
Connexin Gap Junction Channels and Hemichannels: Insights from High-Resolution Structures

 ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Major Insights from Recent High Resolution Structures of Cx Channels and Hemichannels
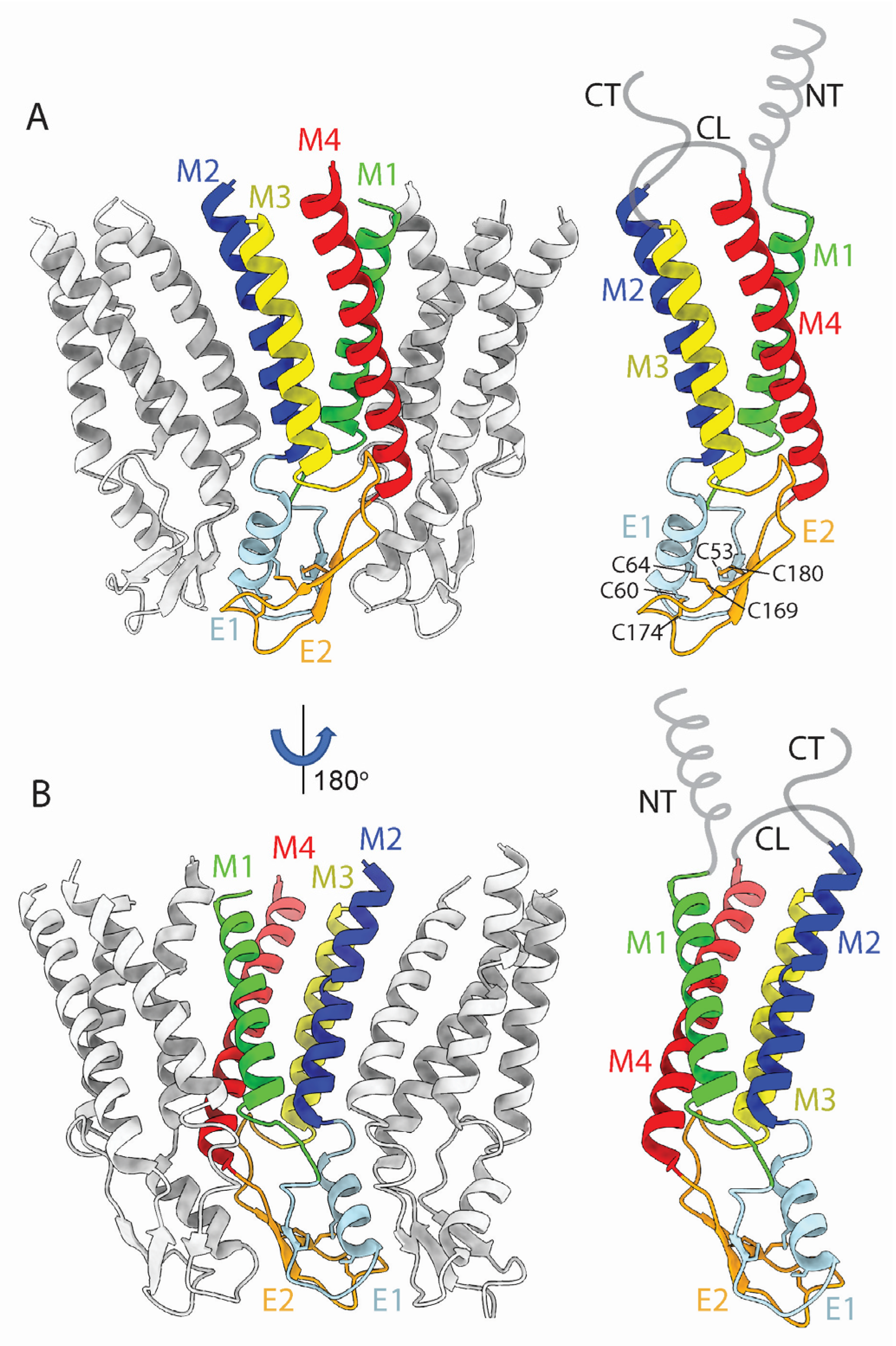
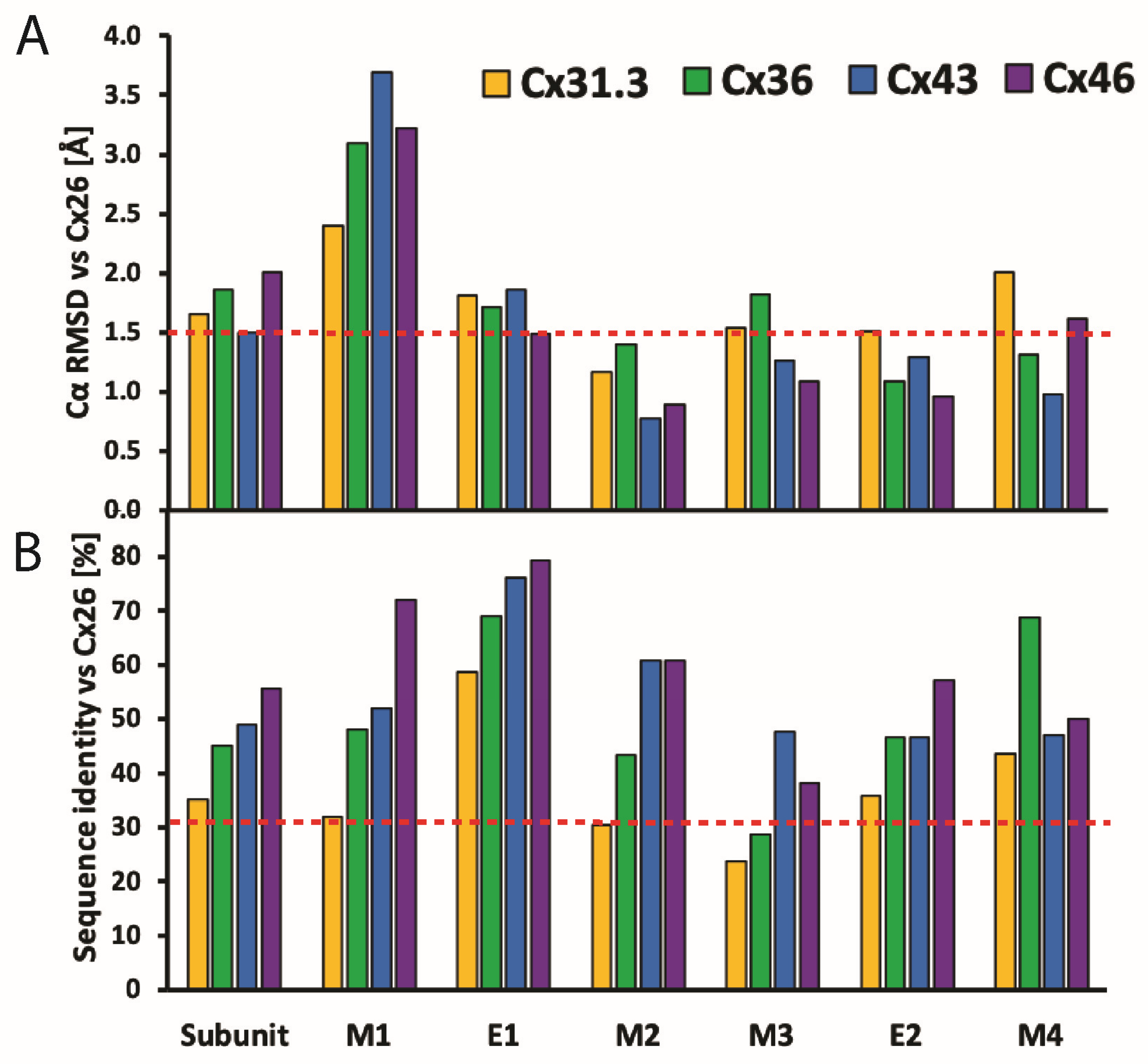
2.1. Conservation of the TMD and ECD Architectures across Seven Cx Isoforms

2.2. Sequence Diversity and Disorder in the CL and CT Domains
2.3. Gating in High Ca2+ Mediated by an Electrostatic Mechanism
2.4. Gating at Acidic pH Mediated by a Steric “Ball-and-Chain” Mechanism
2.5. The NT Visualized in the Channel Pore Adopts Multiple Conformations
2.6. Visualization of Lipid-like Densities in Unexpected Places, within the Pore and/or the TMD Helices

2.7. Functional Implications for Gating and Permeation
2.8. A Summary of Structure/Function Correlations and Insights from High-Resolution Structures
3. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Lucaciu, S.A.; Leighton, S.E.; Hauser, A.; Yee, R.; Laird, D.W. Diversity in connexin biology. J. Biol. Chem. 2023, 299, 105263. [Google Scholar] [CrossRef] [PubMed]
- Revel, J.P.; Karnovsky, M.J. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J. Cell Biol. 1967, 33, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; Laird, D.W. Recent advances in connexin gap junction biology. Fac. Rev. 2022, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 2007, 94, 120–143. [Google Scholar] [CrossRef] [PubMed]
- Söhl, G.; Willecke, K. An update on connexin genes and their nomenclature in mouse and man. Cell Commun. Adhes. 2003, 10, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Koval, M.; Molina, S.A.; Burt, J.M. Mix and match: Investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 2014, 588, 1193–1204. [Google Scholar] [CrossRef]
- Syrjanen, J.; Michalski, K.; Kawate, T.; Furukawa, H. On the molecular nature of large-pore channels. J. Mol. Biol. 2021, 433, 166994. [Google Scholar] [CrossRef] [PubMed]
- Güiza, J.; Solis, F.; Valenzuela, B.; Arancibia, D.; Zamorano, P.; Gonzalez, J.; Saavedra, J.; Neely, A.; Salgado, M.; Martinez, A.D.; et al. Unnexin is a protein subunit of a large-pore channel expressed by unicellular organisms. Proc. Natl. Acad. Sci. USA 2023, 120, e2307898120. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.M.; Gilula, N.B. The gap junction communication channel. Cell 1996, 84, 381–388. [Google Scholar] [CrossRef]
- Yeager, M.; Harris, A.L. Gap junction channel structure in the early 21st century: Facts and fantasies. Curr. Opin. Cell Biol. 2007, 19, 521–528. [Google Scholar] [CrossRef]
- Belousov, A.B.; Fontes, J.D. Neuronal gap junctions: Making and breaking connections during development and injury. Trends Neurosci. 2013, 36, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Desplantez, T.; Dupont, E.; Severs, N.J.; Weingart, R. Gap junction channels and cardiac impulse propagation. J. Membr. Biol. 2007, 218, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.C.; Purdy, M.D.; Baker, K.A.; Acharya, C.; McIntire, W.E.; Stevens, R.C.; Zhang, Q.; Harris, A.L.; Abagyan, R.; Yeager, M. An electrostatic mechanism for Ca2+-mediated regulation of gap junction channels. Nat. Commun. 2016, 7, 8770. [Google Scholar] [CrossRef]
- Khan, A.K.; Jagielnicki, M.; McIntire, W.E.; Purdy, M.D.; Dharmarajan, V.; Griffin, P.R.; Yeager, M. A steric “ball-and-chain” mechanism for pH-mediated regulation of gap junction channels. Cell Rep. 2020, 31, 107482. [Google Scholar] [CrossRef] [PubMed]
- Verselis, V.K.; Ginter, C.S.; Bargiello, T.A. Opposite voltage gating polarities of two closely related connexins. Nature 1994, 368, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W.; Naus, C.C.; Lampe, P.D. SnapShot: Connexins and Disease. Cell 2017, 170, 1260–1260.e1. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W.; Lampe, P.D. Cellular mechanisms of connexin-based inherited diseases. Trends Cell Biol. 2022, 32, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Cisterna, B.A.; Arroyo, P.; Puebla, C. Role of connexin-based gap junction channels in communication of myelin sheath in Schwann cells. Front. Cell Neurosci. 2019, 13, 69. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Nicholson, B.J. The role of connexins in ear and skin physiology—Functional insights from disease-associated mutations. Biochim. Biophys. Acta 2013, 1828, 167–178. [Google Scholar] [CrossRef]
