
Communications between Neutrophil–Endothelial Interaction in Immune Defense against Bacterial Infection

 , , , , and
, , , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Cooperative Behaviors of Neutrophil and Endothelial Cell
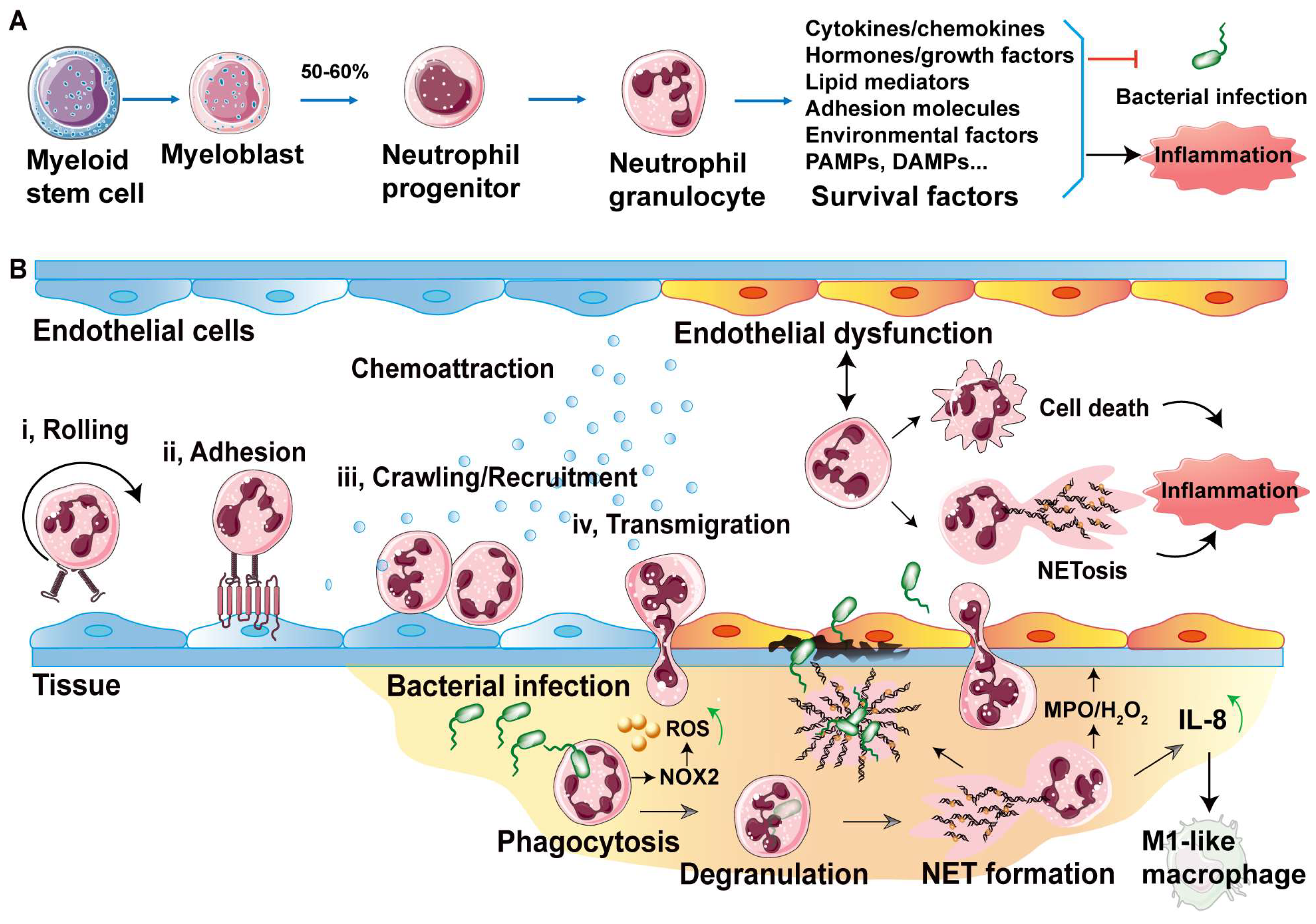
2.1. Neutrophil Generation and Circulating
2.2. Neutrophil Rolling and Adhesion on Endothelium
2.3. Neutrophil Transendothelial Migration Strategies
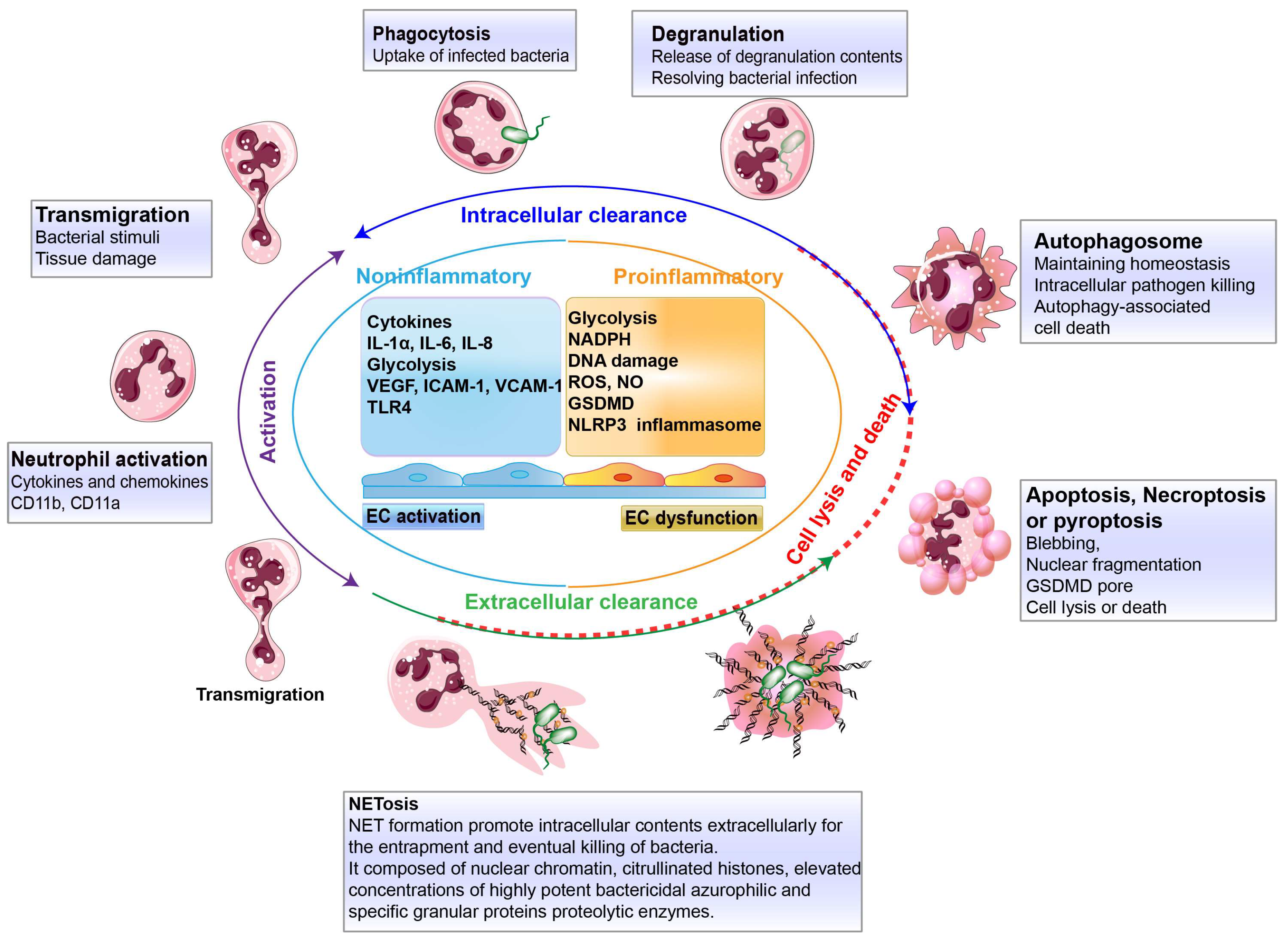
3. Defense Bacterial Infection of Transendothelial Neutrophils
3.1. Clearance Strategies by Transendothelial Neutrophils
3.2. Intracellular Clearance by Neutrophil Neutralization
3.3. Extracellular Clearance by Neutrophil Extracellular Traps
3.4. Neutrophil Death
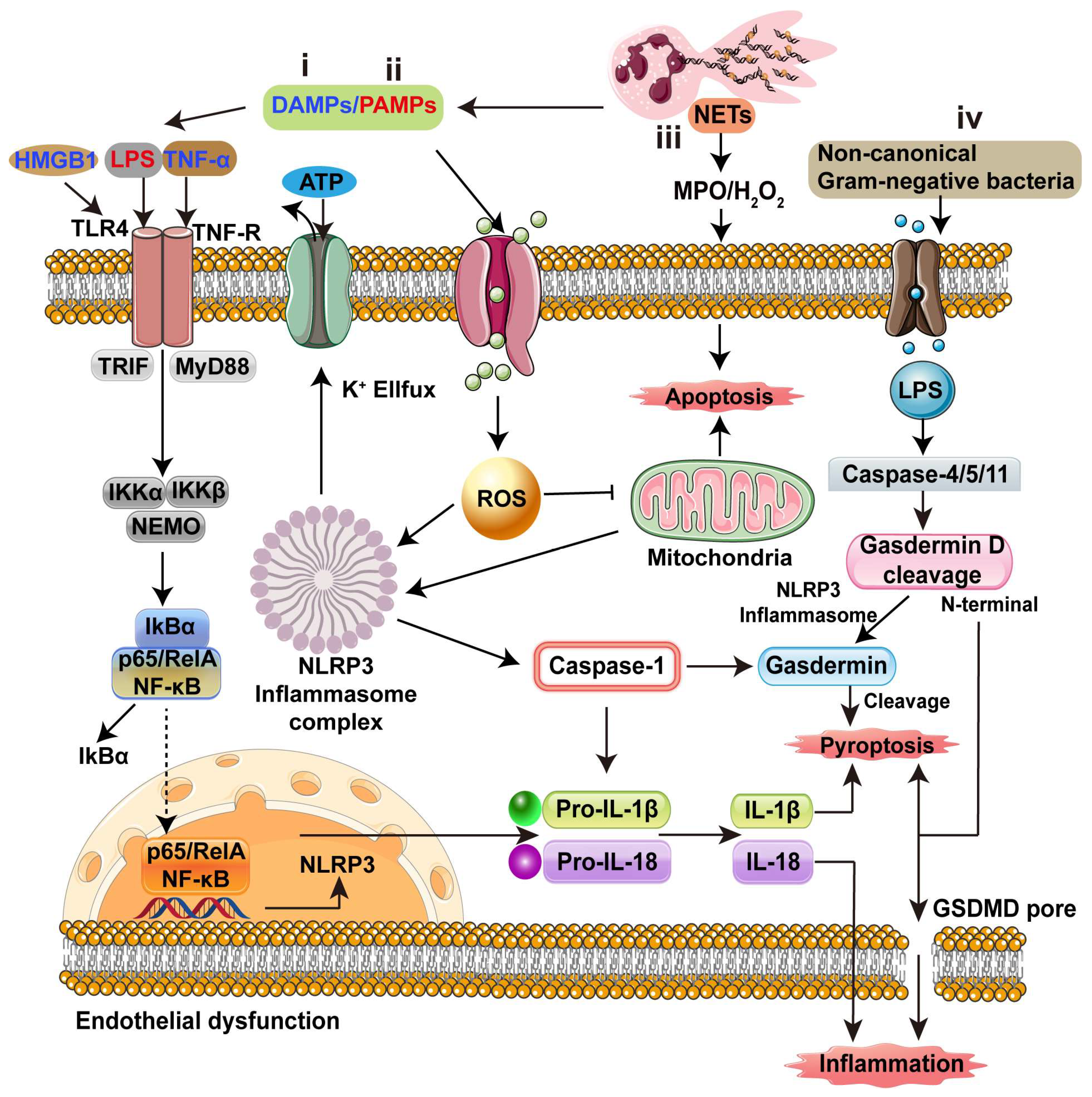
4. Inflammatory Cascades in Endothelial Dysfunction and Potential Targets
4.1. Inflammatory Cascades
4.2. Potential Targets
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-HIAA | 5-hydroxyindoleacetic acid |
| CD95L | CD95-ligand |
| DAMPs | Damage-associated molecular patterns |
| ESAM | Endothelial cell-selective adhesion molecule |
| ESL1 | E-selectin ligand 1 |
| GPR35 | G-protein-coupled receptor |
| GSH | Glutathione |
| ICAM-1 | Endothelial intercellular adhesion molecule-1 |
| IL-1α | Interleukin 1α |
| JAM | Junctional adhesion molecule |
| LPS | Lipopolysaccharide |
| NAC | N-acetylcysteine |
| NETs | Neutrophil extracellular traps |
| OSMR | OSM receptors |
| PAMPs | Pathogen-associated molecular patterns |
| PECAM-1 | Platelet endothelial cell adhesion molecule 1 |
| PSGL1 | P-selectin ligand 1 |
| PTX3 | Pentraxin 3 |
| TNF-α | Tumor necrosis factor-α |
| VLA-4 | Very late antigen 4 |
References
- Bird, L. Innate immunity: Sensing bacterial messages. Nat. Rev. Immunol. 2018, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Naz, F.; Petri, W.A. Host Immunity and Immunization Strategies for Clostridioides difficile Infection. Clin. Microbiol. Rev. 2023, 36, e0015722. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Riera Romo, M.; Pérez-Martínez, D.; Castillo Ferrer, C. Innate immunity in vertebrates: An overview. Immunology 2016, 148, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.D.; Schwarz, M.A. Cell-Cell Communication Breakdown and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gulculer, G.S.; Golbach, L.; Block, H.; Zarbock, A.; Martin-Villalba, A. Endothelial cell-derived CD95 ligand serves as a chemokine in induction of neutrophil slow rolling and adhesion. Elife 2016, 5, e18542. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, H.; Yago, T.; Liu, Z.; McEver, R.P. Endothelial signaling by neutrophil-released oncostatin M enhances P-selectin-dependent inflammation and thrombosis. Blood Adv. 2019, 3, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Wu, X.; Zhao, Z.; Deng, Q.; Chen, Y.; Yin, L. Endothelial cell-targeting, ROS-ultrasensitive drug/siRNA co-delivery nanocomplexes mitigate early-stage neutrophil recruitment for the anti-inflammatory treatment of myocardial ischemia reperfusion injury. Acta Biomater. 2022, 143, 344–355. [Google Scholar] [CrossRef]
