
Characterization and Phylogenetic Analysis of the Complete Mitochondrial Genome of Triplophysa microphthalma


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Genome Sequencing and Assembly
2.3. Sequence Annotation and Analysis
2.4. Phylogenetic Analysis
3. Results
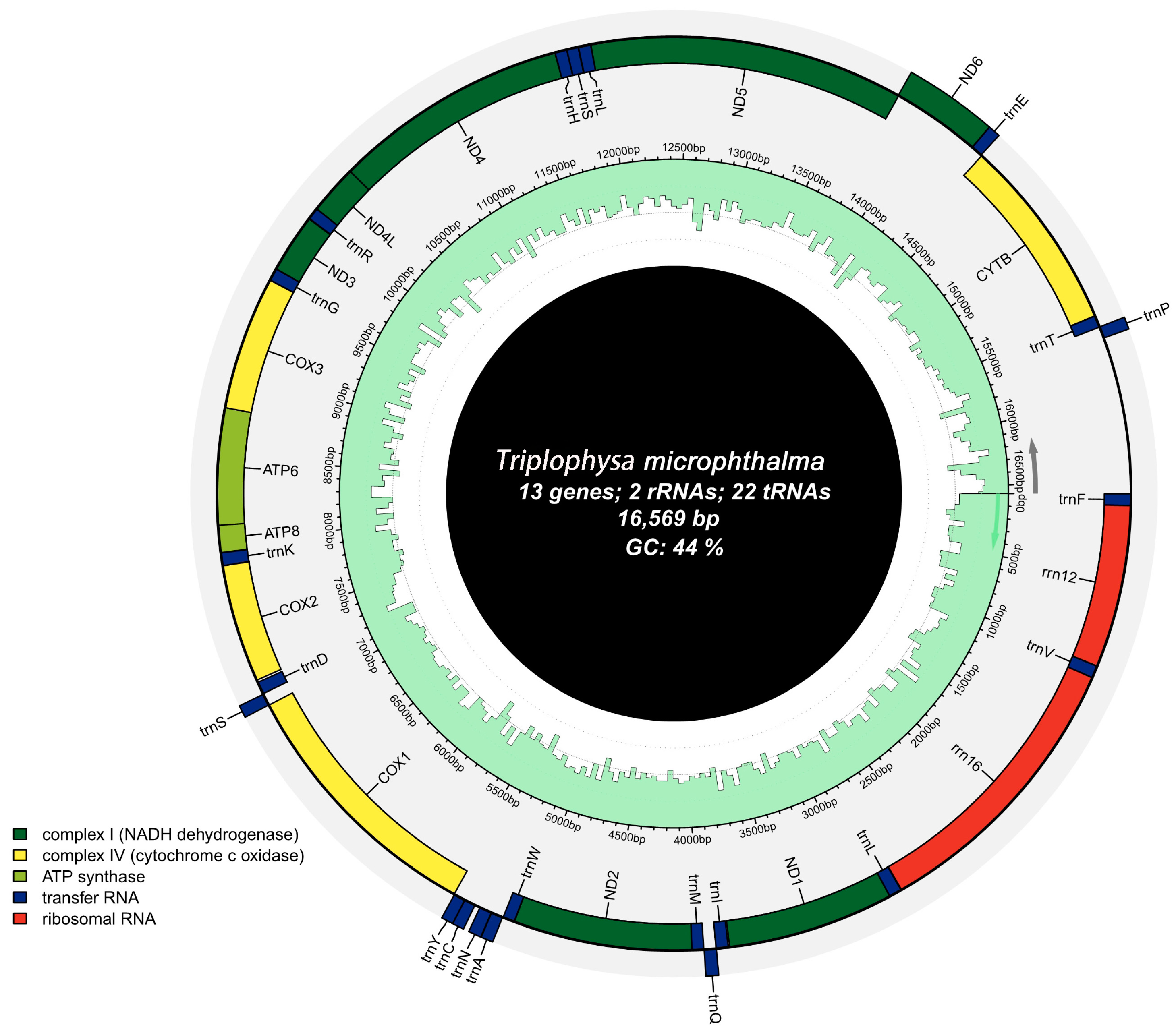
3.1. Mitochondrial Structural Characteristics
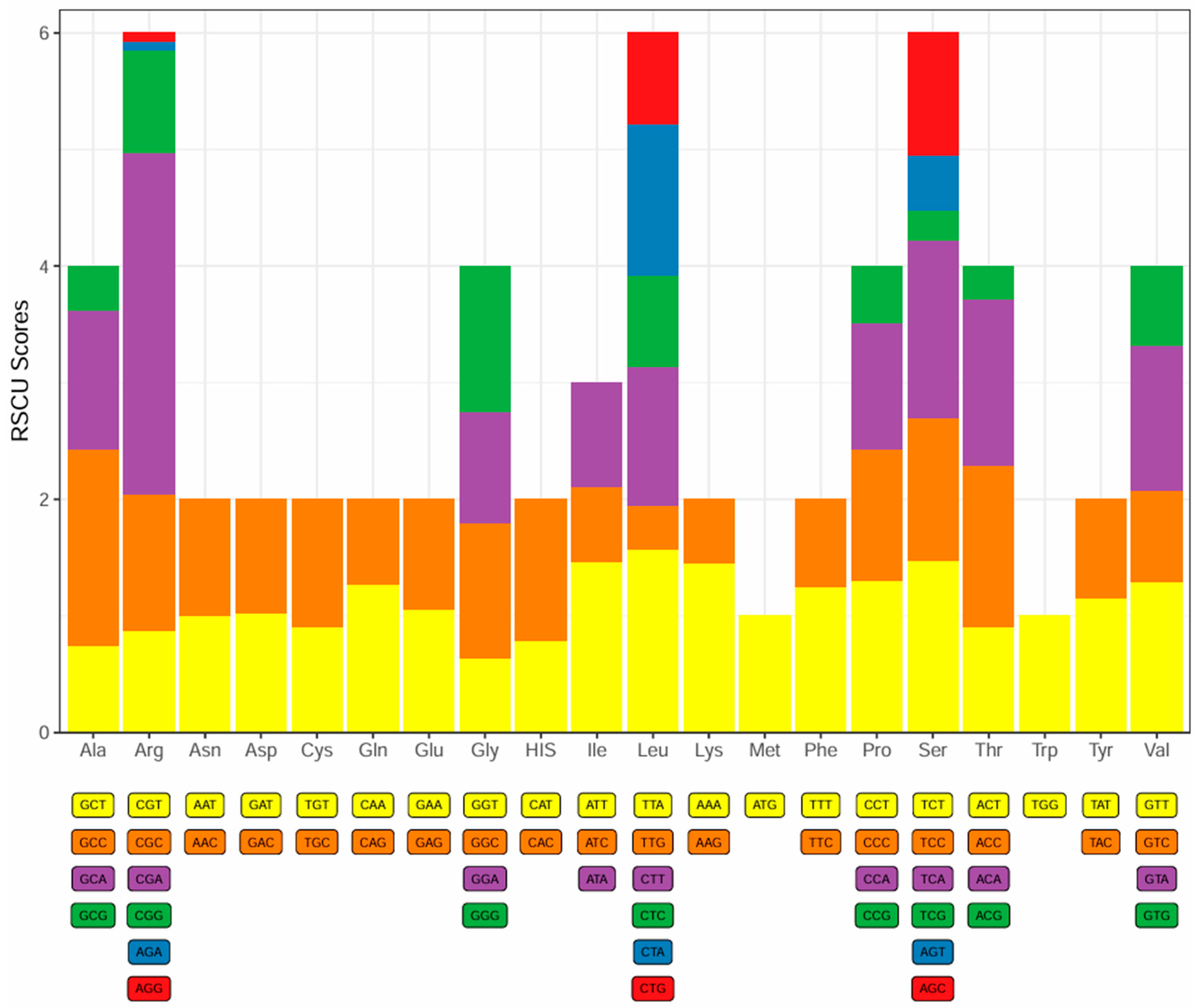
3.2. Protein-Coding Genes and Codon Usage
3.3. Transporter RNA, Ribosomal RNA, and Protein-Coding Gene Codon Usage