- Falk, M.M.; Bell, C.L.; Kells Andrews, R.M.; Murray, S.A. Molecular mechanisms regulating formation, trafficking and processing of annular gap junctions. BMC Cell Biol. 2016, 17 (Suppl. S1), 22. [Google Scholar] [CrossRef]
- Yeager, M.; Unger, V.M.; Falk, M.M. Synthesis, assembly and structure of gap junction intercellular channels. Curr. Opin. Struct. Biol. 1998, 8, 517–524. [Google Scholar] [CrossRef]
- Falk, M.M.; Baker, S.M.; Gumpert, A.M.; Segretain, D.; Buckheit, R.W., 3rd. Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol. Biol. Cell 2009, 20, 3342–3352. [Google Scholar] [CrossRef]
- Fallon, R.F.; Goodenough, D.A. Five-hour half-life of mouse liver gap-junction protein. J. Cell Biol. 1981, 90, 521–526. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 1998, 83, 629–635. [Google Scholar] [CrossRef]
- Berthoud, V.M.; Minogue, P.J.; Laing, J.G.; Beyer, E.C. Pathways for degradation of connexins and gap junctions. Cardiovasc. Res. 2004, 62, 256–267. [Google Scholar] [CrossRef]
- Unwin, P.N.; Ennis, P.D. Two configurations of a channel-forming membrane protein. Nature 1984, 307, 609–613. [Google Scholar] [CrossRef]
- Fleishman, S.J.; Unger, V.M.; Yeager, M.; Ben-Tal, N. A Cα model for the transmembrane α helices of gap junction intercellular channels. Mol. Cell 2004, 15, 879–888. [Google Scholar] [CrossRef]
- Unger, V.M.; Kumar, N.M.; Gilula, N.B.; Yeager, M. Three-dimensional structure of a recombinant gap junction membrane channel. Science 1999, 283, 1176–1180. [Google Scholar] [CrossRef]
- Oshima, A.; Tani, K.; Hiroaki, Y.; Fujiyoshi, Y.; Sosinsky, G.E. Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc. Natl. Acad. Sci. USA 2007, 104, 10034–10039. [Google Scholar] [CrossRef]
- Maeda, S.; Nakagawa, S.; Suga, M.; Yamashita, E.; Oshima, A.; Fujiyoshi, Y.; Tsukihara, T. Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature 2009, 458, 597–602. [Google Scholar] [CrossRef]
- Kwon, T.; Harris, A.L.; Rossi, A.; Bargiello, T.A. Molecular dynamics simulations of the Cx26 hemichannel: Evaluation of structural models with Brownian dynamics. J. Gen. Physiol. 2011, 138, 475–493. [Google Scholar] [CrossRef]
- Unwin, P.N.; Ennis, P.D. Calcium-mediated changes in gap junction structure: Evidence from the low angle X-ray pattern. J. Cell Biol. 1983, 97, 1459–1466. [Google Scholar] [CrossRef]
- Myers, J.B.; Haddad, B.G.; O’Neill, S.E.; Chorev, D.S.; Yoshioka, C.C.; Robinson, C.V.; Zuckerman, D.M.; Reichow, S.L. Structure of native lens connexin 46/50 intercellular channels by cryo-EM. Nature 2018, 564, 372–377. [Google Scholar] [CrossRef]
- Brotherton, D.H.; Savva, C.G.; Ragan, T.J.; Dale, N.; Cameron, A.D. Conformational changes and CO2-induced channel gating in connexin26. Structure 2022, 30, 697–706.e4. [Google Scholar] [CrossRef]
- Khan, A.K.; Jagielnicki, M.; Bennett, B.C.; Purdy, M.D.; Yeager, M. Cryo-EM structure of an open conformation of a gap junction hemichannel in lipid bilayer nanodiscs. Structure 2021, 29, 1040–1047.e3. [Google Scholar] [CrossRef]
- Lee, H.J.; Jeong, H.; Hyun, J.; Ryu, B.; Park, K.; Lim, H.H.; Yoo, J.; Woo, J.S. Cryo-EM structure of human Cx31.3/GJC3 connexin hemichannel. Sci. Adv. 2020, 6, eaba4996. [Google Scholar] [CrossRef]
- Qi, C.; Lavriha, P.; Bayraktar, E.; Vaithia, A.; Schuster, D.; Pannella, M.; Sala, V.; Picotti, P.; Bortolozzi, M.; Korkhov, V.M. Structures of wild-type and selected CMT1X mutant connexin 32 gap junction channels and hemichannels. Sci. Adv. 2023, 9, eadh4890. [Google Scholar] [CrossRef]
- Lee, S.N.; Cho, H.J.; Jeong, H.; Ryu, B.; Lee, H.J.; Kim, M.; Yoo, J.; Woo, J.S.; Lee, H.H. Cryo-EM structures of human Cx36/GJD2 neuronal gap junction channel. Nat. Commun. 2023, 14, 1347. [Google Scholar] [CrossRef]
- Lee, H.J.; Cha, H.J.; Jeong, H.; Lee, S.N.; Lee, C.W.; Kim, M.; Yoo, J.; Woo, J.S. Conformational changes in the human Cx43/GJA1 gap junction channel visualized using cryo-EM. Nat. Commun. 2023, 14, 931. [Google Scholar] [CrossRef]
- Qi, C.; Acosta Gutierrez, S.; Lavriha, P.; Othman, A.; Lopez-Pigozzi, D.; Bayraktar, E.; Schuster, D.; Picotti, P.; Zamboni, N.; Bortolozzi, M.; et al. Structure of the connexin-43 gap junction channel in a putative closed state. Elife 2023, 12, RP87616. [Google Scholar] [CrossRef]