- Filippi, M.D. Neutrophil transendothelial migration: Updates and new perspectives. Blood 2019, 133, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Surmiak, M.; Kosałka-Węgiel, J.; Polański, S.; Sanak, M. Endothelial cells response to neutrophil-derived extracellular vesicles miRNAs in anti-PR3 positive vasculitis. Clin. Exp. Immunol. 2021, 204, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Winneberger, J.; Schöls, S.; Lessmann, K.; Rández-Garbayo, J.; Bauer, A.T.; Mohamud Yusuf, A.; Hermann, D.M.; Gunzer, M.; Schneider, S.W.; Fiehler, J.; et al. Platelet endothelial cell adhesion molecule-1 is a gatekeeper of neutrophil transendothelial migration in ischemic stroke. Brain Behav. Immun. 2021, 93, 277–287. [Google Scholar] [CrossRef] [PubMed]
- De Giovanni, M.; Tam, H.; Valet, C.; Xu, Y.; Looney, M.R.; Cyster, J.G. GPR35 promotes neutrophil recruitment in response to serotonin metabolite 5-HIAA. Cell 2022, 185, 815–830.e819. [Google Scholar] [CrossRef] [PubMed]
- Akbar, N.; Braithwaite, A.T.; Corr, E.M.; Koelwyn, G.J.; van Solingen, C.; Cochain, C.; Saliba, A.E.; Corbin, A.; Pezzolla, D.; Møller Jørgensen, M.; et al. Rapid neutrophil mobilization by VCAM-1+ endothelial cell-derived extracellular vesicles. Cardiovasc. Res. 2023, 119, 236–251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Qu, M.; Li, W.; Wu, D.; Cata, J.P.; Miao, C. Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis. Clin. Transl. Med. 2023, 13, e1170. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H. Inflammation and thrombosis: Roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J. Thromb. Haemost. 2018, 16, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Nepal, S.; Tsukasaki, Y.; Hecquet, C.M.; Soni, D.; Rehman, J.; Tiruppathi, C.; Malik, A.B. Neutrophil Activation of Endothelial Cell-Expressed TRPM2 Mediates Transendothelial Neutrophil Migration and Vascular Injury. Circ. Res. 2017, 121, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, H.; He, S.; Mu, X.; Hu, G.; Dong, H. Endothelial-Derived Interleukin-1α Activates Innate Immunity by Promoting the Bactericidal Activity of Transendothelial Neutrophils. Front. Cell Dev. Biol. 2020, 8, 590. [Google Scholar] [CrossRef]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef]
- Lawrence, S.M.; Corriden, R.; Nizet, V. How Neutrophils Meet Their End. Trends Immunol. 2020, 41, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 2010, 12, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Kienle, K.; Lammermann, T. Neutrophil swarming: An essential process of the neutrophil tissue response. Immunol. Rev. 2016, 273, 76–93. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophils Stepping through (to the Other Side). Immunity 2018, 49, 992–994. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil Diversity in Health and Disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Hawez, A.; Ding, Z.; Wang, Y.; Rönnow, C.F.; Rahman, M.; Thorlacius, H. E3 Ubiquitin Ligase Midline 1 Regulates Endothelial Cell ICAM-1 Expression and Neutrophil Adhesion in Abdominal Sepsis. Int. J. Mol. Sci. 2022, 24, 705. [Google Scholar] [CrossRef] [PubMed]