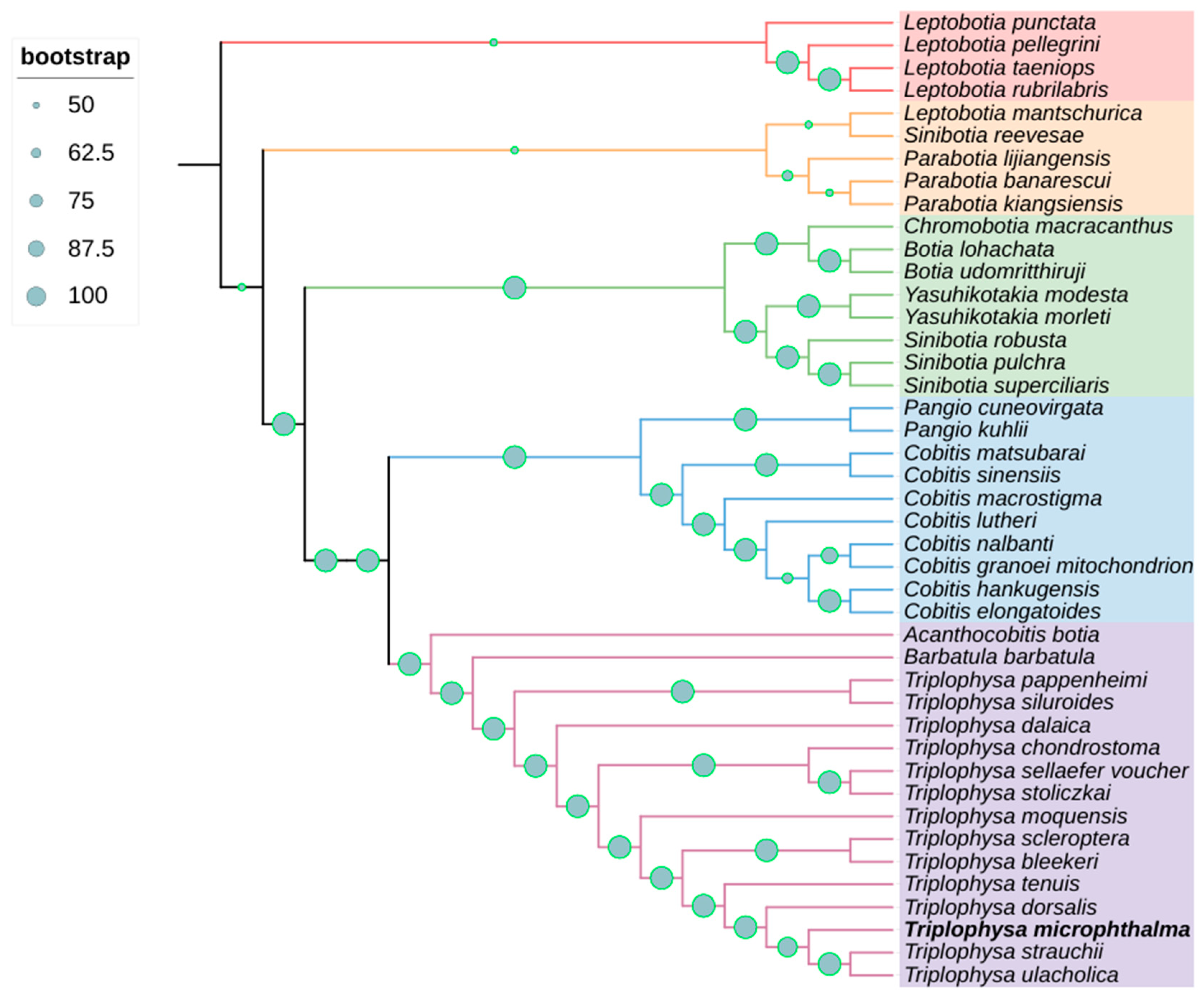
3.4. Phylogenetic Relationships
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ke, Z.; Zhou, K.; Hou, M.; Luo, H.; Li, Z.; Pan, X.; Zhou, J.; Jing, T.; Ye, H. Characterization of the Complete Mitochondrial Genome of the Elongate Loach and Its Phylogenetic Implications in Cobitidae. Animals 2023, 13, 3841. [Google Scholar] [CrossRef] [PubMed]
- The Biodiversity Committee of Chinese Academy of Sciences. Catalogue of Life China: 2024 Annual Checklist, Beijing, China; Species 2000 China, Catalogue of Life China, CoL China (sp2000.org.cn); The Biodiversity Committee of Chinese Academy of Sciences: Beijing, China, 2024. [Google Scholar]
- Zhang, C.G.; Zhao, Y.H.; Xing, Y.C.; Zhou, W.; Tang, W.Q. Species Diversity and Distribution of Inland Fishes in China; Science Press: Beijing, China, 2016. [Google Scholar]
- Kottelat, M. Conspectus Cobitidum: An Inventory of the Loaches of the World (Teleostei: Cypriniformes: Cobitoidei). 2012. Available online: https://api.semanticscholar.org/CorpusID:82341263 (accessed on 8 August 2024).