- Flores, J.A.; Haddad, B.G.; Dolan, K.A.; Myers, J.B.; Yoshioka, C.C.; Copperman, J.; Zuckerman, D.M.; Reichow, S.L. Connexin-46/50 in a dynamic lipid environment resolved by CryoEM at 1.9 A. Nat. Commun. 2020, 11, 4331. [Google Scholar] [CrossRef]
- Bai, D.; Yue, B.; Aoyama, H. Crucial motifs and residues in the extracellular loops influence the formation and specificity of connexin docking. Biochim. Biophys. Acta Biomembr. 2018, 1860, 9–21. [Google Scholar] [CrossRef]
- Hoh, J.H.; John, S.A.; Revel, J.P. Molecular cloning and characterization of a new member of the gap junction gene family, connexin-31. J. Biol. Chem. 1991, 266, 6524–6531. [Google Scholar] [CrossRef]
- Hennemann, H.; Schwarz, H.J.; Willecke, K. Characterization of gap junction genes expressed in F9 embryonic carcinoma cells: Molecular cloning of mouse connexin31 and -45 cDNAs. Eur. J. Cell Biol. 1992, 57, 51–58. [Google Scholar]
- Nakagawa, S.; Gong, X.Q.; Maeda, S.; Dong, Y.; Misumi, Y.; Tsukihara, T.; Bai, D. Asparagine 175 of connexin32 is a critical residue for docking and forming functional heterotypic gap junction channels with connexin26. J. Biol. Chem. 2011, 286, 19672–19681. [Google Scholar] [CrossRef]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System; Version 2.5; Schrodinger, L.L.C.: New York, NY, USA, 2015. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Hervé, J.C.; Derangeon, M.; Sarrouilhe, D.; Giepmans, B.N.; Bourmeyster, N. Gap junctional channels are parts of multiprotein complexes. Biochim. Biophys. Acta 2012, 1818, 1844–1865. [Google Scholar] [CrossRef]
- Lampe, P.D.; Lau, A.F. The effects of connexin phosphorylation on gap junctional communication. Int. J. Biochem. Cell Biol. 2004, 36, 1171–1186. [Google Scholar] [CrossRef]
- Gilleron, J.; Carette, D.; Chevallier, D.; Segretain, D.; Pointis, G. Molecular connexin partner remodeling orchestrates connexin traffic: From physiology to pathophysiology. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 407–423. [Google Scholar] [CrossRef]
- Thévenin, A.F.; Kowal, T.J.; Fong, J.T.; Kells, R.M.; Fisher, C.G.; Falk, M.M. Proteins and mechanisms regulating gap-junction assembly, internalization, and degradation. Physiology 2013, 28, 93–116. [Google Scholar] [CrossRef]
- Sorgen, P.L.; Trease, A.J.; Spagnol, G.; Delmar, M.; Nielsen, M.S. Protein(-)Protein interactions with Connexin 43: Regulation and Function. Int. J. Mol. Sci. 2018, 19, 1428. [Google Scholar] [CrossRef]
- Evans, W.H.; Martin, P.E. Gap junctions: Structure and function (Review). Mol. Membr. Biol. 2002, 19, 121–136. [Google Scholar] [CrossRef]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef]
- Sorgen, P.L.; Duffy, H.S.; Sahoo, P.; Coombs, W.; Delmar, M.; Spray, D.C. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J. Biol. Chem. 2004, 279, 54695–54701. [Google Scholar] [CrossRef]
- Sáez, J.C.; Retamal, M.A.; Basilio, D.; Bukauskas, F.F.; Bennett, M.V. Connexin-based gap junction hemichannels: Gating mechanisms. Biochim. Biophys. Acta 2005, 1711, 215–224. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, S.; Zhang, Z.; Hou, M.; Du, C.; Zhao, Z.; Vogel, H.; Li, Z.; Yan, K.; Zhang, X.; et al. Cryo-EM structure of human heptameric pannexin 2 channel. Nat. Commun. 2023, 14, 1118. [Google Scholar] [CrossRef]
- González-Nieto, D.; Gómez-Hernández, J.M.; Larrosa, B.; Gutiérrez, C.; Muñoz, M.D.; Fasciani, I.; O’Brien, J.; Zappala, A.; Cicirata, F.; Barrio, L.C. Regulation of neuronal connexin-36 channels by pH. Proc. Natl. Acad. Sci. USA 2008, 105, 17169–17174. [Google Scholar] [CrossRef]
- Ek-Vitorín, J.F.; Calero, G.; Morley, G.E.; Coombs, W.; Taffet, S.M.; Delmar, M. PH regulation of connexin43: Molecular analysis of the gating particle. Biophys. J. 1996, 71, 1273–1284. [Google Scholar] [CrossRef]
- Delmar, M.; Coombs, W.; Sorgen, P.; Duffy, H.S.; Taffet, S.M. Structural bases for the chemical regulation of Connexin43 channels. Cardiovasc. Res. 2004, 62, 268–275. [Google Scholar] [CrossRef]
- Anumonwo, J.M.; Taffet, S.M.; Gu, H.; Chanson, M.; Moreno, A.P.; Delmar, M. The carboxyl terminal domain regulates the unitary conductance and voltage dependence of connexin40 gap junction channels. Circ. Res. 2001, 88, 666–673. [Google Scholar] [CrossRef]
- Oliveira-Castro, G.M.; Loewenstein, W.R. Junctional membrane permeability: Effects of divalent cations. J. Membr. Biol. 1971, 5, 51–77. [Google Scholar] [CrossRef]