- Schunk, S.J.; Triem, S.; Schmit, D.; Zewinger, S.; Sarakpi, T.; Becker, E.; Hütter, G.; Wrublewsky, S.; Küting, F.; Hohl, M.; et al. Interleukin-1α Is a Central Regulator of Leukocyte-Endothelial Adhesion in Myocardial Infarction and in Chronic Kidney Disease. Circulation 2021, 144, 893–908. [Google Scholar] [CrossRef]
- Liu, X.; Dong, H.; Wang, M.; Gao, Y.; Zhang, T.; Hu, G.; Duan, H.; Mu, X. IL-1alpha-induced microvascular endothelial cells promote neutrophil killing by increasing MMP-9 concentration and lysozyme activity. Immunol. Res. 2016, 64, 133–142. [Google Scholar] [CrossRef]
- Shao, Y.; Li, L.; Liu, L.; Yang, Y.; Huang, J.; Ji, D.; Zhou, Y.; Chen, Y.; Zhu, Z.; Sun, B. CD44/ERM/F-actin complex mediates targeted nuclear degranulation and excessive neutrophil extracellular trap formation during sepsis. J. Cell. Mol. Med. 2022, 26, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Hulthe, J.; Fagerberg, B. Baseline ICAM-1 and VCAM-1 are increased in initially healthy middle-aged men who develop cardiovascular disease during 6.6 years of follow-up. Angiology 2009, 60, 108–114. [Google Scholar] [CrossRef]
- Harding, M.G.; Zhang, K.; Conly, J.; Kubes, P. Neutrophil crawling in capillaries; a novel immune response to Staphylococcus aureus. PLoS Pathog. 2014, 10, e1004379. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Kimmey, J.M. Neutrophil Recruitment in Pneumococcal Pneumonia. Front. Cell. Infect. Microbiol. 2022, 12, 894644. [Google Scholar] [CrossRef] [PubMed]
- Margraf, A.; Ley, K.; Zarbock, A. Neutrophil Recruitment: From Model Systems to Tissue-Specific Patterns. Trends Immunol. 2019, 40, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Babes, L.; Rahn, J.J.; Ahn, B.Y.; Goring, K.R.; King, J.C.; Lau, A.; Petri, B.; Hao, X.; Chojnacki, A.K.; et al. Dipeptidase-1 Is an Adhesion Receptor for Neutrophil Recruitment in Lungs and Liver. Cell 2019, 178, 1205–1221.e1217. [Google Scholar] [CrossRef] [PubMed]
- Kameritsch, P.; Renkawitz, J. Principles of Leukocyte Migration Strategies. Trends Cell Biol. 2020, 30, 818–832. [Google Scholar] [CrossRef]
- Bixel, M.G.; Li, H.; Petri, B.; Khandoga, A.G.; Khandoga, A.; Zarbock, A.; Wolburg-Buchholz, K.; Wolburg, H.; Sorokin, L.; Zeuschner, D.; et al. CD99 and CD99L2 act at the same site as, but independently of, PECAM-1 during leukocyte diapedesis. Blood 2010, 116, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Kumar, S. Neutrophil and remnant clearance in immunity and inflammation. Immunology 2022, 165, 22–43. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, N.V.; Ksenofontov, A.L.; Serebryakova, M.V.; Stadnichuk, V.I.; Gaponova, T.V.; Baratova, L.A.; Sud’ina, G.F.; Galkina, S.I. Neutrophils Release Metalloproteinases during Adhesion in the Presence of Insulin, but Cathepsin G in the Presence of Glucagon. Mediat. Inflamm. 2018, 2018, 1574928. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, J.M.; Sabbione, F.; Morelli, M.P.; Tateosian, N.L.; Castello, F.A.; Amiano, N.O.; Palmero, D.; Levi, A.; Ciallella, L.; Colombo, M.I.; et al. Neutrophil autophagy during human active tuberculosis is modulated by SLAMF1. Autophagy 2021, 17, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.L.; Xie, J.; Zhao, Z.Z.; Li, P.; Liu, Q.; Guo, Y.; Meng, Y.; Wan, X.J.; Bian, J.J.; Deng, X.M.; et al. PD-L1 maintains neutrophil extracellular traps release by inhibiting neutrophil autophagy in endotoxin-induced lung injury. Front. Immunol. 2022, 13, 949217. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, H.; Fu, X.; Han, L.; Zhang, H.; Zhang, L.; Zhao, J.; Xiao, D.; Li, H.; Li, P. Autophagy-driven neutrophil extracellular traps: The dawn of sepsis. Pathol. Res. Pract. 2022, 234, 153896. [Google Scholar] [CrossRef] [PubMed]
- Dunham-Snary, K.J.; Surewaard, B.G.; Mewburn, J.D.; Bentley, R.E.; Martin, A.Y.; Jones, O.; Al-Qazazi, R.; Lima, P.A.; Kubes, P.; Archer, S.L. Mitochondria in human neutrophils mediate killing of Staphylococcus aureus. Redox Biol. 2022, 49, 102225. [Google Scholar] [CrossRef] [PubMed]
- Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules 2019, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production through Interleukin-1alpha and Cathepsin G. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, K.; Yang, D.; Oppenheim, J.J. Interleukin-8: An evolving chemokine. Cytokine 2022, 153, 155828. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wu, W.; Xia, Y.; Wang, X.; Liang, J. Platelet-derived extracellular vesicles promote endothelial dysfunction in sepsis by enhancing neutrophil extracellular traps. BMC Immunol. 2023, 24, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Dehaini, D.; Zhang, Y.; Zhou, J.; Chen, X.; Zhang, L.; Fang, R.H.; Gao, W.; Zhang, L. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotechnol. 2018, 13, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.E.; Kiani, M.F. Experimental Approaches to Evaluate Leukocyte-Endothelial Cell Interactions in Sepsis and Inflammation. Shock 2020, 53, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Reglero-Real, N.; Pérez-Gutiérrez, L.; Yoshimura, A.; Rolas, L.; Garrido-Mesa, J.; Barkaway, A.; Pickworth, C.; Saleeb, R.S.; Gonzalez-Nuñez, M.; Austin-Williams, S.N.; et al. Autophagy modulates endothelial junctions to restrain neutrophil diapedesis during inflammation. Immunity 2021, 54, 1989–2004.e1989. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Garabet, L.; Henriksson, C.E.; Lozano, M.L.; Ghanima, W.; Bussel, J.; Brodin, E.; Fernández-Pérez, M.P.; Martínez, C.; González-Conejero, R.; Mowinckel, M.C.; et al. Markers of endothelial cell activation and neutrophil extracellular traps are elevated in immune thrombocytopenia but are not enhanced by thrombopoietin receptor agonists. Thromb. Res. 2020, 185, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Lee, H.; Lee, H.K.; Kim, I.D.; Lee, J.K. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol. Commun. 2019, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Suarez, J.S.; Minaai, M.; Li, S.; Gaudino, G.; Pass, H.I.; Carbone, M.; Yang, H. HMGB1 as a therapeutic target in disease. J. Cell. Physiol. 2021, 236, 3406–3419. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kumari, T.; Barazia, A.; Jha, V.; Jeong, S.Y.; Olson, A.; Kim, M.; Lee, B.K.; Manickam, V.; Song, Z.; et al. Neutrophil DREAM promotes neutrophil recruitment in vascular inflammation. J. Exp. Med. 2022, 219, e20211083. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Peng, Z.; Wang, J. Trelagliptin Alleviates Lipopolysaccharide (LPS)-Induced Inflammation and Oxidative Stress in Acute Lung Injury Mice. Inflammation 2021, 44, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Baselet, B.; Sonveaux, P.; Baatout, S.; Aerts, A. Pathological effects of ionizing radiation: Endothelial activation and dysfunction. Cell. Mol. Life Sci. 2019, 76, 699–728. [Google Scholar] [CrossRef] [PubMed]
- Hirano, S.I.; Ichikawa, Y.; Sato, B.; Yamamoto, H.; Takefuji, Y.; Satoh, F. Potential Therapeutic Applications of Hydrogen in Chronic Inflammatory Diseases: Possible Inhibiting Role on Mitochondrial Stress. Int. J. Mol. Sci. 2021, 22, 2549. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, L.; Wang, M.; Wang, H.; Zhang, H.; Chen, Y.; Wang, X.; Gong, J.; Zhang, J.J.; Adcock, I.M.; et al. Mitochondrial ROS and NLRP3 inflammasome in acute ozone-induced murine model of airway inflammation and bronchial hyperresponsiveness. Free Radic. Res. 2019, 53, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Dominic, A.; Le, N.T.; Takahashi, M. Loop Between NLRP3 Inflammasome and Reactive Oxygen Species. Antioxid. Redox Signal. 2022, 36, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between NLRP3 inflammasome and autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Paget, C.; Doz-Deblauwe, E.; Winter, N.; Briard, B. Specific NLRP3 Inflammasome Assembling and Regulation in Neutrophils: Relevance in Inflammatory and Infectious Diseases. Cells 2022, 11, 1188. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Demon, D.; Vande Walle, L.; Paerewijck, O.; Zecchin, A.; Bosseler, L.; Santoni, K.; Planès, R.; Ribo, S.; Fossoul, A.; et al. GSDMD drives canonical inflammasome-induced neutrophil pyroptosis and is dispensable for NETosis. EMBO Rep. 2022, 23, e54277. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, M.; Minns, M.; Greenberg, E.N.; Diaz-Aponte, J.; Pestonjamasp, K.; Johnson, J.L.; Rathkey, J.K.; Abbott, D.W.; Wang, K.; Shao, F.; et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat. Commun. 