- Guo, Y.; Zhang, R.M.; Cai, L.G. Xinjiang of Fishery; Xinjiang Science and Technology Press: Urumqi, China, 2012; pp. 148–149. ISBN 978-7-5466-1529-5. (In Chinese) [Google Scholar]
- Goodsell, D.S. Mitochondrion. Biochem. Mol. Biol. Educ. 2010, 38, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. A mitogenomic perspective on the basal teleostean phylogeny: Resolving higher-level relationships with longer DNA sequences. Mol. Phylogenet. Evol. 2001, 20, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.G. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol. Evol. 1989, 4, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Chen, W.; Qin, H.; Zhao, Z.; Liao, J.; Chen, H.; Jiang, L.; Dayananda, B. The mitochondrial genome and phylogenetic analysis of Rhacophorus rhodopus. Sci. Rep. 2022, 12, 13693. [Google Scholar] [CrossRef]
- Myers, E.A.; Mulcahy, D.G.; Falk, B.; Johnson, K.; Carbi, M.; de Queiroz, K. Interspecific Gene Flow and Mitochondrial Genome Capture during the Radiation of Jamaican Anolis Lizards (Squamata; Iguanidae). Syst. Biol. 2022, 71, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Qiu, G.; Cao, C.; Wang, X.; Jiang, L.; Zhang, T.; Geng, Z.; Jin, S. Mitochondrial genome and phylogenetic analysis of Chaohu duck. Gene 2023, 851, 147018. [Google Scholar] [CrossRef]
- Jia, X.; Liu, W.; Zhang, Y.; Wang, H.; Li, X.; Yu, Q. Characterization and Phylogenetic Evolution of Mitochondrial Genome in Tibetan Chicken. Anim. Biotechnol. 2022, 33, 1371–1377. [Google Scholar] [CrossRef]
- Song, N.; Lin, A.; Zhao, X. Insight into higher-level phylogeny of Neuropterida: Evidence from secondary structures of mitochondrial rRNA genes and mitogenomic data. PLoS ONE 2018, 13, e0191826. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef] [PubMed]
- Massouras, A.; Parts, L.; Antonovsky, I.; George, P.; Yoon, J.; Glockner, G.; Jones, W.; Hauser, P.; Gisladottir, B.; Lehrach, H.; et al. Primer-initiated sequence synthesis to detect and assemble structural variants. Nat. Methods 2010, 7, 485–486. [Google Scholar] [CrossRef] [PubMed]
- Seberg, H.E.; Van Otterloo, E.; Cornell, R.A. Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment. Cell Melanoma Res. 2017, 30, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, P.D.; Zhang, D.Z.; Zhang, H.B.; Tang, B.P.; Liu, Q.N. Mitochondrial genome of the yellow catfish Pelteobagrus fulvidraco and insights into Bagridae phylogenetics. Genomics 2018, 111, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.Q.; Wang, M.H.; Li, D.; Tang, S.; Zhang, T.Q.; Bian, W.J.; Chen, X.H. Complete mitochondrial genome of Odontobutis haifengensis (Perciformes, Odontobutiae): A unique rearrangement of tRNAs and additional non-coding regions identified in the genus Odontobutis. Genomics 2018, 110, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, V.R.; Singha, H.S.; Kumar, R.G.; Gopalakrishnan, A.; Nagarajan, M. Characterization of the complete mitochondrial genome of Barilius malabaricus and its phylogenetic implications. Genomics 2020, 112, 2154–2163. [Google Scholar] [CrossRef] [PubMed]