- Phelan, P.; Bacon, J.P.; Davies, J.A.; Stebbings, L.A.; Todman, M.G.; Avery, L.; Baines, R.A.; Barnes, T.M.; Ford, C.; Hekimi, S.; et al. Innexins: A family of invertebrate gap-junction proteins. Trends Genet. 1998, 14, 348–349. [Google Scholar] [CrossRef]
- Iwatsuki, N.; Petersen, O.H. Acetylcholine-like effects of intracellular calcium application in pancreatic acinar cells. Nature 1977, 268, 147–149. [Google Scholar] [CrossRef]
- Dahl, G.; Isenberg, G. Decoupling of heart muscle cells: Correlation with increased cytoplasmic calcium activity and with changes of nexus ultrastructure. J. Membr. Biol. 1980, 53, 63–75. [Google Scholar] [CrossRef]
- Spray, D.C.; Stern, J.H.; Harris, A.L.; Bennett, M.V. Gap junctional conductance: Comparison of sensitivities to H and Ca ions. Proc. Natl. Acad. Sci. USA 1982, 79, 441–445. [Google Scholar] [CrossRef]
- Paul, D.L.; Ebihara, L.; Takemoto, L.J.; Swenson, K.I.; Goodenough, D.A. Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J. Cell Biol. 1991, 115, 1077–1089. [Google Scholar] [CrossRef]
- Pfahnl, A.; Dahl, G. Gating of cx46 gap junction hemichannels by calcium and voltage. Pflugers Arch. 1999, 437, 345–353. [Google Scholar] [CrossRef]
- Ebihara, L.; Steiner, E. Properties of a nonjunctional current expressed from a rat connexin46 cDNA in Xenopus oocytes. J. Gen. Physiol. 1993, 102, 59–74. [Google Scholar] [CrossRef]
- Harris, A.L.; Contreras, J.E. Motifs in the permeation pathway of connexin channels mediate voltage and Ca2+ sensing. Front. Physiol. 2014, 5, 113. [Google Scholar] [CrossRef]
- Lopez, W.; Ramachandran, J.; Alsamarah, A.; Luo, Y.; Harris, A.L.; Contreras, J.E. Mechanism of gating by calcium in connexin hemichannels. Proc. Natl. Acad. Sci. USA 2016, 113, E7986–E7995. [Google Scholar] [CrossRef]
- Lopez, W.; Gonzalez, J.; Liu, Y.; Harris, A.L.; Contreras, J.E. Insights on the mechanisms of Ca2+ regulation of connexin26 hemichannels revealed by human pathogenic mutations (D50N/Y). J. Gen. Physiol. 2013, 142, 23–35. [Google Scholar] [CrossRef]
- Sanchez, H.A.; Villone, K.; Srinivas, M.; Verselis, V.K. The D50N mutation and syndromic deafness: Altered Cx26 hemichannel properties caused by effects on the pore and intersubunit interactions. J. Gen. Physiol. 2013, 142, 3–22. [Google Scholar] [CrossRef]
- Müller, D.J.; Hand, G.M.; Engel, A.; Sosinsky, G.E. Conformational changes in surface structures of isolated connexin 26 gap junctions. EMBO J. 2002, 21, 3598–3607. [Google Scholar] [CrossRef]
- Turin, L.; Warner, A.E. Intracellular pH in early Xenopus embryos: Its effect on current flow between blastomeres. J. Physiol. 1980, 300, 489–504. [Google Scholar] [CrossRef]
- Spray, D.C.; Harris, A.L.; Bennett, M.V. Gap junctional conductance is a simple and sensitive function of intracellular pH. Science 1981, 211, 712–715. [Google Scholar] [CrossRef]
- Locke, D.; Bian, S.; Li, H.; Harris, A.L. Post-translational modifications of connexin26 revealed by mass spectrometry. Biochem. J. 2009, 424, 385–398. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Bezanilla, F. Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 1977, 70, 567–590. [Google Scholar] [CrossRef]
- Hoshi, T.; Zagotta, W.N.; Aldrich, R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 1990, 250, 533–538. [Google Scholar] [CrossRef]
- Zhou, M.; Morais-Cabral, J.H.; Mann, S.; MacKinnon, R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 2001, 411, 657–661. [Google Scholar] [CrossRef]
- Oshima, A.; Tani, K.; Toloue, M.M.; Hiroaki, Y.; Smock, A.; Inukai, S.; Cone, A.; Nicholson, B.J.; Sosinsky, G.E.; Fujiyoshi, Y. Asymmetric configurations and N-terminal rearrangements in connexin26 gap junction channels. J. Mol. Biol. 2011, 405, 724–735. [Google Scholar] [CrossRef][Green Version]
- Beyer, E.C.; Lipkind, G.M.; Kyle, J.W.; Berthoud, V.M. Structural organization of intercellular channels II. Amino terminal domain of the connexins: Sequence, functional roles, and structure. Biochim. Biophys. Acta 2012, 1818, 1823–1830. [Google Scholar] [CrossRef]
- Batir, Y.; Bargiello, T.A.; Dowd, T.L. Structural studies of N-terminal mutants of Connexin 26 and Connexin 32 using (1)H NMR spectroscopy. Arch. Biochem. Biophys. 2016, 608, 8–19. [Google Scholar] [CrossRef]
- Purnick, P.E.; Benjamin, D.C.; Verselis, V.K.; Bargiello, T.A.; Dowd, T.L. Structure of the amino terminus of a gap junction protein. Arch. Biochem. Biophys. 2000, 381, 181–190. [Google Scholar] [CrossRef]