2020, 11, 2212. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, K.M.; Creed, S.; Gan, P.Y.; Holdsworth, S.R. Supervised Machine Learning for Semi-Quantification of Extracellular DNA in Glomerulonephritis. J. Vis. Exp. 2020, 160, e61180. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Y.; Tu, Q.; Yao, L.; Li, J.; Yang, Z. Alpha-linolenic acid pretreatment alleviates NETs-induced alveolar macrophage pyroptosis by inhibiting pyrin inflammasome activation in a mouse model of sepsis-induced ALI/ARDS. Front. Immunol. 2023, 14, 1146612. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, X.; Chatterjee, V.; Meegan, J.E.; Beard, R.S., Jr.; Yuan, S.Y. Role of Neutrophil Extracellular Traps and Vesicles in Regulating Vascular Endothelial Permeability. Front. Immunol. 2019, 10, 1037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, J.; Zhou, Y.; Qu, M.; Wang, Y.; Guo, K.; Shen, R.; Sun, Z.; Cata, J.P.; Yang, S.; et al. Neutrophil extracellular traps mediate m(6)A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int. J. Biol. Sci. 2022, 18, 3337–3357. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.M.S.; Wanderley, C.W.S.; Veras, F.P.; Sonego, F.; Nascimento, D.C.; Gonçalves, A.V.; Martins, T.V.; Cólon, D.F.; Borges, V.F.; Brauer, V.S.; et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood 2021, 138, 2702–2713. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Immler, R.; Pruenster, M.; Sellmayr, M.; Li, C.; von Brunn, A.; von Brunn, B.; Ehmann, R.; Wölfel, R.; Napoli, M.; et al. Soluble uric acid inhibits β2 integrin-mediated neutrophil recruitment in innate immunity. Blood 2022, 139, 3402–3417. [Google Scholar] [CrossRef] [PubMed]
- Baiula, M.; Greco, R.; Ferrazzano, L.; Caligiana, A.; Hoxha, K.; Bandini, D.; Longobardi, P.; Spampinato, S.; Tolomelli, A. Integrin-mediated adhesive properties of neutrophils are reduced by hyperbaric oxygen therapy in patients with chronic non-healing wound. PLoS ONE 2020, 15, e0237746. [Google Scholar] [CrossRef]
- Chavakis, T.; Athanasopoulos, A.; Rhee, J.S.; Orlova, V.; Schmidt-Wöll, T.; Bierhaus, A.; May, A.E.; Celik, I.; Nawroth, P.P.; Preissner, K.T. Angiostatin is a novel anti-inflammatory factor by inhibiting leukocyte recruitment. Blood 2005, 105, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Willeke, T.; Scharffetter-Kochanek, K.; Gaehtgens, P.; Walzog, B. A role for beta2 integrin (CD11/CD18)-mediated tyrosine signaling in extravasation of human polymorphonuclear neutrophils. Biorheology 2001, 38, 89–100. [Google Scholar] [PubMed]
