue, M. Characterization of two-component system citB family in Salmonella pullorum. Int. J. Mol. Sci. 2022, 23, 10201. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, S.K.; Oates, C.W.; Raivio, T.L. Characterization of the induction and cellular role of the baeSR two-component envelope stress response of Escherichia coli. J. Bacteriol. 2011, 193, 3367–3375. [Google Scholar] [CrossRef]
- Guffey, A.A.; Loll, P.J. Regulation of resistance in vancomycin-resistant Enterococci: The vanRS two-component system. Microorganisms 2021, 9, 2026. [Google Scholar] [CrossRef]
- Arias, C.A.; Courvalin, P.; Reynolds, P.E. vanC cluster of vancomycin-resistant Enterococcus gallinarum BM4174. Antimicrob. Agents Chemother. 2000, 44, 1660–1666. [Google Scholar] [CrossRef]
- Binda, E.; Cappelletti, P.; Marinelli, F.; Marcone, G.L. Specificity of induction of glycopeptide antibiotic resistance in the producing actinomycetes. Antibiotics 2018, 7, 36. [Google Scholar] [CrossRef]
- Gök, Ş.M.; Türk Dağı, H.; Kara, F.; Arslan, U.; Fındık, D. Investigation of antibiotic resistance and virulence factors of enterococcus faecium and enterococcus faecalis strains isolated from clinical samples. Mikrobiyol. Bul. 2020, 54, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Ahmad-Mansour, N.; Loubet, P.; Pouget, C.; Dunyach-Remy, C.; Sotto, A.; Lavigne, J.-P.; Molle, V. Staphylococcus aureus toxins: An update on their pathogenic properties and potential treatments. Toxins 2021, 13, 677. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wang, C.; Lu, W.; Lu, H.; Chen, H.; Tan, C. BaeSR activates type VI secretion system expression in porcine extra-intestinal pathogenic Escherichia coli to enhance bacterial resistance to zinc stress. Microb. Pathog. 2020, 147, 104357. [Google Scholar] [CrossRef]
- Qi, C.; Sun, F.; Wei, Q.; Xu, J.; Li, R.; Zhang, L.; Lu, F.; Jiang, X.; Fu, H.; Zhang, C.; et al. Quantitative phosphoproteomics reveals the effect of baeSR and acrB genes on protein phosphorylation in Salmonella enterica serovar typhimurium. Res. Microbiol. 2022, 173, 103886. [Google Scholar] [CrossRef] [PubMed]
- Baranova, N.; Nikaido, H. The baeSR two-component regulatory system activates transcription of the yegMNOB (mdtABCD) transporter gene cluster in Escherichia coli and increases its resistance to novobiocin and deoxycholate. J. Bacteriol. 2002, 184, 4168–4176. [Google Scholar] [CrossRef]
- Nishino, K.; Nikaido, E.; Yamaguchi, A. Regulation of multidrug efflux systems involved in multidrug and metal resistance of Salmonella enterica serovar Typhimurium. J. Bacteriol. 2007, 189, 9066–9075. [Google Scholar] [CrossRef]
- Pakula, K.K.; Hansen, L.H.; Vester, B. Combined effect of the Cfr methyltransferase and ribosomal protein L3 mutations on resistance to ribosome-targeting antibiotics. Antimicrob. Agents Chemother. 2017, 61, 10–1128. [Google Scholar] [CrossRef]
- Sukhum, K.V.; Vargas, R.C.; Boolchandani, M.; D’souza, A.W.; Patel, S.; Kesaraju, A.; Walljasper, G.; Hegde, H.; Ye, Z.; Valenzuela, R.K.; et al. Manure microbial communities and resistance profiles reconfigure after transition to manure pits and differ from those in fertilized field soil. mBio 2021, 12, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z. Comparison of microbial communities and the antibiotic resistome between prawn mono- and poly-culture systems. Ecotoxicol. Environ. Saf. 2021, 207, 111310. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, J.; Wei, F.; Chen, X.; Li, M.; Li, C.; Xia, S.; Zhang, G.; You, W.; Cong, X.; et al. Microbial functional communities and the antibiotic resistome profile in a high-selenium ecosystem. Chemosphere 2023, 311 Pt 1, 136858. [Google Scholar] [CrossRef]
- Rutherford, S.T.; Bassler, B.L. Bacterial quorum sensing: Its role in virulence and possibilities for its control. Cold. Spring. Harb. Perspect. Med. 2012, 2, a012427. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Bassler, B.L. Bacterial quorum sensing in complex and dynamically changing environments. Nat. Rev. Microbiol. 2019, 17, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, M.; Diggle, S.P.; Greenberg, E.P. Progress in and promise of bacterial quorum sensing research. Nature 2017, 551, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Das, B.; Balakrish Nair, G.; Basak, S. Dynamics in genome evolution of Vibrio cholerae. Infect. Genet. Evol. 2014, 23, 32–41. [Google Scholar] [CrossRef]
- Liao, W.; Liu, Y.; Zhang, W. Virulence evolution, molecular mechanisms of resistance and prevalence of ST11 carbapenem-resistant Klebsiella pneumoniae in China: A review over the last 10 years. J. Glob. Antimicrob. Resist. 2020, 23, 174–180. [Google Scholar] [CrossRef]
- Zhu, Y.; Huang, W.E.; Yang, Q. Clinical perspective of antimicrobial resistance in bacteria. Infect. Drug Resist. 2022, 15, 735–746. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Lu, X.; Xing, Z.; Teng, X.; Wang, S.; Liu, T.; Zheng, L.; Wang, X.; Qu, J. Macrogenomics Reveals Effects on Marine Microbial Communities during Oplegnathus punctatus Enclosure Farming. Biology 2024, 13, 618. https://doi.org/10.3390/biology13080618
Wang L, Lu X, Xing Z, Teng X, Wang S, Liu T, Zheng L, Wang X, Qu J. Macrogenomics Reveals Effects on Marine Microbial Communities during Oplegnathus punctatus Enclosure Farming. Biology. 2024; 13(8):618. https://doi.org/10.3390/biology13080618
Chicago/Turabian StyleWang, Lijun, Xiaofei Lu, Zhikai Xing, Xindong Teng, Shuang Wang, Tianyi Liu, Li Zheng, Xumin Wang, and Jiangyong Qu. 2024. "Macrogenomics Reveals Effects on Marine Microbial Communities during Oplegnathus punctatus Enclosure Farming" Biology 13, no. 8: 618. https://doi.org/10.3390/biology13080618
APA StyleWang, L., Lu, X., Xing, Z., Teng, X., Wang, S., Liu, T., Zheng, L., Wang, X., & Qu, J. (2024). Macrogenomics Reveals Effects on Marine Microbial Communities during Oplegnathus punctatus Enclosure Farming. Biology, 13(8), 618. https://doi.org/10.3390/biology13080618