- Bukauskas, F.F.; Verselis, V.K. Gap junction channel gating. Biochim. Biophys. Acta 2004, 1662, 42–60. [Google Scholar] [CrossRef]
- Oh, S.; Rivkin, S.; Tang, Q.; Verselis, V.K.; Bargiello, T.A. Determinants of gating polarity of a connexin 32 hemichannel. Biophys. J. 2004, 87, 912–928. [Google Scholar] [CrossRef]
- Oh, S.; Verselis, V.K.; Bargiello, T.A. Charges dispersed over the permeation pathway determine the charge selectivity and conductance of a Cx32 chimeric hemichannel. J. Physiol. 2008, 586, 2445–2461. [Google Scholar] [CrossRef]
- Purnick, P.E.; Oh, S.; Abrams, C.K.; Verselis, V.K.; Bargiello, T.A. Reversal of the gating polarity of gap junctions by negative charge substitutions in the N-terminus of connexin 32. Biophys. J. 2000, 79, 2403–2415. [Google Scholar] [CrossRef]
- Tong, J.J.; Liu, X.; Dong, L.; Ebihara, L. Exchange of gating properties between rat cx46 and chicken cx45.6. Biophys. J. 2004, 87, 2397–2406. [Google Scholar] [CrossRef]
- Peracchia, C.; Peracchia, L.L. Inversion of both gating polarity and CO2 sensitivity of voltage gating with D3N mutation of Cx50. Am. J. Physiol. Cell Physiol. 2005, 288, C1381–C1389. [Google Scholar] [CrossRef]
- Xin, L.; Nakagawa, S.; Tsukihara, T.; Bai, D. Aspartic acid residue D3 critically determines Cx50 gap junction channel transjunctional voltage-dependent gating and unitary conductance. Biophys. J. 2012, 102, 1022–1031. [Google Scholar] [CrossRef]
- Yue, B.; Haddad, B.G.; Khan, U.; Chen, H.; Atalla, M.; Zhang, Z.; Zuckerman, D.M.; Reichow, S.L.; Bai, D. Connexin 46 and connexin 50 gap junction channel properties are shaped by structural and dynamic features of their N-terminal domains. J. Physiol. 2021, 599, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Kraujalis, T.; Gudaitis, L.; Kraujaliene, L.; Snipas, M.; Palacios-Prado, N.; Verselis, V.K. The Amino Terminal Domain and Modulation of Connexin36 Gap Junction Channels by Intracellular Magnesium Ions. Front. Physiol. 2022, 13, 839223. [Google Scholar] [CrossRef]
- Sanchez, H.A.; Slavi, N.; Srinivas, M.; Verselis, V.K. Syndromic deafness mutations at Asn 14 differentially alter the open stability of Cx26 hemichannels. J. Gen. Physiol. 2016, 148, 25–42. [Google Scholar] [CrossRef]
- Valdez Capuccino, J.M.; Chatterjee, P.; Garcia, I.E.; Botello-Smith, W.M.; Zhang, H.; Harris, A.L.; Luo, Y.; Contreras, J.E. The connexin26 human mutation N14K disrupts cytosolic intersubunit interactions and promotes channel opening. J. Gen. Physiol. 2019, 151, 328–341. [Google Scholar] [CrossRef] [PubMed]
- García, I.E.; Villanelo, F.; Contreras, G.F.; Pupo, A.; Pinto, B.I.; Contreras, J.E.; Perez-Acle, T.; Alvarez, O.; Latorre, R.; Martinez, A.D.; et al. The syndromic deafness mutation G12R impairs fast and slow gating in Cx26 hemichannels. J. Gen. Physiol. 2018, 150, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Oshima, A.; Tani, K.; Hiroaki, Y.; Fujiyoshi, Y.; Sosinsky, G.E. Projection structure of a N-terminal deletion mutant of connexin 26 channel with decreased central pore density. Cell Commun. Adhes. 2008, 15, 85–93. [Google Scholar] [CrossRef]
- Shao, Q.; Liu, Q.; Lorentz, R.; Gong, X.Q.; Bai, D.; Shaw, G.S.; Laird, D.W. Structure and functional studies of N-terminal Cx43 mutants linked to oculodentodigital dysplasia. Mol. Biol. Cell 2012, 23, 3312–3321. [Google Scholar] [CrossRef]
- Drożdżyk, K.; Sawicka, M.; Bahamonde-Santos, M.I.; Jonas, Z.; Deneka, D.; Albrecht, C.; Dutzler, R. Cryo-EM structures and functional properties of CALHM channels of the human placenta. Elife 2020, 9, e55853. [Google Scholar] [CrossRef]
- Syrjanen, J.L.; Michalski, K.; Chou, T.H.; Grant, T.; Rao, S.; Simorowski, N.; Tucker, S.J.; Grigorieff, N.; Furukawa, H. Publisher Correction: Structure and assembly of calcium homeostasis modulator proteins. Nat. Struct. Mol. Biol. 2020, 27, 305. [Google Scholar] [CrossRef]
- Burendei, B.; Shinozaki, R.; Watanabe, M.; Terada, T.; Tani, K.; Fujiyoshi, Y.; Oshima, A. Cryo-EM structures of undocked innexin-6 hemichannels in phospholipids. Sci. Adv. 2020, 6, eaax3157. [Google Scholar] [CrossRef] [PubMed]