- Martinod, K.; Fuchs, T.A.; Zitomersky, N.L.; Wong, S.L.; Demers, M.; Gallant, M.; Wang, Y.; Wagner, D.D. PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood 2015, 125, 1948–1956. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Wang, Y.; Esmon, N.L.; Esmon, C.T. Regulated endothelial protein C receptor shedding is mediated by tumor necrosis factor-alpha converting enzyme/ADAM17. J. Thromb. Haemost. 2007, 5, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Kant, R.; Halder, S.K.; Fernández, J.A.; Griffin, J.H.; Milner, R. Activated Protein C Attenuates Experimental Autoimmune Encephalomyelitis Progression by Enhancing Vascular Integrity and Suppressing Microglial Activation. Front. Neurosci. 2020, 14, 333. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Wang, X.; Chen, F.; Yang, C.; Shi, L.; Xu, W.; Wang, K.; Liu, B.; Wang, C.; Sun, D.; et al. Neutrophil extracellular traps aggravate intestinal epithelial necroptosis in ischaemia-reperfusion by regulating TLR4/RIPK3/FUNDC1-required mitophagy. Cell Prolif. 2024, 57, e13538. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.A.; Minarchick, V.C.; Zahavi, M.; Rysenga, C.E.; Sturm, K.A.; Hoy, C.K.; Sarosh, C.; Knight, J.S.; Demoruelle, M.K. Ginger intake suppresses neutrophil extracellular trap formation in autoimmune mice and healthy humans. JCI Insight 2023, 8, e172011. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; Laterre, P.F.; Francois, B.; LaRosa, S.P.; Angus, D.C.; Mira, J.P.; Wittebole, X.; Dugernier, T.; Perrotin, D.; Tidswell, M.; et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: The ACCESS randomized trial. JAMA 2013, 309, 1154–1162. [Google Scholar] [CrossRef]
- Savov, J.D.; Brass, D.M.; Lawson, B.L.; McElvania-Tekippe, E.; Walker, J.K.; Schwartz, D.A. Toll-like receptor 4 antagonist (E5564) prevents the chronic airway response to inhaled lipopolysaccharide. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2005, 289, L329–L337. [Google Scholar] [CrossRef] [PubMed]
- Pylaeva, E.; Bordbari, S.; Spyra, I.; Decker, A.S.; Häussler, S.; Vybornov, V.; Lang, S.; Jablonska, J. Detrimental Effect of Type I IFNs During Acute Lung Infection with Pseudomonas aeruginosa Is Mediated through the Stimulation of Neutrophil NETosis. Front. Immunol. 2019, 10, 2190. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, A.; Lykkesfeldt, J. Does vitamin C enhance nitric oxide bioavailability in a tetrahydrobiopterin-dependent manner? In vitro, in vivo and clinical studies. Nitric Oxide 2014, 36, 51–57. [Google Scholar] [CrossRef]
- Fowler, A.A., 3rd; Truwit, J.D.; Hite, R.D.; Morris, P.E.; DeWilde, C.; Priday, A.; Fisher, B.; Thacker, L.R., 2nd; Natarajan, R.; Brophy, D.F.; et al. Effect of Vitamin C Infusion on Organ Failure and Biomarkers of Inflammation and Vascular Injury in Patients with Sepsis and Severe Acute Respiratory Failure: The CITRIS-ALI Randomized Clinical Trial. JAMA 2019, 322, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, J.; Zhang, P.; Peng, B.; Li, C.; Ming, Y.; Liu, H. Berberine protects hepatocyte from hypoxia/reoxygenation-induced injury through inhibiting circDNTTIP2. PeerJ 2023, 11, e16080. [Google Scholar] [CrossRef] [PubMed]