- Garey, J.R.; Wolstenholme, D.R. Platyhelminth mitochondrial DNA: Evidence for early evolutionary origin of a tRNAserAGN that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J. Mol. Evol. 1989, 28, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Pavan-Kumar, A.; Singh, S.; Mishra, A.; Suman, S.; Gireesh-Babu, P.; Chaudhari, A.; Shen, K.N.; Borsa, P. Characterization of mitochondrial genome of Indian Ocean blue-spotted maskray, Neotrygon indica and its phylogenetic relationship within Dasyatidae Family. Int. J. Biol. Macromol. 2022, 223, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Mar-Silva, A.F.; Arroyave, J.; Díaz-Jaimes, P. The complete mitochondrial genome of the Mexican-endemic cavefish Ophisternon infernale (Synbranchiformes, Synbranchidae): Insights on patterns of selection and implications for synbranchiform phylogenetics. ZooKeys 2022, 1089, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Zeng, Y.; Li, J.; Zhang, F.; Liu, Y.; Gong, J. The complete mitochondrial genome sequence and phylogenetic analysis of Gnathopogon herzensteini (Cypriniformes, Cyprinidae, Gobioninae). Biologia 2021, 76, 1087–1094. [Google Scholar] [CrossRef]
- Asakawa, S.; Kumazawa, Y.; Araki, T.; Himeno, H.; Miura, K.I.; Watanabe, K. Strand-specific nucleotide composition bias in echinoderm and vertebrate mitochondrial genomes. J. Mol. Evol. 1991, 32, 511–520. [Google Scholar] [CrossRef]
- Cui, L.; Cao, R.; Dong, Y.; Gao, X.; Cen, J.; Lu, S. The first complete mitochondrial genome of the flathead Cociella crocodilus (Scorpaeniformes: Platycephalidae) and the phylogenetic relationships within Scorpaeniformes based on whole mitogenomes. Genes 2019, 10, 533. [Google Scholar] [CrossRef]
- Zhang, H.; Fang, W.; Zhao, X.; Jiang, X.; Stroiński, A.; Qin, D. Comparative analysis of the complete mitochondrial genomes of five species of Ricaniidae (Hemiptera: Fulgoromorpha) and phylogenetic implications. Biology 2022, 11, 92. [Google Scholar] [CrossRef]
- Yang, N.; Li, Y.; Liu, Z.; Chen, Q.; Shen, Y. The complete mitochondrial genome of Cobitis macrostigma (Cypriniformes: Cobitidae: Cobitinae) and a phylogenetic implication for its closely related species. Biologia 2020, 75, 393–399. [Google Scholar] [CrossRef]
- Ma, Q.; Zhang, T.L.; Chen, L.; Tang, Q.Y. The Complete Mitochondrial Genomes of Parabotia kiangsiensis (Cypriniformes: Botiidae). Mitochondrial DNA 2020, 5, 3629–3631. [Google Scholar] [CrossRef]
- Ohtsuki, T.; Kawai, G.; Watanabe, K. The minimal tRNA: Unique structure of Ascaris suum mitochondrial tRNA (Ser)(UCU) having a short T arm and lacking the entire D arm. FEBS Lett. 2002, 514, 37–43. [Google Scholar] [CrossRef]
- Wei, L.; Liu, J.G.; Liu, F.; Chu, L.X. Comparison of mitochondrial genome of six Felidae. J. Chuzhou Univ. 2013, 15, 80–87. (In Chinese) [Google Scholar]
- Yu, P.; Cao, T.; Zhao, C.P.; Liu, Y.F.; Shi, L.G.; Zhang, L.L.; Hou, G.Y. Complete mitochondrial genome sequence and analysis of Wuzhishan pig. Acta Agric. Borealioccidentalis Sin. 2015, 24, 16–22. (In Chinese) [Google Scholar]
- Liu, S.Q.; Zhang, J.B.; Tang, Q.Y.; Liu, H.Z. Phylogenetic relationships among Cobitoidea based on mitochondrial ND4 and ND5 gene sequences. Dongwuxue Yanjiu 2010, 31, 221–229. (In Chinese) [Google Scholar] [PubMed]