- Brohawn, S.G.; Campbell, E.B.; MacKinnon, R. Physical mechanism for gating and mechanosensitivity of the human TRAAK K+ channel. Nature 2014, 516, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Brohawn, S.G. How ion channels sense mechanical force: Insights from mechanosensitive K2P channels TRAAK, TREK1, and TREK2. Ann. N. Y. Acad. Sci. 2015, 1352, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Rossi, A.R.; Harris, A.L. Computational Studies of Molecular Permeation through Connexin26 Channels. Biophys. J. 2016, 110, 584–599. [Google Scholar] [CrossRef]
- Jiang, W.; Lin, Y.C.; Botello-Smith, W.; Contreras, J.E.; Harris, A.L.; Maragliano, L.; Luo, Y.L. Free energy and kinetics of cAMP permeation through connexin26 via applied voltage and milestoning. Biophys. J. 2021, 120, 2969–2983. [Google Scholar] [CrossRef]
- Harris, A.L.; Locke, D. Permeability of Connexin Channels. In Connexins: A Guide; Harris, A.L., Locke, D., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 165–206. [Google Scholar]
- Srinivas, M.; Costa, M.; Gao, Y.; Fort, A.; Fishman, G.I.; Spray, D.C. Voltage dependence of macroscopic and unitary currents of gap junction channels formed by mouse connexin50 expressed in rat neuroblastoma cells. J. Physiol. 1999, 517 Pt 3, 673–689. [Google Scholar] [CrossRef]
- Bukauskas, F.F. Neurons and β-cells of the pancreas express connexin36, forming gap junction channels that exhibit strong cationic selectivity. J. Membr. Biol. 2012, 245, 243–253. [Google Scholar] [CrossRef]
- Veenstra, R.D. Size and selectivity of gap junction channels formed from different connexins. J. Bioenerg. Biomembr. 1996, 28, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Bukauskas, F.F.; Bukauskiene, A.; Verselis, V.K. Conductance and permeability of the residual state of connexin43 gap junction channels. J. Gen. Physiol. 2002, 119, 171–185. [Google Scholar] [CrossRef]
- Oh, S.; Abrams, C.K.; Verselis, V.K.; Bargiello, T.A. Stoichiometry of transjunctional voltage-gating polarity reversal by a negative charge substitution in the amino terminus of a connexin32 chimera. J. Gen. Physiol. 2000, 116, 13–31. [Google Scholar] [CrossRef]
- Kopec, W.; Rothberg, B.S.; de Groot, B.L. Molecular mechanism of a potassium channel gating through activation gate-selectivity filter coupling. Nat. Commun. 2019, 10, 5366. [Google Scholar] [CrossRef] [PubMed]
- Spray, D.C.; Harris, A.L.; Bennett, M.V. Equilibrium properties of a voltage-dependent junctional conductance. J. Gen. Physiol. 1981, 77, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L.; Spray, D.C.; Bennett, M.V. Kinetic properties of a voltage-dependent junctional conductance. J. Gen. Physiol. 1981, 77, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Bukauskas, F.F.; Bukauskiene, A.; Bennett, M.V.; Verselis, V.K. Gating properties of gap junction channels assembled from connexin43 and connexin43 fused with green fluorescent protein. Biophys. J. 2001, 81, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Paulauskas, N.; Pranevicius, M.; Pranevicius, H.; Bukauskas, F.F. A stochastic four-state model of contingent gating of gap junction channels containing two “fast” gates sensitive to transjunctional voltage. Biophys. J. 2009, 96, 3936–3948. [Google Scholar] [CrossRef] [PubMed]
- Trexler, E.B.; Bennett, M.V.; Bargiello, T.A.; Verselis, V.K. Voltage gating and permeation in a gap junction hemichannel. Proc. Natl. Acad. Sci. USA 1996, 93, 5836–5841. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Rubin, J.B.; Bennett, M.V.; Verselis, V.K.; Bargiello, T.A. Molecular determinants of electrical rectification of single channel conductance in gap junctions formed by connexins 26 and 32. J. Gen. Physiol. 1999, 114, 339–364. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Voltage-sensing and substate rectification: Moving parts of connexin channels. J. Gen. Physiol. 2002, 119, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Verselis, V.K.; Srinivas, M. Divalent cations regulate connexin hemichannels by modulating intrinsic voltage-dependent gating. J. Gen. Physiol. 2008, 132, 315–327. [Google Scholar] [CrossRef]
- Ebihara, L.; Liu, X.; Pal, J.D. Effect of external magnesium and calcium on human connexin46 hemichannels. Biophys. J. 2003, 84, 277–286. [Google Scholar] [CrossRef]
- Verselis, V.K.; Trelles, M.P.; Rubinos, C.; Bargiello, T.A.; Srinivas, M. Loop gating of connexin hemichannels involves movement of pore-lining residues in the first extracellular loop domain. J. Biol. Chem. 2009, 284, 4484–4493. [Google Scholar] [CrossRef] [PubMed]
- Kühlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y. Single-particle cryo-EM-How did it get here and where will it go. Science 2018, 361, 876–880. [Google Scholar] [CrossRef]
- Callaway, E. Revolutionary cryo-EM is taking over structural biology. Nature 2020, 578, 201. [Google Scholar] [CrossRef]
- Callaway, E. The revolution will not be crystallized: A new method sweeps through structural biology. Nature 2015, 525, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Papasergi-Scott, M.M.; Pérez-Hernandez, G.; Batebi, H.; Gao, Y.; Eskici, G.; Seven, A.B.; Panova, O.; Hilger, D.; Casiraghi, M.; He, F.; et al. Time-resolved cryo-EM of G protein activation by a GPCR. bioRxiv 2023. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Jin, X.; Medina, C.B.; Leonhardt, S.A.; Kiessling, V.; Bennett, B.C.; Shu, S.; Tamm, L.K.; Yeager, M.; Ravichandran, K.S.; et al. A quantized mechanism for activation of pannexin channels. Nat. Commun. 2017, 8, 14324. [Google Scholar] [CrossRef]
- Adair, B.D.; Xiong, J.P.; Yeager, M.; Arnaout, M.A. Cryo-EM structures of full-length integrin αIIbβ3 in native lipids. Nat. Commun. 2023, 14, 4168. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Zhao, C.; MacKinnon, R. Membrane protein isolation and structure determination in cell-derived membrane vesicles. Proc. Natl. Acad. Sci. USA 2023, 120, e2302325120. [Google Scholar] [CrossRef]
- Singh, D.; Lampe, P.D. Identification of connexin-43 interacting proteins. Cell Commun. Adhes. 2003, 10, 215–220. [Google Scholar] [CrossRef]
- Young, L.N.; Villa, E. Bringing structure to cell biology with cryo-electron tomography. Annu. Rev. Biophys. 2023, 52, 573–595. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Corzo, C.; Chang, M.R.; Shang, J.; Lam, V.Q.; Brust, R.; Blayo, A.L.; Bruning, J.B.; Kamenecka, T.M.; Kojetin, D.J.; et al. Chemical Crosslinking Mass spectrometry reveals the conformational landscape of the activation helix of PPARgamma; a model for ligand-dependent antagonism. Structure 2018, 26, 1431–1439.e6. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Strutzenberg, T.; Pascal, B.D.; Griffin, P.R. Protein dynamics and conformational changes explored by hydrogen/deuterium exchange mass spectrometry. Curr. Opin. Struct. Biol. 2019, 58, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Lerch, M.T.; Yang, Z.; Brooks, E.K.; Hubbell, W.L. Mapping protein conformational heterogeneity under pressure with site-directed spin labeling and double electron-electron resonance. Proc. Natl. Acad. Sci. USA 2014, 111, E1201–E1210. [Google Scholar] [CrossRef] [PubMed]
- Dalmas, O.; Hyde, H.C.; Hulse, R.E.; Perozo, E. Symmetry-constrained analysis of pulsed double electron-electron resonance (DEER) spectroscopy reveals the dynamic nature of the KcsA activation gate. J. Am. Chem. Soc. 2012, 134, 16360–16369. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Huang, C.; Emery, B.P.; Sedgwick, A.C.; Bull, S.D.; He, X.P.; Tian, H.; Yoon, J.; Sessler, J.L.; James, T.D. Forster resonance energy transfer (FRET)-based small-molecule sensors and imaging agents. Chem. Soc. Rev. 2020, 49, 5110–5139. [Google Scholar] [CrossRef] [PubMed]
- Stryer, L.; Haugland, R.P. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. USA 1967, 58, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Russel, D.; Lasker, K.; Webb, B.; Velázquez-Muriel, J.; Tjioe, E.; Schneidman-Duhovny, D.; Peterson, B.; Sali, A. Putting the pieces together: Integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 2012, 10, e1001244. [Google Scholar] [CrossRef] [PubMed]
- Sali, A. From integrative structural biology to cell biology. J. Biol. Chem. 2021, 296, 100743. [Google Scholar] [CrossRef]
- Purdy, M.D.; Bennett, B.C.; McIntire, W.E.; Khan, A.K.; Kasson, P.M.; Yeager, M. Function and dynamics of macromolecular complexes explored by integrative structural and computational biology. Curr. Opin. Struct. Biol. 2014, 27, 138–148. [Google Scholar] [CrossRef]
- Acosta, M.L.; Mat Nor, M.N.; Guo, C.X.; Mugisho, O.O.; Coutinho, F.P.; Rupenthal, I.D.; Green, C.R. Connexin therapeutics: Blocking connexin hemichannel pores is distinct from blocking pannexin channels or gap junctions. Neural Regen. Res. 2021, 16, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W.; Lampe, P.D. Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 2018, 17, 905–921. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cx | Gene | Structures | Technique | Res. [A] | Symmetry | Solvent | Conformation | Features | Gating | Mutations | Refs. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 26 | GJB2 | GJC | X-ray | 3.5 | C2, C6 NCS | Detergent: UDM | Open | NT | [30] | ||