- Meziani, F.; Kremer, H.; Tesse, A.; Baron-Menguy, C.; Mathien, C.; Mostefai, H.A.; Carusio, N.; Schneider, F.; Asfar, P.; Andriantsitohaina, R. Human serum albumin improves arterial dysfunction during early resuscitation in mouse endotoxic model via reduced oxidative and nitrosative stresses. Am. J. Pathol. 2007, 171, 1753–1761. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Basic Steps | Molecules | Reference | |
|---|---|---|---|
| Neutrophils | Endothelial Cells | ||
| i, Rolling | PSG1, PTX3 | P-selectin | [20] |
| PSGL1, ESL1, CD44 | E-selectin | [20,31] | |
| L-selectin | E-selectin | [20,23] | |
| CD95 | CD95L | [6] | |
| ii, Adhesion | β2 integrins LAF-1(αLβ2) | ICAM-1 | [7,20] |
| Mac-1 (αMβ2) | ICAM-2 | [26,27] | |
| VLA-4 | VCAM-1 | [32] | |
| OSM | OSMR | [7] | |
| iii, Crawling/Recruitment | β2 integrins Mac-1 (α4β2) | ICAM-1 | [33,34,35] |
| DPEP1 ligand | DPEP1 | [36] | |
| GPR35 | 5-HIAA | [12] | |
| iv, Migration | β2 integrins Mac-1 (α4β2), LFA-1 | ICAM-1 and ICAM-2 | [9,37] |
| PECAM-1, JAM-A, CD99 | PECAM-1, JAM-ABC, ESAM, VE-Cadherin, CD99, CD99L2 | [11,38] | |
| Potential Targets | Compounds | Mechanisms | Reference |
|---|---|---|---|
| Inhibit β2-integrin-mediated neutrophil recruitment | Soluble uric acid | Regulates intracellular pH and cytoskeletal dynamics | [75] |
| hyperbaric oxygen | Reduces integrin-mediated adhesive properties of neutrophils | [76] | |
| Angiostatin | The kringles 1–3 and its kringle 4 directly interact with leukocyte beta1- and beta2-integrins | [77] | |
| herbimycin A | Inhibits of beta2 integrin-mediated tyrosine signaling | [78] | |
| Inhibit NET formation | C34 | Inhibits TLR4 | [82] |
| 6-gingerol | Inhibits phosphodiesterase | [83] | |
| nNIF and NRPs | Blocks NET formation in vitro and in mouse models of infection and systemic inflammation | [84] | |
| DNase1 | Preferentially cleaves protein-free DNA and degrades the scaffold of NET structures | [85] | |
| ROS scavengers | Berberine | Reduces H/R-induced apoptosis and ROS production | [89] |
| HSA | Reduces ROS and RNS production | [90] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Z.; Lv, R.; Zhao, Y.; Cai, Z.; Si, X.; Zhang, Q.; Liu, X. Communications between Neutrophil–Endothelial Interaction in Immune Defense against Bacterial Infection. Biology 2024, 13, 374. https://doi.org/10.3390/biology13060374
Sun Z, Lv R, Zhao Y, Cai Z, Si X, Zhang Q, Liu X. Communications between Neutrophil–Endothelial Interaction in Immune Defense against Bacterial Infection. Biology. 2024; 13(6):374. https://doi.org/10.3390/biology13060374
Chicago/Turabian StyleSun, Zhigang, Ruoyi Lv, Yanxin Zhao, Ziwen Cai, Xiaohui Si, Qian Zhang, and Xiaoye Liu. 2024. "Communications between Neutrophil–Endothelial Interaction in Immune Defense against Bacterial Infection" Biology 13, no. 6: 374. https://doi.org/10.3390/biology13060374
APA StyleSun, Z., Lv, R., Zhao, Y., Cai, Z., Si, X., Zhang, Q., & Liu, X. (2024). Communications between Neutrophil–Endothelial Interaction in Immune Defense against Bacterial Infection. Biology, 13(6), 374. https://doi.org/10.3390/biology13060374