- Qin, Q.; Chen, X.; Liu, Q.; Wang, L.; Zhang, Y.; Zhao, K. Characterization of the Complete Mitochondrial Genome of Schizothorax kozlovi (Cypriniformes, Cyprinidae, Schizothorax) and Insights into the Phylogenetic Relationships of Schizothorax. Animals 2024, 14, 721. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Chen, Y. Biogeography and molecular phylogeny of the genus Schizothorax (Teleostei: Cyprinidae) in China inferred from cytochrome b sequences. J. Biogeogr. 2006, 33, 1448–1460. [Google Scholar] [CrossRef]
- Briolay, J.; Galtier, N.; Brito, R.M.; Bouvet, Y. Molecular phylogeny of Cyprinidae inferred from cytochrome b DNA sequences. Mol. Phylogenet. Evol. 1998, 9, 100–108. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Genus | Species | Assession Number |
|---|---|---|---|
| Cobitidae | Sinibotia | Sinibotia reevesae | NC 030322.1 |
| Sinibotia robusta | NC 027853.1 | ||
| Sinibotia pulchra | KT362179.1 | ||
| Sinibotia superciliaris | JX683724.1 | ||
| Leptobotia | Leptobotia mantschurica | AB242170.1 | |
| Leptobotia elongata | OR818399.1 | ||
| Leptobotia microphthalma | KY307846.1 | ||
| Leptobotia punctata | MH644033.1 | ||
| Leptobotia pellegrini | NC 031602.1 | ||
| Leptobotia taeniops | AP013304.1 | ||
| Leptobotia rubrilabris | KY307847.1 | ||
| Parabotia | Parabotia lijiangensis | MT323118.1 | |
| Parabotia banarescui | NC 026127.1 | ||
| Parabotia kiangsiensis | MT850132.1 | ||
| Chromobotia | Chromobotia macracanthus | AB242163.1 | |
| Botia | Botia lohachata | KP729183.1 | |
| Botia udomritthiruji | AP011349.1 | ||
| Yasuhikotakia | Yasuhikotakia modesta | KY131962.1 | |
| Yasuhikotakia morleti | NC 031600.1 | ||
| Pangio | Pangio cuneovirgata | NC 031594.1 | |
| Pangio kuhlii | NC 031599.1 | ||
| Cobitis | Cobitis matsubarai | NC 029441.1 | |
| Cobitis sinensiis | NC 007229.1 | ||
| Cobitis macrostigma | MK156771.1 | ||
| Cobitis lutheri | NC 022717.1 | ||
| Cobitis nalbanti | MH349461.1 | ||
| Cobitis granoei mitochondrion | NC 023473.1 | ||
| Cobitis hankugensis | MN841275.1 | ||
| Cobitis elongatoides | NC 023947.1 | ||
| Nemacheilidae | Acanthocobitis | Acanthocobitis botia | AP012139.1 |
| Barbatula | Barbatula barbatula | CM074126.1 | |
| Triplophysa | Triplophysa microphthalma | PP979136 | |
| Triplophysa pappenheimi | NC 033972.1 | ||
| Triplophysa siluroides | NC 024611.1 | ||
| Triplophysa dalaica | KT213590.1 | ||
| Triplophysa chondrostoma | KT213589.1 | ||
| Triplophysa sellaefer voucher | KY851112.1 | ||
| Triplophysa stoliczkai | JQ663847.1 | ||
| Triplophysa moquensis | KT213597.1 | ||
| Triplophysa scleroptera | KT213602.1 | ||
| Triplophysa bleekeri | JQ686729.1 | ||
| Triplophysa tenuis | KT224363.1 | ||
| Triplophysa dorsalis | KT213591.1 | ||
| Triplophysa strauchii | KP979754.1 | ||
| Triplophysa ulacholica | KT259194.1 |
| Gene | Position | Size | Intergenic Nucleotides | Codon | Strand | ||
|---|---|---|---|---|---|---|---|
| From | To | Start | Stop | ||||
| trnF | 1 | 69 | 69 | 0 | H | ||
| rrnS | 70 | 1021 | 952 | 0 | H | ||
| trnV | 1022 | 1093 | 72 | 0 | H | ||
| rrnL | 1094 | 2768 | 1675 | 0 | H | ||