| 26 | GJB2 | GJC | X-ray | 3.3–3.8 | C2, C3 | Detergent: FA3 | Open, Ca2+-bound | Ca2+ | [13] | ||
| 26 | GJB2 | GJC, pseudo-HC | CryoEM | 1.9–2.2 | C6, D6 | Detergent: DDM | Partially closed | NT, lipids or detergents, waters | PCO2 | [34] | |
| 26 | GJB2 | GJC | CryoEM | 4–7.5 | D6 | Amphipol: A8–35 | Open, Closed | NT | pH | [14] | |
| 26 | GJB2 | HC | CryoEM | 4.2 | C6 | Nanodisc: Soy Lipids | Open | N176Y | [35] | ||
| 31.3 | GJC3 | HC | CryoEM | 2.3–2.6 | C6 | Detergent: LMNG | Partially closed | NT, lipids or detergent, waters | Ca2+ | R15G | [36] |
| 32 | GJB1 | GJC, HC | CryoEM | 2.1–3.7 | D6, C6 | Detergent: Digitonin | Open, Partially closed | NT, lipids or detergent, waters | W3S, R22G | [37] | |
| 36 | GJD2 | GJC, asymmetric GJC | CryoEM | 2.2–7.2 | D6, C6, C1 | Detergent: LMNG/CHS Nanodisc: Soy Lipids | Open, Lipid-occluded | NT, lipids or detergent, waters | +BRIL Δ1–8 +BRILΔ1–16 | [38] | |
| 43 | GJA1 | GJC, asymmetric GJC, pseudo-HC | CryoEM | 2.4–4 | D6, C6, C1 | Detergent: LMNG/CHS, GDN Nanodisc: Soy Lipids, POPE/CHS | Open, Partially closed | NT, lipids or detergent, waters | Δ257–382 | [39] | |
| 43 | GJA1 | GJC, HC | CryoEM | 2.3–4 | D6, C6 | Detergent: Digitonin Nanodisc: POPC | Partially closed | NT, lipids or detergent, | [40] | ||
| 46/50 | GJA3, GJA8 | GJC | CryoEM | 3.4–3.5 | D6 | Amphipol: A8–35 | Open | NT | [33] | ||
| 46/50 | GJA3, GJA8 | GJC | CryoEM | 1.9–2.5 | D6 | Nanodisc: DMPC | Open | NT, lipids or detergent, waters | [41] |
| AA Number | (β) | NT Conformations | pI | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AA Number | (α) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | |||
| Cx26 | GJB2 | O, PC | 8.4 | M | - | D | W | G | T | L | Q | T | I | L | G | G | - | V | N | K | H | S | T | S | I | G | K |
| Cx31.3 | GJC3 | PC | 11.7 | M | - | C | G | R | F | L | R | R | L | L | A | E | - | E | S | R | R | S | T | P | V | G | R |
| Cx32 | GJB1 | O, PC | 10.8 | M | - | N | W | T | G | L | Y | T | L | L | S | G | - | V | N | R | H | S | T | A | I | G | R |
| Cx36 | GJD2 | O | 5.5 | M | G | E | W | T | I | L | E | R | L | L | E | A | A | V | Q | Q | H | S | T | M | I | G | R |
| Cx43 | GJA1 | O, PC | 8.2 | M | G | D | W | S | A | L | G | K | L | L | D | K | - | V | Q | A | Y | S | T | A | G | G | K |
| Cx46 | GJA3 | O | 5.4 | M | G | D | W | S | F | L | G | R | L | L | E | N | - | A | Q | E | H | S | T | V | I | G | K |
| Cx50 | GJA8 | O | 4.4 | M | G | D | W | S | F | L | G | N | I | L | E | E | - | V | N | E | H | S | T | V | I | G | R |
| Open Conformation | |||
|---|---|---|---|
| Cx Isoform | Pore Diameter (Å) | GJ/HC | References |
| Cx26 | 15 | GJ | [30] |
| Cx32 | 15 | HC | [37] |
| Cx36 | 15 | GJ | [38] |
| Cx43 | 14 | GJ | [39] |
| Cx46 | 16 | GJ | [41] |
| Cx50 | 15 | GJ | [41] |
| Partially closed conformation | |||
| Cx31.3 | 12 | HC | [36] |
| Cx32 W3S | 10 | HC | [37] |
| Cx32 R22G | 10 | HC | [37] |
| Cx36 | 11 | GJ | [38] |
| Cx43 | 10 | GJ | [39] |
| Cx43 | 10 | GJ | [39] |
| Cx43 | 10 | GJ | [40] |
| Cx46 | 10 | GJ | [41] |
| Cx50 | 9 | HC | [41] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jagielnicki, M.; Kucharska, I.; Bennett, B.C.; Harris, A.L.; Yeager, M. Connexin Gap Junction Channels and Hemichannels: Insights from High-Resolution Structures. Biology 2024, 13, 298. https://doi.org/10.3390/biology13050298
Jagielnicki M, Kucharska I, Bennett BC, Harris AL, Yeager M. Connexin Gap Junction Channels and Hemichannels: Insights from High-Resolution Structures. Biology. 2024; 13(5):298. https://doi.org/10.3390/biology13050298
Chicago/Turabian StyleJagielnicki, Maciej, Iga Kucharska, Brad C. Bennett, Andrew L. Harris, and Mark Yeager. 2024. "Connexin Gap Junction Channels and Hemichannels: Insights from High-Resolution Structures" Biology 13, no. 5: 298. https://doi.org/10.3390/biology13050298
APA StyleJagielnicki, M., Kucharska, I., Bennett, B. C., Harris, A. L., & Yeager, M. (2024). Connexin Gap Junction Channels and Hemichannels: Insights from High-Resolution Structures. Biology, 13(5), 298. https://doi.org/10.3390/biology13050298