| trnL2 | 2771 | 2845 | 75 | 2 | H | ||
| nad1 | 2846 | 3820 | 975 | 0 | ATG | TAA | H |
| trnI | 3828 | 3899 | 72 | 7 | H | ||
| trnQ | 3898 | 3968 | 71 | −2 | L | ||
| trnM | 3970 | 4038 | 69 | 1 | H | ||
| nad2 | 4039 | 5083 | 1045 | 0 | ATG | T-- | H |
| trnW | 5084 | 5153 | 70 | 0 | H | ||
| trnA | 5156 | 5224 | 69 | 2 | L | ||
| trnN | 5226 | 5298 | 73 | 1 | L | ||
| trnC | 5330 | 5395 | 66 | 31 | L | ||
| trnY | 5396 | 5463 | 68 | 0 | L | ||
| cox1 | 5465 | 7015 | 1551 | 1 | GTG | TAA | H |
| trnS2 | 7016 | 7086 | 71 | 0 | L | ||
| trnD | 7089 | 7161 | 73 | 2 | H | ||
| cox2 | 7175 | 7865 | 691 | 13 | ATG | T-- | H |
| trnK | 7866 | 7941 | 76 | 0 | H | ||
| atp8 | 7943 | 8110 | 168 | 1 | ATG | TAA | H |
| atp6 | 8101 | 8784 | 684 | −10 | ATG | TAA | H |
| cox3 | 8784 | 9567 | 784 | −1 | ATG | T-- | H |
| trnG | 9568 | 9640 | 73 | 0 | H | ||
| nad3 | 9641 | 9987 | 347 | 0 | ATG | GA- | H |
| trnR | 9990 | 10,059 | 70 | 2 | H | ||
| nad4L | 10,060 | 10,356 | 297 | 0 | ATG | TAA | H |
| nad4 | 10,350 | 11,731 | 1382 | −7 | ATG | TA- | H |
| trnH | 11,732 | 11,801 | 70 | 0 | H | ||
| trnS1 | 11,802 | 11,869 | 68 | 0 | H | ||
| trnL1 | 11,871 | 11,943 | 73 | 1 | H | ||
| nad5 | 11,944 | 13,782 | 1839 | 0 | ATG | TAG | H |
| nad6 | 13,779 | 14,300 | 522 | −4 | ATG | TAA | L |
| trnE | 14,301 | 14,369 | 69 | 0 | L | ||
| cytb | 14,374 | 15,514 | 1141 | 4 | ATG | T-- | H |
| trnT | 15,515 | 15,585 | 71 | 0 | H | ||
| trnP | 15,584 | 15,653 | 70 | −2 | L | ||
| D-loop | 15,654 | 16,569 | 916 | H | |||
| Overlap: | 6 | Gap: | 13 | ||||
| Regions | Size (bp) | T (U) | C | A | G | AT (%) | GC (%) | GT (%) | AT Skew | GC Skew |
|---|---|---|---|---|---|---|---|---|---|---|
| PCGs | 11,418 | 30.3 | 26.2 | 25.5 | 17.9 | 55.8 | 44.1 | 48.2 | −0.085 | −0.188 |
| Control Region | 916 | 33.1 | 20.3 | 34.1 | 12.6 | 67.2 | 32.9 | 45.7 | 0.015 | −0.236 |
| rRNAs | 2627 | 21.7 | 23.5 | 32.7 | 22.1 | 54.4 | 45.6 | 43.8 | 0.203 | −0.031 |
| tRNAs | 1558 | 27.9 | 20.8 | 28.4 | 23.0 | 56.3 | 43.8 | 50.9 | 0.009 | 0.050 |
| Full genome | 16,569 | 28.2 | 25.7 | 28.1 | 18.0 | 56.3 | 43.7 | 46.2 | −0.003 | −0.177 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, P.; Guo, W.; Wei, C.; Wang, X.; Wang, Y.; Wang, J. Characterization and Phylogenetic Analysis of the Complete Mitochondrial Genome of Triplophysa microphthalma. Biology 2024, 13, 608. https://doi.org/10.3390/biology13080608
Yang P, Guo W, Wei C, Wang X, Wang Y, Wang J. Characterization and Phylogenetic Analysis of the Complete Mitochondrial Genome of Triplophysa microphthalma. Biology. 2024; 13(8):608. https://doi.org/10.3390/biology13080608
Chicago/Turabian StyleYang, Ping, Wei Guo, Chao Wei, Xin Wang, Yixuan Wang, and Jia Wang. 2024. "Characterization and Phylogenetic Analysis of the Complete Mitochondrial Genome of Triplophysa microphthalma" Biology 13, no. 8: 608. https://doi.org/10.3390/biology13080608
APA StyleYang, P., Guo, W., Wei, C., Wang, X., Wang, Y., & Wang, J. (2024). Characterization and Phylogenetic Analysis of the Complete Mitochondrial Genome of Triplophysa microphthalma. Biology, 13(8), 608. https://doi.org/10.3390/biology13080608